药物化学第二章-药物设计的基本原理和方法
药物化学
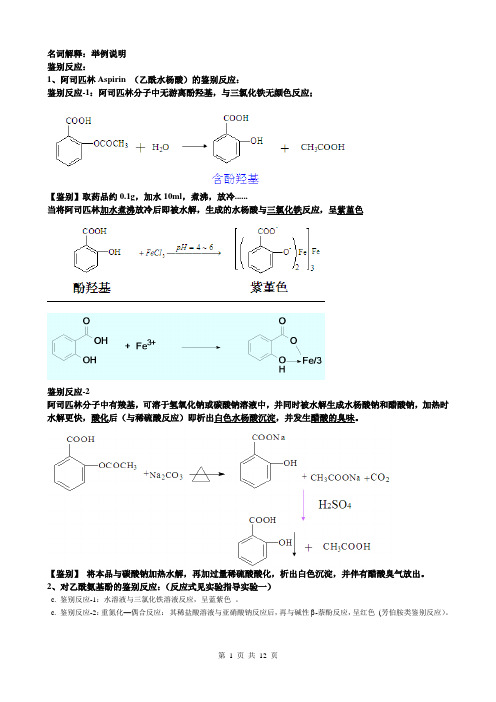
名词解释:举例说明鉴别反应:1、阿司匹林Aspirin (乙酰水杨酸)的鉴别反应:鉴别反应-1:阿司匹林分子中无游离酚羟基,与三氯化铁无颜色反应;【鉴别】取药品约0.1g,加水10ml,煮沸,放冷......当将阿司匹林加水煮沸放冷后即被水解,生成的水杨酸与三氯化铁反应,呈紫堇色鉴别反应-2阿司匹林分子中有羧基,可溶于氢氧化钠或碳酸钠溶液中,并同时被水解生成水杨酸钠和醋酸钠,加热时水解更快,酸化后(与稀硫酸反应)即析出白色水杨酸沉淀,并发生醋酸的臭味。
【鉴别】将本品与碳酸钠加热水解,再加过量稀硫酸酸化,析出白色沉淀,并伴有醋酸臭气放出。
2、对乙酰氨基酚的鉴别反应:(反应式见实验指导实验一)c. 鉴别反应-1:水溶液与三氯化铁溶液反应,呈蓝紫色。
c. 鉴别反应-2:重氮化—偶合反应:其稀盐酸溶液与亚硝酸钠反应后,再与碱性 -萘酚反应,呈红色(芳伯胺类鉴别反应)。
3、羟基保泰松(3,5-吡唑烷二酮类)的鉴别反应: 酸水解后重排,呈芳伯氨反应,与亚硝酸钠试液作用生成黄色重氮盐,再与β-萘酚偶合生成橙色沉淀4、 吲哚美辛的鉴别反应:本品的氢氧化钠溶液与重铬酸钾溶液和硫酸反应,呈紫色 ;(芳基乙酸类) 与亚硝酸钠和盐酸反应,呈绿色,放置后渐变黄色。
5、吗啡的鉴别反应:(1) 吗啡+中性FeCl 3 →蓝色 (2)吗啡+铁氰化钾→伪吗啡 + 亚铁氰化钾亚铁氰化铁 —— 蓝色可待因无此反应,可用作鉴别(3) 吗啡+甲醛硫酸试液→蓝紫色(Marquis 反应)(4) 吗啡+钼酸铵硫酸溶液→紫色,继变蓝色,最后变为绿色(Fröhde 反应)(5)对吗啡要进行杂质的限量检查6、肾上腺素(Epinephrine )的鉴别反应:(1)溶于稀盐酸后,与过氧化氢试液反应被氧化,显血红色。
(2)在pH3-3.5时与碘试液反应,再加硫代硫酸钠试液使过量碘的颜色消退,溶液呈红色。
(3)与三氯化铁试液反应,即显翠绿色(酚羟基与铁离子络合呈色);再加氨试液后变为紫色,最后变为紫红色。
药物设计基本原理和方法

❖ 主要内容: 先导化合物的发现 先导化合的优化
一、新药开发的两阶段
❖先导化合物的发现 (Lead Generation) ❖先导化合物优化
NCE
(Lead Optimization)
两者相辅相成
lead discovery
lead optimization
先导化合物Lead compound
❖ 经研究后发现是由于异烟肼具有 抑制单胺氧化酶的副作用,于是 以异烟肼为先导化合物,发展了 单胺氧化酶抑制剂类抗抑郁药, 异丙烟肼是其中一例。
CONHNH2
N 异 烟 肼 Iso n ia z id
C O N H N H C H (C H 3 )2
N 异 丙 烟 肼 Ip r o n ia z id
❖The structure of the lead compound is then modified by synthesis to amplify the desired activity and to minimize or eliminate the unwanted properties.
❖原型药物(Prototype Drug) ❖ 随之出现了大量的“Me-too”药物
A me-too drug is a compound that is structurally very similar to already known drugs, with only minor pharmacological differences.
二、先导化合物的发现
Approaches for lead discovery
1 改进和优化已有药物 2 筛选途径 3 利用自然界生物资源 4 合理药物设计
药物化学考试重点

2019药物化学第二章新药研究的基本原理与方法一、先导化合物的发现(选择)1.天然产物:青蒿素、β内酰胺酶抑制剂克拉维酸、HMG-COA还原酶抑制剂他汀类、猪胰岛素2.现有药物:(1)副作用:氯丙嗪由抗组胺药异丙嗪镇静副作用发展而来;磺胺类降糖和利尿由抗菌药发展而来(2)代谢:羟布宗是保泰松的活性代谢物;奥沙西泮是地西泮的活性代谢物(3)现有突破性药物:me too ,兰索拉唑由奥美拉唑发展而来3.活性内源性物质:避孕药的先导化合物是甾体激素黄体酮;抗炎药吲哚美辛先导化合物是炎性介质5-羟色胺4.组合化学和高通量筛选5.计算机靶向筛选二、先导化合物的优化(简答)在新药研究过程中:发现的先导化合物可能存在某些缺陷如活性不够高,化学结构不稳定,毒性较大,选择性不高,药代动力学性质不合理等,需要对先导化合物进行结构修饰或改造,使之成为理想的药物,这一过程称为先导化合物的优化。
先导化合物的优化方法:传统的药物化学方法和现代的方法。
1.传统的药物化学方法1)利用生物电子等排体原理优化先导化合物生物电子等排体是具有相似的分子形状和体积、相似的电荷分布并由此表现出相似的物理性质(如疏水性),对同一靶标产生相似或拮抗的生物活性的分子或基团。
分为经典和非经典的生物电子等排体。
经典的生物电子等排包括外层价电子相同的原子或基团,元素周期表中同主族的元素,以及环等价体。
非经典的生物电子等排体是具有相似的空间排列、电性或其他性质的分子或基团,相互替换会产生相似或相反生物活性的分子或基团。
利用生物电子等排体对先导化合物中的某一个基团逐个进行替换得到一系列的新化合物,是药物化学家设计研究药物的经典方法,有许多成功例子。
例如将H2受体拮抗剂西味替丁 (aimetidine)结构中的咪唑环用呋喃环和噻唑环替换得到雷尼替丁( rnitidine )和法莫替丁 ( famotidine) ,它们的H2受体拮抗作用均比西咪替丁强。
2)通过前药设计优化先导化合物。
药物化学期末复习

绪论1、药物化学(Medicinal Chemistry)是关于药物的发现、发展和确证,并在分子水平上研究药物作用方式的一门学科。
2、药物是对疾病具有预防、治疗和诊断作用或用以调节机体生理功能的物质。
3、根据药物的来源和性质不同,可以分为中药或天然药物、化学药物和生物药物。
4、化学药物是一类既有药物的功效,同时又有确切的化学结构的物质。
5、药物化学的三个时期:以天然产物为主的发现时期、以合成药物为主的发展时期、药物分子设计时期。
6、1899年,阿司匹林上市,标志着药物化学学科的形成。
第一章:新药研究和开发概论1、新化学实体(New Chemical Entities)是指在以前的文献中没有报道过的新化合物。
而有可能成为药物的新化学实体则需要时能够以安全和有效的方法治疗疾病的新化合物。
2、通常新药的发现分为4个主要的阶段:靶分子的确定和选择、靶分子的优化、先导化合物的发现和先导化合物的优化。
3、药品质量的主要含义是:A、药物的疗效和毒副作用,B、药物的纯度。
4、药品质量标准中,有两个重要的指征:一是药物的纯度,即有效成分的含量;二是药物的杂质限度。
5、药物的商品名通常是针对药物的最终产品,即剂量和剂型已确定的含有一种或多种药物活性成分的药品。
含同样活性成分的同一药品,每个企业应有自己的商品名,不得冒用、顶替别人的药品商品名称。
6、药物的通用名:也称为国际非专利药品名称,是世界卫生组织推荐使用的名称,通常是指有活性的药物物质,而不是最终产品,因此是药学研究人员和医务人员使用的共同名称,所以一个药物只有一个药品通用名,比商品名使用起来更为方便。
第二章:药物设计的基本原理和方法1、目前新药设计的靶点集中在受体、酶、核酸、离子通道和基因等上。
2、先导化合物(Lead Compound):通过各种途径得到的具有一定生理活性的化学物质。
3、先导化合物的发现方法和途径:a、从天然产物活性成分中发现先导化合物;b、通过分子生物学途径发现先导化合物;c、通过随机机遇发现先导化合物;d、从代谢产物中发现先导化合物;e、从临床药物的副作用或者老药新用途中发现新药;f、从药物合成中间体中发现先导化合物;g、通过计算机辅助药物筛选寻找先导化合物。
第二章+新药研究的基本原理与方法

1
* 早期寻找新药的方法多是基于经验和尝 试,通过大量化合物的筛选与偶然发现。
* 随着生命科学的相关学科在上世纪后半 期的迅速发展,定量构效关系、合理药物 设计、计算机辅助药物设计、组合化学、 高通量筛选等新技术、新方法不断涌现, 新药设计学也应运而生。
•上•0午7:74时6 46分21秒
•23
第三种情况是只有特异性的优势构象才产生最大活性
第四种情况是等效构象(Conformational equivalence),又称 构象的等效性,是指药物没有相同的骨架,但有相同的药效团, 并有相同的药理作用和最广义的相似构象
•上•0午7:74时6 46分21秒
药物与受体以共价键结合时,形成不可逆复合物 在大多数情况下,药物与受体的结合是可逆的
•上•0午7:74时6 46分21秒
•14
2.药物的各功能基团对药效的影响
在基本母体上至少含有6种功能基,各功能基分 别有不同的性质,对其活性、毒性、药代动力学等可 产生不同的影响。
卤素是强的吸电子基,引入卤素,可影响药物的 电荷分布,从而增强与受体的电性结合作用
31
三、通过随机机遇发现 (From Accidentally discover )
1929年青霉素的发现
英国医生 Fleming发现已接种金黄色葡萄球菌的平面皿被霉 菌污染,污染物邻近细菌明显遭到溶菌。他联想到可能是霉菌 的代谢产物对金黄色葡萄球菌有抑制作用,因此把这种霉菌放 在培养液中培养,结果培养液有明显的抑制革兰氏阳性菌的作 用。从此揭开了青霉素研究的序幕。
药物化学第二章-药物设计的基本原理和方法

§ 2. 先导化合物的优化
Lead Optimization
先导化合物的优化
Izant等人于1984年首次提出反义寡核苷酸技术,该技术是根据
核酸间碱基互补原理,利用一小段外源性的人工或生物合成的特
异互补RNA或DNA片断,与靶细胞中的mRNA或DNA通过碱基
互补结合,通过这种寡核苷酸键抑制或封闭其基因的表达。与反
义寡核苷酸相似的是反义DNA,是用一小段人工会成的约8~23
碱基组成的脱氧核苷酸单链,与靶mRNA形成碱基配对的DNA-
S
可旋转键的数量不超过10个。(删去)
ADMET
ADMET (药物的吸收、分配、代谢、排泄 和毒性)药物动力学方法是当代药物设计和 药物筛选中十分重要的方法。
A:吸收 Absorption D:分配 Distribution M:代谢 Metabolism E :排泄 Excretion T: 毒性 Toxcity
3.综合技术平台
目前最快速的发现先导化合物的途径是被各国称为综合技术平台的方法, 简单说就是用液相串联质谱( LC MS/MS)作为化合物的分离和分析结构 的工具,与药理学、组合化学的高通量筛选、计算机辅助设计、分子生物学、 受体(酶)学,及化学基因组学等学科结合起来,可迅速而大量地确定具有 不同活性药物的基本母核(scaffold),作为先导化合物。
药物进入体内后发生的代谢过程实质上是药物在体内 发生的化学转化过程。 代谢失活:体内代谢的结果主要是产物降低或失去 活性,排出体外 代谢活化:有些药物却发生代谢产物活化或产生其 它新的作用,转化为保留活性、毒副作用小的代谢 物,这样的代谢产物可成为新的先导化合物。
药物化学第2章 新药研究的基本原理与方法题库

第2章新药研究的基本原理与方法选择题每题1分
第2章新药研究的基本原理与方法填空题1每空1分
填空题2 每空1分
填空题3每空1分
第2章新药研究的基本原理与方法概念题每题2分
第2章新药研究的基本原理与方法问答与讨论题每题6分
前列腺素E2(PGE2)为结晶固体,但室温稳定期短,几个月内可迅速分解,不稳定因素是C-11位羟基易在酸性条件下,发生消除反应生成前列腺素A2(PGA 2) 这也是其口服无效的主因。
请设计两种较为稳定的衍生物。
举例说明根据受体结构进行药物分子设计
HIV蛋白水解酶催化机理
根据催化机理设计的HIV蛋白水解酶抑制剂
第2章新药研究的基本原理与方法合成/代谢/反应/设计题每题6分。
药物化学知识点总结

友情提示。
▪总论部分1篇,4章,主要内容:▪第一章:药物化学及发展过程▪第二章:药效及药代▪第三章:药物分子设计的基本原理和方法▪第四章:药物的研发程序▪需要掌握的内容:▪ 1 基本概念先导化合物的来源、电子等排体、前药和生物前体、药物代谢、影响药效的因素,药效团等。
▪ 2 药物优化的基本程序、构效关系、定量构效关系。
▪需要了解的内容:▪ 1 药物开发的基本程序▪ 2 计算机技术在药物设计中的应用分子模拟,先导化合物的虚拟筛选,定量构效关系(2DQSAR、3DQSAR),Hansch分析法、Docking程序、CoMFA程序。
▪个论部分3篇,14章,主要内容:▪第2篇与中枢系统有关的药物▪第一章:麻醉药▪第二章:镇静催眠和抗癫痫药▪第三章:精神神经疾病治疗药物▪第四章:镇痛药▪需要掌握的内容:▪ 1 局麻药的结构类型,盐酸普鲁卡因、利多卡因的合成路线。
▪ 2 巴比妥类、苯二氮桌类催眠镇静药的作用靶点、结构特征。
苯巴比妥、地西泮的合成方法。
了解治疗癫痫病药物的种类。
▪ 3 掌握治疗精神病的代表性药物:氯丙嗪、奋乃静、氯普噻吨、氟哌啶醇、奥氮平的结构式及合成路线。
▪ 4 了解抗抑郁药的类型和主要药物。
▪ 5 了解吗啡类镇痛药物的简化过程,合成镇痛药的种类。
▪ 6 掌握盐酸哌替啶、芬太尼的合成路线。
▪第3篇作用靶点是外周组织、器官上的受体、功能酶等的各类药物。
▪第五章:非甾体抗炎药▪ 1 了解花生四烯酸的代谢途径,前列腺素、白三烯与炎症的关系,非甾体抗炎药的作用靶点。
▪ 2 掌握代表性药物阿司匹林、保泰松、双氯芬酸钠、吲哚美辛、布洛芬、萘普生、吡洛昔康的结构式及合成路线。
▪ 3 了解非甾体抗炎药的进展。
▪第六章:拟胆碱药和抗胆碱药▪ 1 了解胆碱的生化来源及生理作用,胆碱受体和疾病的关系,拟胆碱药物的用途。
▪ 2 掌握盐酸苯海索的合成方法,了解肌松药的基本结构。
▪第七章:作用于肾上腺素能受体的药物▪ 1 掌握内源性物质去甲肾上腺素、肾上腺素、多巴胺的结构式及构型,了解其来源和生理作用。
药物设计的基本原理和方法

所选择参数之间不能有相关性,要有比较大的差异,并且生物活性数据的变化幅度应大于一个对数单位(即大于10倍),否则得不到足够的信息;
2 所设计化合物的物理化学性质差异要大
Hansch方法的一般操作过程
*
Hansch方程除了研究定量构效关系外, 还能用来解释药物作用机理,推测和描述可能的受体模型,研究除活性以外的其他药代动力学定量关系
分子对接法(Docking)
通过生长、旋转等得到基本骨架,按照受体的腔穴,定出靶标边界,这是一级结构的生成。从有关数据库搜索与受体受点结合的原子或原子团,设计新的化合物
*
二、间接药物设计(Indirect Drug Design)
间接药物设计法 受体的三维结构并不清楚
以小分子的构效关系为基础,从一组小分子化合物的结构和生物活性数据出发,研究结构与活性关系的规律
是一种新药设计的研究方法,可以作为先导化合物优化的一种手段。也是计算机辅助药物设计的一个重要内容
A=f(C)
生物活性
化合物的结构特征
由于学科的限制,并没有成功地将此关系用于药物设计
发展建立了三种定量构效关系的研究方法
*
20世纪 60年代 定量构效关系: 并根据信息进一步 结构参数 找出结构与活性间的
分子疏水性参数IogP,即分子的脂水分配系数(partition coefficient),表示分子的疏水性
化合物在有机相和水相中分配平衡时的量(摩尔)浓度Co和CW之比值,P=CO/CW
P值一般较大,常用IogP表示
当分子中有该取代基时I为1,当分子中没有该取代基时I为0。
logP的测定:
*
药物化学第二章思维导图

第二章新药研究的基本原理与方法概论新药上市临床前药学研究药效学药代动力学安全性等临床Ⅰ:健康志愿者Ⅱ:患者Ⅲ:大规模、多中心的临床实验新药发现治疗靶分子的确定和选择靶分子的优化先导化合物的发现先导化合物的优化药物的化学结构与生物活性的关系理化性质与生物活性脂水分配系数与生物活性酸碱性与生物活性解离度药物-受体相互作用化学键的作用化学键离子键氢键疏水键范德华力离子-偶极键及偶极-偶极键电荷转移复合物金属配合物立体化学的作用几何异构光学异构构象异构官能团的作用烷基卤素羟基与巯基磺酸基和羧基氨基和酰胺基醚键药物产生药效的两个主要决定因素:药物的理化性质以及药物和受体的相互作用先导化合物的发现从天然产物得到先导化合物植物微生物海洋动植物爬行类两栖类动物例:从植物黄花蒿中分离出含有过氧桥的倍半萜内酯化合物以现有药物作为先导化合物由药物副作用发现先导化合物通过药物的代谢研究发现先导化合物以现有突破性药物作为先导化合物用活性内源性物质作为先导化合物内源性物质神经递质受体酶利用组合化学和高通量筛选得到先导化合物先导化合物的优化现代的方法传统的药物化学方法生物电子等排体经典非经典前药设计分类载体前药生物前药目的和应用提高生物利用度和生物膜通透性提高前药的靶向性设计一个前药(部位指向性药物运输)设计一种前药(部位特异性药物释放)改善药物的水溶性、稳定性、克服不良气味或理化性质以适应制剂的需要软药设计定量构效关系计算机辅助设计自学。
药物设计的基本原理和方法 PPT

先导化合物发现的方法和途径
以现有的药物作为先导化合物
1.由药物副作用发现先导化合物 基于抗结核药异烟肼的副作用,发展了单胺氧化酶抑制剂类抗抑郁
药,如异丙烟肼。
二、新药设计与开发
Drug design and development
先导化合物发现的方法和途径
二、新药设计与开发
Drug design and developmentΒιβλιοθήκη 先导化合物发现的方法和途径
利用计算机进行靶向筛选得到先导化合物
以生物靶点为基础,利用计算机软件对化合物进行靶向合理筛选和从 头设计已成为发现先导化合物的一个重要手段。
二、新药设计与开发
Drug design and development
二、新药设计与开发
Drug design and development
先导化合物发现的方法和途径
用活性内源物质作为先导化合物
根据对生理病理的了解来研究新药,通常是针对与该生理活动有关的 酶或受体来设计药物,被称为合理药物设计。内源性神经递质、受体或酶 的底物就是初始的先导化合物。例如以炎症介质5-羟色胺为先导化合物研 发了抗炎药吲哚美辛。
高通量筛选(High-throughput screening)是以随机筛选和广泛筛选 为基础的。高通量筛选是利用近二、三十年来生物化学、分子生物学、分 子药理学和生物技术的研究成果,将已阐明影响生命过程的一些环节的酶、 受体、离子通道等作为药物作用的靶标进行分离、纯化和鉴定,由此建立 分子、细胞水平的高特异性的体外筛选模型,具有灵敏度高、特异性强、 需用药量少、快速筛选的特点。
二、新药设计与开发
Drug design and development
第二章 药物设计的基本原理和方法

O 青蒿素
H H CH3 OCOCH2CH2COOH 青蒿琥酯
2.微生物来源
美伐他汀和洛伐他汀,来源于青霉菌属、红
曲霉菌和土曲霉菌,羟甲戊二酰辅酶A还原
酶抑制剂的Lead Compound。后开发了人工合 成的阿托伐他汀(No. 1)
HO O H3C H3C H O H O H CH3 H3C H3C
NH2 N HO N O O HO HO
CHO HO OH OH CH2OH HO O O HO N
NH
. HCl
NH3.H2O MeOH
. HCl
N
D-阿拉伯糖
安西他滨
阿糖胞苷
第二节 分子的互补性-先导化合物的 发现
又称:基于结构的药物设计 一、分子识别 配体与受体相互作用的本质是分子识别。 分子识别是由于两个分子的多个特定的原子或基团性质的 互补性和空间的适配所驱动的,这种特异性的本质是双方 的互补性。 药物与受体的分子识别和相互作用,大都形成非共价键作 用,与维持机体的生物大分子的空间结构的键合力在本质 上是相似的(氢键、静电引力和疏水作用)。 药物和受体分子构象变化,诱导契合。
H2N
N=N NH2
SO2NH2
H2N
SO2NH2
活性代谢产物---磺胺 先导化合物
H2N
SO2NH N O CH3
磺胺甲恶唑 磺胺类抗菌药
从代谢产物开发的新药:
(1)非索那定
(2)诺阿司咪唑
(3)地氯雷他定
五、从临床药物的副作用或者老药新用中发(From SideEffect or New Purpose of Medicine)
1、 1929年青霉素的发现
2、异丙肾上腺素:β -受体激动剂,结构改造,发现 β -受体阻断剂--普萘洛尔,
药物化学与药物合成

药物化学与药物合成药物化学作为一门独立的学科,致力于研究药物的化学结构以及其在化学反应中的性质与行为。
药物合成则是利用化学反应来合成新的药物分子,以满足药物学领域对于新药的需求。
本文将介绍药物化学的基本原理和方法,以及药物合成的相关技术。
一、药物化学的基本原理和方法1. 药物化学的定义与意义药物化学是研究药物与化学反应的关系,探索药物化学结构与活性的相互关联,以及通过化学手段改进药物分子的活性和属性。
药物化学的研究结果对药物设计和合成具有重要意义。
2. 药物分子的化学结构药物分子通常由多个功能团组成,这些功能团赋予了药物分子生物活性。
药物的分子结构对于其在生物体内的吸收、分布、代谢和排泄等性质起着决定性作用。
3. 药物的性质与行为药物分子的化学性质和物理性质决定了其在药物研发和应用中的表现。
例如,药物的溶解度、稳定性、光学活性等特性对于药物的研究和设计具有重要影响。
4. 药物化学的方法和技术药物化学研究常常采用合成化学、结构优化以及定量构效关系等方法和技术。
通过合理设计和合成化合物,进行活性测试和构效关系研究,以获得更加活性和具备良好生物利用度的药物分子。
二、药物合成的相关技术1. 药物合成的目标和挑战药物合成的目标是通过化学合成来制备具有特定活性的药物分子。
合成路线的设计和选择需要考虑效率、选择性、稳定性和经济性等因素,以克服药物合成中所面临的各种挑战。
2. 药物合成的关键步骤药物合成过程中的关键步骤包括反应的选择和优化、中间体的合成、保护基的引入与去除、立体选择性的控制等。
这些步骤需要合理的合成策略和有效的合成方法来实现。
3. 药物合成的新技术与进展近年来,药物合成领域涌现出许多新技术和方法,如催化化学、微流控技术、多组分反应和多步骤合成等。
这些新技术的应用使得药物合成更加高效、经济,并且有助于解决一些传统合成方法的困难和瓶颈。
4. 药物合成的绿色化与可持续发展随着对环境保护和可持续发展的要求日益提高,药物合成中的绿色合成概念也越来越重要。
药物化学(第二版)第2章 药物设计的基本原理和方法

先导化合物的优化可分为:
(1)传统的化学方法 (2)现代的方法: 包括CADD和3DQSAR(在第三章介绍)
先导化合物优化的一般方法
一、烷基链或环的结构改造
二、生物电子等排原理
三、前药原理
四、软药
五、硬药
六、孪药
七、定量构效关系研究
一、烷基链或环的结构改造
1、成环或开环 镇痛药吗啡→哌替啶
N
4 -苯 基 哌 啶 类
2、插烯原理(vinylogues)(烷基链局部减少双键 或引入双键) 抗癫痫药胡椒碱→桂皮酰胺类的衍生物
O O 胡 椒 碱 N O 桂 皮 酰 胺 衍 生 物 X O N H R
3、烃链的同系化原理 利福平(甲基哌嗪)→利福喷汀(环戊基哌嗪)
二、生物电子等排(Bioisosteris)
安西他滨(环胞苷)是阿糖胞苷的中间体, 后发现安西他滨不仅具有抗肿瘤作用,且 副作用轻,体内代谢比阿糖胞苷慢,故作 用时间长,治疗各种白血病。
C H O H O O H O H C H O H 2 H O N O O N N H
.H C l
. O N H 3H 2
N H 2 N H O O N O H O
发展为理想的药物,这一过程称为先导化
合物的优化。
第一节
先导化合物发现的方法和途径
Approaches for lead discovery
一、从天然药物的活性成分中获得
(From Active Component of Natural Resources)
(1)植物来源 p17
青蒿素:来自于中药黄花蒿,以此为先导 物,发现蒿甲醚、青蒿琥酯等; 紫杉醇:来自于红豆杉树皮,以此为先导 物,发现多西他赛等。
药物设计的基本原理和方法2-三节

第三节 先导化合物的优化 ( Lead Optimization)或称为药物设计
寻找
药物设计的目的
活性高、选择性强、毒副作用小的新药
在发现了先导化合物后,就要对先导化合物进行合理的结构修饰, 这种过程和方法称为先导化合物的优化(lead optimization)或称为 药物设计。
寻找
优化的目的
更理想的理化性质, 或者具更良好的药物动力学性质, 或者提高了生物利用度, 或者选择性更强而毒副作用减弱。
第一,用生物电子等排体替代时,往往可以得到相 似的药理活 性。通过药物设计可以得到新的化学实体或类似物。
如 N-甲基四氢吡啶甲酸甲酯, 3位是脂肪链状化合物,具有抗炎 活性。它的3位杂环状生物具有相同的抗炎活性.
2019/2/15
11
将组胺结构中的咪唑环分别用吡啶、吡唑、三唑环替代时, 生物活性没有改变,这四种含氮杂环互为生物电子等排体。
那么,插烯后的化合物表达为: A-BI=B2-EI=E2 减少双健及插烯规则后来被广泛用在会成上,在药物 设计中,常用来优化先导化合物。
当减少或插入一个及多个双键后,药物的构型、分子形状和性质发 生改变,可影响药物与受体的作用,进而对其生物活性产生影响。
2019/2/15 4
如胡椒碱是从民间验方得到的抗癫痫有效成分,全合成有 一定困难,经减少一个双键得到栓皮酰胺类的衍生物,合 成简单,而且增强了抗癫痫的活性。
2019/2/15
14
后面内容直接删除就行 资料可以编辑修改使用 资料可以编辑修改使用
主要经营:网络软件设计、图文设计制作、发布 广告等 公司秉着以优质的服务对待每一位客户,做到让 客户满意!
致力于数据挖掘,合同简历、论文写作、PPT设 计、计划书、策划案、学习课件、各类模板等方 方面面,打造全网一站式需求
Y02第二章—新药研究的基本原理与方法

(二)通过药物的代谢研究发现先导化合物
药物通过体内代谢过程,可能被活化,也可能被 失活,甚至转化成有毒的化合物。 在药物研究中,可以选择其活化形式或考虑可以 避免代谢失活或毒化的结构来作为药物研究的先 导物。
采用这类先导化合物,得到优秀的药物的可能性 较大,甚至直接得到比原来药物更好的药物。
优化,先导化合物的发现和先导化合物的优化四
个阶段。
2
临床试验概述
I期—健康志愿者(抗肿瘤药为患者);评价安全性、 耐受性、药代动力学性质等。 II期—患者;主要考察有效性,也考察适应症、治 疗方案以及安全性(不良反应)。 III期—患者;随机、双盲对照;大规模(300)、多 中心;考察有效性和安全性。 完成III期临床后,提交材料,进行新药注册申请 (New Drug Application, NDA)。 IV期—上市后应用研究阶段;考察在广泛使用条 件下的疗效和不良反应,评价在普通或者特殊人 群中使用的利益与风险关系以及改进给药剂量等。
32
载体前药原理是通过共价键把活性药物与载体连 接,从而改变药物的理化性质,然后在酶的作用 下释放出活性药物。
33
载体前药的制备通常是利用活性化合物和药物 分子中含有的极性官能团来合成前药。 含有醇或羧酸基团的药物,最常见的前药形式 是酯; 胺类可采用形成酰胺、亚胺、偶氮、胺甲基化 等形式来制备前药; 含羰基的药物可通过席夫碱(schiff base)、肟、 缩醛或缩酮等的形成来制备前药。
同时配合高通量筛选,寻找先导化合物。
20
高通量筛选是利用生物化学、分子生物学、分 子药理学和生物技术的研究成果,对已阐明影 响生命过程的一些环节的酶、受体、离子通道 等,被用作药物作用的靶标进行分离、纯化和 鉴定,由此建立起来的分子、细胞水平的高特 异性的体外筛选模型,具有灵敏度高、特异性 强、用药量少、快速筛选的特点。
药物设计原理

药物设计原理药物设计是指根据疾病的特性和机制,结合药物化学、药理学、生物化学等知识,设计和合成具有治疗作用的化合物。
药物设计的目标是找到既能有效治疗疾病又能减少不良反应的药物。
在药物设计的过程中,需要考虑分子的结构、亲和性、药代动力学、毒性等因素,以确保药物的安全性和有效性。
首先,药物设计需要充分理解疾病的发病机制和生物学特性。
只有深入了解疾病的本质,才能有针对性地设计药物。
例如,针对癌症的药物设计就需要了解癌细胞的生长特点和代谢途径,以便找到能够干扰癌细胞生长的药物靶点。
其次,药物设计需要考虑药物分子的结构特征。
药物分子的结构对其生物活性和药代动力学特性具有重要影响。
通过合理设计分子结构,可以提高药物的亲和性和选择性,降低毒性和不良反应。
例如,通过改变分子的立体构型或功能团的位置,可以影响药物与靶点的结合方式,从而改变药物的生物活性。
另外,药物设计还需要考虑药物的药代动力学特性。
药代动力学研究药物在体内的吸收、分布、代谢和排泄过程,这些过程直接影响药物的药效和毒性。
因此,在药物设计中需要考虑如何提高药物的生物利用度、延长药物在体内的半衰期,以及减少药物代谢和排泄的影响。
最后,药物设计需要进行严格的毒性评价和安全性评估。
药物的毒性是药物研发中需要重点关注的问题,合理设计药物结构和控制药物剂量可以降低药物的毒性。
此外,还需要对药物进行全面的安全性评估,确保药物在临床使用中不会出现严重的不良反应和毒性效应。
综上所述,药物设计是一个综合性的学科,需要深入理解疾病的生物学特性,合理设计药物分子的结构,考虑药物的药代动力学特性,以及进行严格的毒性评价和安全性评估。
只有在这些方面都得到充分考虑和实践,才能设计出安全有效的药物,为临床治疗提供更多的选择和希望。
药物化学作业【精选文档】

药物化学第一部分;绪论及药学基本概念6,2绪论第一章:新药研究与开发概论第二章:药物设计的基本原理和方法第三章:药物的结构和生物活性第四章:药物代谢第二部分:中枢神经系统用药9, 6第五章:镇定催眠药和抗癫痫药第六章:精神神经病治疗药第七章:神经退行性疾病治疗药第八章:镇痛药第三部分:外周神经用药10,8第九章:局部麻醉药第十章:拟胆碱和抗胆碱药第十一章:组胺受体拮抗剂及抗过敏和抗溃疡药第十二章:作用于肾上腺能受体的药物第四部分;与血有关疾病用药7, 4第十三章:抗高血压药和利尿药第十四章:心脏疾病药物和血脂调节药第十六章:降血糖药和骨质疏松治疗药第五部分:抗病毒及抗菌及抗寄生虫药9,7第十七章:合成抗菌药第十八章:抗病毒药第十九章:抗生素第二十一章:抗寄生虫药第六部分;其他7, 5第十五章:甾体激素药物第二十二章:非甾体抗炎药第二十章:抗肿瘤药第二十三章:维生素药物化学习题第一部分:绪论及药学基本概念 61. 什么是药物?什么是药物化学?药物化学的主要任务?2.什么是先导化合物?发现先导物主要有哪些途径,举出二种从天然产物中发现先导物的例子。
3.什么是前体药物(前药)举例说明。
4.应用前药原理进行化学结构修饰的目的主要有哪些?5.为什么药物的脂水分配系数在一定范围内,才能显示最佳的药效?6.为什么药物的解离度对药效有影响?7.药物分子中电子密度分布是否均匀,对药效有何影响?8.药物分子与受体作用的键合形式主要有哪些?9.立体异构对药效的影响主要包括那些?10.举出至少三个对映异构体的药理活性有显著差异的例子。
11. 什么是药物代谢?举例说明药物代谢所涉及的反应类型。
12. 药物代谢在药物开发中有哪些主要用途?第二部分:中枢神经系统用药91.写出苯二氮卓类的基本结构及化学命名2.地西泮在体内胃肠道发生什么样的化学变化?对其生物利用度有何影响?3.奥沙西泮与地西泮在化学结构上有何区别,如何进行鉴别?4.艾司唑仑化学结构与地西泮有何区别,对活性有何影响?5.写出巴比妥类药物的基本结构,为什么巴比妥酸无活性?6.为什么苯巴比妥显弱酸性,可与碱成盐?7.苯妥英钠属哪种结构类型?写出卡马西平的结构及用途?8.写出抗精神病药物主要结构类型,各举出一例药物。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
七、通过计算机辅助药物筛选寻找先导化合物
利用计算机辅助药物筛选的方法称为虚拟筛选(Virtual screening)。 用一系列“基于知识的滤片”对虚拟库“筛选”,以 “浓缩”出能够满足预定标准的化合物。 这些滤片包括类药性(drug like)、药代动力学性质、 毒性、知识产权问题以及与受体的互补性或与配体的相 似性等,是通过数据库搜寻和计算化学实现的。
CH3 CH3 COOH
动物
1965年,Ferreira从巴西毒蛇毒液中分离出 九肽替普罗肽(谷-色-脯-精-脯-谷-亮-脯-脯), 具有降血压作用,不能口服。 以此为先导物,发现了血管紧张素转化酶 (ACE)抑制剂卡托普利,开创了一类新的抗 高血压药物。
海洋生物
海葵毒素是从海洋生物中分离出的一种剧毒物质, 肽类毒素,具有强心作用,有望成为新型心血管药物 和抗癌化疗药物。
高通量筛选
分子水平和细胞水平的实验方法 (筛选模型)是实现高通量筛选药 物筛选的技术基础。 高通量筛选的特点
快速-每天筛选数万药次 微量-筛选样品需要量为微克级 灵敏-准确判断筛选样品的活性和 选择性(自动化操作系统) 经济-筛选费用低
2.反义寡核苷酸
反义寡核苷酸( Antisenseoligo nucleotides,ASON)是发现先 导化合物的新途径之一。 Izant等人于1984年首次提出反义寡核苷酸技术,该技术是根据 核酸间碱基互补原理,利用一小段外源性的人工或生物合成的特 异互补RNA或DNA片断,与靶细胞中的mRNA或DNA通过碱基 互补结合,通过这种寡核苷酸键抑制或封闭其基因的表达。与反 义寡核苷酸相似的是反义DNA,是用一小段人工会成的约8~23 碱基组成的脱氧核苷酸单链,与靶mRNA形成碱基配对的DNAmRNA 杂交链,从而封闭某一特定基因片段。 反义核酸
反义寡核苷酸(Antisense oligonucleotides)
反义寡核苷酸的分子大小是设计的重要环节。
12-25个碱基范围,15-20较佳,超过25难以通过细胞 膜
反义寡核苷酸作为药物的条件
制备方法简便、经济 具有一定的稳定性 具有较强的细胞通透性 能在靶细胞内保持一定的浓度 能在靶细胞内特点位点作用 不与其他生物大分子作用
植物
南美洲古柯叶
H3C N O O O H O CH3
可卡因(Cocaine)
普鲁卡因(Procaine)
HN H3C
H3C O O β-优卡因 N O O
NH2
CH3 β-Eucaine
植物
从中药青蒿中分离抗疟有效成分 青蒿素(Artemisinin)为新型结构的 倍半萜过氧化物
对耐疟原虫有极高的杀灭作用
抗肿瘤药物安西他滨(环胞苷)的发现
阿糖胞苷( Cytarabie)是干扰DNA合成的抗肿瘤药 物,其合成是以D-阿拉伯糖为起始原料,经多步反应生 成环胞苷,再用氨水开环得到的。后来发现其中间环胞苷 不仅具有抗肿瘤作用,且副作用轻,在体内代谢速度比阿 糖胞苷慢,故作用时间长,可用于各种白血病的治疗。
药物化学
第二章 药物设计的基本原理和方法
Basic Principles of Drug Design
本章主要内容
先导化合物的发现 先导化合的优化
药物设计
药物化学的根本任务是设计和发现新药。 新药设计的目的是寻找高效、低毒的新化学实体(NCE).
药物设计方法学
60年代前 经典的药物设计方法阶段 60年代后 分子水平阶段 (从细胞和分子水平认识疾病和药物作用机制)
百浪多息(Prontosil)
50多种磺胺类抗菌药
五、 从临床药物的副作用或者老药新用途中
发现新药
药物对机体有多种药理作用
用于治疗的称治疗作用 其他的作用通常称为毒副作用
药物的毒副作用可能对另一种疾病有治疗作用
可从已知药物的毒副作用出发找到新药 或将毒副作用与治疗作用分开而获得新药
一、从天然产物活性成分中发现先导物 天然生物活性物质来源广泛 植物 微生物 动物(陆地) 海洋生物 矿物
目前临床应用的不少药物是直接从植物 中提取到的。
可直接作为药物使用,同时又是良好的先导化合物, 可发展多种合成和半合成类的药物。
天然生物活性物质的特点
新颖的结构类型(分子多样性) 独特的药理活性 资源有限及地域性差异 有效成分含量很低 大多数结构复杂,作用强度不同
异丙嗪(抗过敏药)
氯丙嗪(安定)
吩噻嗪类抗精神病药物
六、从药物合成的中间体中发现先导化合物
一些药物合成的中间体,由于与目的化合物结构上有相似 性,经过筛选也可发现先导化合物。如早期在寻找抗结核 药物时,Fox设计合成硫代缩氨脲的衍生物,其合成路线 如下:
异烟醛与硫代氨基脲缩合得到目的物,将合成过程的中间体 异烟肼同时进行药理活性实验时,发现异烟肼的抗结核活性超过 目的物,故放弃目的物的研究,将异烟肼推上临床。
modified by synthesis to amplify the desired activity and to minimize or eliminate the unwanted properties.
§ 1. 先导化合物发现的方法和途径
Approaches for lead discovery
虚 拟 库 类 药 原 则 药 代 性 质
潜 在 毒 性
专 利 指 导
受 体 结 构
设 计 库
类药性
Lipinski归纳的“类药5规则”(Rule of Five),概括了类药的最低标准:
相对分子质量在500以下; 计算的脂水分配系数(正辛醇-水系统)ClogP值小于5; 氢键的给体不超过5个:X-H(OH、NH2、RNH) 氢键的接受体不超过10个:带孤对电子的N、O、F、S
基于结构的药物设计
血管紧张素转化酶(ACE)抑制剂
2-甲基丙酰基与S1′结合。
His
吡咯环与S2′结合
S1'
Glu O His N O Zn2+ HS NH O Glu Ser Arg O OH N NH H CH 3 N H O O H 2N NH O NH 2 HO
S2'
Ty r
巯基与Zn2+结 合
可旋转键的数量不超过10个。(删去)
ADMET
ADMET (药物的吸收、分配、代谢、排泄 和毒性)药物动力学方法是当代药物设计和 药物筛选中十分重要的方法。
A:吸收 D:分配 M:代谢 E :排泄 T: 毒性 Absorption Distribution Metabolism Excretion Toxcity
10
疗效比青蒿素高5倍,且毒性比青蒿素低
植物
从红豆杉树皮中分 离出的抗癌药 紫杉醇(含量0.07%)
结构修饰,优化得到半合成的 多西他赛 (Taxotere)
微生物
1929年,英国医生 Fleming发现青霉素, 从此揭开了青霉素研 究的序幕。
为什么服用头孢药物后不能饮酒?
H H H N S O O N
八、通过其他方法得到先导化合物
1.组合化学 组合化学是利用一些基本的小分子单元如氨基 酸、单核苷酸、单糖及各种有机小分子化合物 通过化学或生物合成的方法,系统地反复以共 价键装配成不同的组合,构建结构多样性的化 合物库。 组合化学能高效、大批量地合成化合物,配合 高通量筛选,大大加速了先导化合物的发现速 度。
简称先导物(Lead),又称原型物,是通过各种 途径得到的具有一定生理活性的化学物质。 A chemical structure or series of structures that 有独特的结构 show activity and selectivity in a pharmacological or relevant screen. biochemically 具有一定活性的化合物 (缺陷 ) The structure of the lead compoundT G T A C T G G T T C T A G G T A C A T G A C
碱基配对是 反义核酸作 用的基础
抑制mRNA剪接
mRNA
反义寡核苷酸(Antisense oligonucleotides)
这种可与DNA或信使 RNA结合的互补链称作 反义寡核苷酸。 能够与DNA 或信使RNA 发生特异性结合,分 别阻断核酸的转录或 翻译功能,阻止与病 理过程相关的核酸或 蛋白质的生物合成。
化学基因组学是近年发展的基因组与药物设计相交叉的学科, 基本思路是基于靶标活性部位的抑制剂的设计及合成。目前, 随着人类基因组的研究,大量的疾病相关基因被发现,使得药 物作用的靶标分子急剧增加,为药物设计开辟了广阔前景。
129个碳,64个手性中心, 7个可异构双键
二、通过分子生物学途径发现先导化合物 以药物作用靶点的三维结构和生物化学 作用机制为基础进行药物设计。
基于机理的药物设计 (Mechanism-based drug design) 基于结构的药物设计 (Structure-based drug design)
基于机理的药物设计
H1受体拮抗剂类的抗过敏药 H2受体拮抗剂类抗溃疡药物,
如西咪替丁(Cimetidine),用 于溃疡病的治疗。
西咪替丁(Cimetidine)
组胺
西咪替丁
基于结构的药物设计
配体与受体相互作用 的本质是分子识别。 分子识别是由于两个 分子的多个特定的原 子或基团性质的互补 性和空间的适配所驱 动的,这种特异性的 本质是双方的互补性。 药物与受体的分子识 别和相互作用,大都 形成非共价键作用, 与维持机体的生物大 分子的空间结构的键 合力在本质上是相似 的(氢键、静电引力和 疏水作用)。 药物和受体分子构象 变化,诱导契合。