蛋白表达及纯化
蛋白质的表达纯化

完全紧贴。)正常安装则会形成一个密闭的容
器。
蛋白质的表达纯化
5.将底座的开关打开, 并向外拨动透明的架子, 使中间空隙增大,放入 电极架。
6.如图所示将电极架往下 压(约1~2mm),使底面 完全接触。
蛋白质的表达纯化
7.关上底座开关。
8.放入电泳槽内,加上加样 架加样。注意加样架与刚才 制胶所用的梳子齿数必须一 致。
• 3. 细胞裂解液3mL以10-15mL/h 流速上Ni2+-NTA柱,收集流 出液。
• 4.洗脱杂蛋白:用50mL洗涤缓冲液UWB以10-15mL/h流速洗柱, 分别取10ul洗涤开始与结束时的样品用于SDS-PAGE 分析。
• 5.洗脱目标蛋白:用10mL洗脱缓冲液洗柱,每管0.5-1 mL, 共收集6-10管,分别取1蛋0白u质l样的表品达纯用化于SDS-PAGE 分析。
蛋白质的表达纯化
实验步骤
• 一、氯霉素酰基转移酶重组蛋白的诱导 • 1. 接种含有重组氯霉素酰基转移酶蛋白的大肠杆菌
BL21菌株于5mL LB液体培养基中 (含100ug/mL 氨苄 青霉素),37℃震荡培养过夜。 • 2. 转接1mL过夜培养物于100mL(含100ug/mL 氨苄青霉 素)LB液体培养基中,37℃震荡培养至OD600 = 0.6 0.8。取10ul 样品用于SDS-PAGE 分析。 • 3. 加入IPTG至终浓度0.5 mmol/l, 37℃继续培养1-3h. • 4. 12,000rpm 离心10 min, 弃上清,菌体沉淀保存于20℃或-70℃冰箱中。
蛋白质的表达纯化
• 3.制浓缩胶:按所需的浓度配制4%的浓缩胶,配
方如下:
30%Arc- Tris-HCl H2O
Bis
蛋白表达纯化的实验原理

蛋白表达纯化的实验原理蛋白表达与纯化是生物学实验中常用的技术,可以用来研究和生产各种蛋白质。
本文将一步一步回答关于蛋白表达纯化实验的原理和步骤。
第一步:蛋白表达系统的选择蛋白表达系统是指用来制备和表达目标蛋白质的细胞或病毒载体。
常用的表达系统包括细菌、酵母、昆虫和哺乳动物细胞等。
选择表达系统时需要考虑目标蛋白的性质、需求量和表达效率等因素。
第二步:构建表达载体表达载体是将目标蛋白的基因序列插入到细胞或病毒载体中,以实现蛋白表达的工具。
通常采用的方法是将目标基因通过限制性内切酶切割,然后与适当的载体连接。
第三步:细胞转染或感染将构建好的表达载体转染到细胞中,使目标蛋白基因在细胞内进行表达。
对于真核细胞(如哺乳动物细胞)可以通过转染的方式传递质粒DNA,而对于原核细胞(如大肠杆菌)可以通过热激转化或电穿孔等方法进行具体的转染。
第四步:培养表达细胞转染或感染后,需要将细胞培养到合适的条件下,以促进目标蛋白表达。
培养条件包括适宜的培养基、温度、氧气供应和营养物等。
此外,可以考虑添加特定的诱导剂或抑制剂,以调控蛋白表达的级别。
对于细菌目标蛋白表达,通常将细胞培养在含有抗生素的培养基上以选择表达带有目标基因的细菌。
第五步:蛋白表达检测为了确定目标蛋白是否在细胞中表达,可以使用多种方法进行检测。
常用的方法包括Western blot、ELISA、原位杂交、荧光染色等。
同时,可以通过调整培养条件或表达载体的构建来提高蛋白表达的水平。
第六步:蛋白纯化蛋白纯化是从表达系统中提取和纯化目标蛋白的过程。
纯化步骤的选择取决于目标蛋白的性质和所需的纯度。
常见的纯化方法包括亲和纯化、凝胶过滤、离子交换、大小排除层析、亲水性层析等。
此外,还可以使用亲和标签(如His标签、GST标签等)来辅助蛋白质的纯化。
第七步:蛋白质的鉴定和定量通过蛋白纯化后,需要对目标蛋白进行鉴定和定量。
可以使用SDS-PAGE、Western blot、质谱分析等方法来确定蛋白质的分子量和纯度。
蛋白表达纯化流程

第一章蛋白表达纯化一、诱导表达分析1) 把测序正确的表达质粒转化到合适的表达宿主菌中并涂布在相应抗性的LB 平板上培养过夜;2) 分别挑两个单克隆于3ml含抗生素的LB培养基中培养过夜;3) 按1%接种过夜培养的菌液于4ml含抗生素LB培养基中37度培养2-3小时;4) 加入终浓度为0.1mM的IPTG诱导表达3个小时左右,取样做蛋白电泳分析目标蛋白的表达情况。
二、蛋白纯化2.1重组蛋白的提取2.1.1 提取缓冲液20 mM Tris-HCl (pH8.0) 或者其他推荐使用的缓冲体系。
如需溶解难溶蛋白或者包涵体还可加入8 M urea 或 6 M guanidine hydrochloride 。
Note: 处理(His)6融合蛋白时可以加入5–50 mM imidazole 以减少上柱时的非特异性吸附。
2.1.2 方法1)发酵液离心收集菌体(at 7 000–8 000 g for 10 minutes or 1 000–1 500 g for 30minutes at +4 °C)。
2)弃去上清液,加入适当的20 mM Tris-HCl (pH8.0),重悬;3)离心收集菌体同上,弃去上清液,将装有菌体的离心管置于冰中。
4)每ml菌体加入50ul冰冷的提取缓冲液重悬菌体。
5)冰浴中超声裂解菌体(超声2秒,停止6秒),之后取样进行SDS-PAGE电泳分析。
Note:超声破菌应尽量使用最短的时间,长时间的进行可能会破坏蛋白功能。
还应避免产生泡沫,因为这样会使蛋白变性和导致宿主蛋白与融合蛋白协同纯化。
6)离心使细胞碎片沉降(at 12 000 g for 10 minutes at +4 °C)。
7)小心将上清液移到干净的容器中,并取样进行SDS-PAGE电泳分析。
Note:含有8 M urea的样品可以直接电泳上样,但含有6 M guanidine hydrochloride的样品必须换成8 M urea才可上样。
重组蛋白质的表达纯化和结构鉴定

重组蛋白质的表达纯化和结构鉴定在生物医学领域中,重组蛋白质凭借其广泛的应用前景成为了研究热点。
然而,要想获得高纯度的重组蛋白质,并对其结构进行准确的鉴定,需要经历一系列复杂而细致的实验步骤。
本文将从表达、纯化和结构鉴定三个方面介绍重组蛋白质的研究过程。
一、表达重组蛋白质的表达是研究重组蛋白质最初的关键步骤之一。
通常采用大肠杆菌(Escherichia coli)作为表达宿主。
首先,需要将目标基因克隆至表达载体中,确保其与启动子、转录因子等相互配合,并携带一定的标签,如His 标签、GST 标签等。
接着,将修饰好的表达载体转化至大肠杆菌中,采用选择性培养基筛选出目标菌株。
最后,将筛选得到的菌株进行大规模培养,促使目标基因在细胞内表达。
二、纯化获得表达目标蛋白的菌株后,需要将蛋白从细胞中纯化出来。
首先,采用超声波或高压颠破细胞壁,释放出蛋白质。
接着,通过离心等手段将蛋白质与其他细胞组分分离。
此时,可以利用目标蛋白质特异性的亲和层析柱,如镍柱、葡聚糖柱等,吸附目标蛋白质,并通过逆向洗脱等方法,得到高纯度的目标蛋白质。
三、结构鉴定获得纯度较高的重组蛋白质后,需要对其结构进行进一步的鉴定。
常用的结构鉴定方法包括X射线晶体学、核磁共振(NMR)、电子显微镜等。
X射线晶体学是目前应用最广泛的方法,通过将蛋白质结晶并进行X射线衍射实验,得到蛋白质的高分辨率结构。
NMR则通过测量蛋白质中核自旋的相对位置和相互作用关系,获取蛋白质的三维结构信息。
电子显微镜是一种能够获得蛋白质高分辨率结构的技术,主要应用于研究大分子复合物和纤维形态的蛋白质。
除了上述常用技术外,近年来还涌现出一些新的结构鉴定方法,如质谱联用技术、光学显微镜成像、负染电镜等。
这些方法的出现,为蛋白质结构鉴定提供了更多的选择和便利。
由于篇幅所限,本文仅对重组蛋白质的表达、纯化和结构鉴定进行了简要介绍。
事实上,研究重组蛋白质的过程还包括目标基因的设计与合成、蛋白质的功能分析等环节。
蛋白质表达纯化概述

昆虫表达系统的优缺点
• 1,组蛋白具有完整的生物学功能,如蛋白的正确 折叠、二硫键的搭配
•
2,蛋白翻译后的加工修饰;
•
3,表达水平高,可达总蛋白量的50%;
•
4,可容纳大分子的插入片段;
•
5,能同时表达多个基因。主要缺点是外源蛋
白表达处于极晚期病毒启动子的调控之下,这时
由于病毒感染,细胞开始死亡。
2.3 采用什么培养基进行表达?LB,TB,SOB,SOC等等。 2.4 客户是否有Protocol、essay等参考文献 • 客户是否指定用某种纯化工艺:亲和层析、排阻层析、反向层析、疏水层析、离子交
换 2.5 表达前是否要做小量实验(预表达/预实验/小试)。是否要做表达条件的优化筛选?
如果有,需要做温度、IPTG浓度、诱导OD、抗生素浓度(不常用)、诱导时间中的哪 些优化筛选。 2.3 蛋白纯度要求、浓度要求(蛋白定量方法:分光光度法、旋光光度法等)、蛋白总量 要求。是否要去热源(又叫内毒素,常用于抗原抗体的制备以及药物的筛选。)是否 要去除RNA,是否要去除核酸,是否要去除DNA。 2.4 蛋白活性要求(如果用于抗体的制备可以不需要活性。) 2.6 蛋白WB检测:常用于真核细胞表达检测。
2.1是否需要蛋白标签 GST(常用,增加蛋白溶解性)、His(最常用,用于易表达纯化, 性质稳定的蛋白)、Strep(有I、II两种!!实验方法不同。)、Flag (Flag是一种抗体, 特异性高,表达量相对较低,常用于昆虫、哺乳动物细胞的表达)等
2.2 如果有标签,是在3‘端还是在5’端(或者是蛋白标签在N端还是C端)。需不需要 酶切位点(用于后续纯化中去除标签)。是否要进行基因突变,突变位点在哪里?
• 昆虫表达系统是一类应用广泛的真核表达系统,它具有同大多数高等 真核生物相似的翻译后修饰加工以及转移外源蛋白的能力。昆虫杆状 病毒表达系统是目前国内外十分推崇的真核表达系统。利用杆状病毒 结构基因中多角体蛋白的强启动子构建的表达载体,可使很多真核目 的基因得到有效甚至高水平的表达。它具有真核表达系统的翻译后加 工功能,如二硫键的形成、糖基化及磷酸化等,使重组蛋白在结构和 功能上更接近天然蛋白;其最高表达量可达昆虫细胞蛋白总量的50%; 可表达非常大的外源性基因(一200kD);具有在同一个感染昆虫细胞 内同时表达多个外源基因的能力;对脊椎动物是安全的。由于病毒多 角体蛋白在病毒总蛋白中的含量非常高,至今已有很多外源基因在此 蛋白的强大启动子作用下获得高效表达。。常用的杆状病毒包括苜蓿 银纹夜蛾核型多角体病毒(AcNPV)和家蚕型多角体病毒(BmNPV),常 用的宿主细胞则来源于草地夜蛾Sf9细胞,用于表达外源基因的质粒 来源于PUC系列,其含有一个多克隆位点和多角体蛋白启动子。
目的蛋白表达与纯化protocol

目的蛋白表达与纯化protocol小量表达测试:1、挑一单克隆到3ml LB(含抗生素)中,370C,220rpm振荡培养10h~12h。
2、保种:700μl菌液+400μl 80%灭菌甘油,充分混匀后置于-800C保存。
3、扩大培养:以1:100接菌,即取50μl菌液到5ml LB(含抗生素)中,370C,220rpm振荡培养2h~3h至OD值达到0.6。
4、对照取样:取1ml菌液,12000rpm离心1min,弃尽上清,-200C保存。
对照5、另取1ml菌液于一新的试管中,加1/1000 IPTG(终浓度为1mM),370C,220rpm振荡培养3h后收菌:12000rpm离心1min,弃尽上清,-200C保存。
370C样品6、其余约3ml菌液于160C,220rpm继续振荡培养1h后,加0.5/1000 IPTG(终浓度为0.5mM),160C,220rpm振荡培养12~24h(通常18h)后收菌:12000rpm 离心1min,弃尽上清,-200C保存。
160C样品7、Bugbuster分别处理上述三个样品,取20μl总蛋白,其余离心后分离上清和沉淀,进行SDS-PAGE电泳鉴定,上样顺序为:对照370C样品160C样品总蛋白上清沉淀总蛋白上清沉淀总蛋白上清沉淀若目的蛋白在上清中有表达,则可以进行大量表达与纯化,具体操作如下。
大量表达:1、挑一单克隆到7.5ml LB(含抗生素)中,370C,220rpm振荡培养10h~12h。
注意:此步用50ml离心管摇菌!2、扩大培养:以1:100接菌,即取将上述7.5ml菌液全部接到750ml TB(含抗生素)中,370C,220rpm振荡培养4h~5h至OD值达到1.0。
3、对照取样:取1ml菌液,12000rpm离心1min,弃尽上清,-200C保存。
(对照)4、其余约菌液于160C,220rpm继续振荡培养1h后,加0.5/1000 IPTG(终浓度为0.5mM),160C,220rpm振荡培养12~24h(通常18h)后收菌:6000rpm、40C离心10min,弃尽上清,取一点沉淀做表达鉴定(160C样品),其余-200C 保存。
基于生物素化技术的蛋白表达和纯化策略研究

基于生物素化技术的蛋白表达和纯化策略研究随着生物技术的高速发展和生物学研究的深入,对于蛋白表达和纯化的需求也越来越大。
其中,生物素化技术作为一种快速、高效、灵敏的标记技术,正受到越来越多的关注和研究。
本文将重点介绍基于生物素化技术的蛋白表达和纯化策略。
第一部分:生物素化技术的基础原理生物素是一种水溶性维生素,广泛存在于细菌、真菌、植物和动物细胞中,是一种重要的共生信号分子。
生物素化技术即利用生物素和生物素结合蛋白(Biotin-binding protein, BBP)之间的高亲和力,并通过干扰生物素与其它蛋白质的结合来标记和纯化感兴趣的蛋白或酶。
生物素化技术还可以分为两种:一种是利用菌体表达S tagging的生物素化技术(Biotin tag),另一种是利用体外生物素化技术(Biotinylation)。
前者是将生物素连接到蛋白N端或C端附近,后者则是在蛋白N端或C端附近引入一个生物素化底物,随后加入生物素化酶引入生物素,从而实现蛋白标记和纯化。
第二部分:基于生物素化技术的蛋白表达策略1. 菌体表达生物素标记蛋白(Biotin tag)菌体表达生物素标记蛋白(Biotin tag)是基于遗传工程的方法,将BBP结构域融合到目标蛋白的N、C端,在表达和纯化过程中即可利用高亲和力进行标记和纯化。
优点:操作简单、标记和纯化效率高、纯度高。
缺点:蛋白质结构和生物活性可能会受到BBP的融合影响。
2. 利用体外生物素化技术(Biotinylation)表达蛋白利用体外生物素化技术,首先在蛋白的表达载体中引入生物素化底物(Biotin acceptor peptide, BAP),随后在表达过程中加入生物素化酶,即可引入生物素,实现蛋白的标记和纯化。
优点:BBP与蛋白质结合灵活,不影响蛋白结构和生物活性。
缺点:操作复杂,标记和纯化效率可能会受到底物和酶浓度的影响。
第三部分:基于生物素化技术的蛋白纯化策略基于生物素化技术的蛋白纯化策略包括干扰生物素与其它蛋白质的结合、利用高亲和力纯化带标记蛋白和对标记蛋白进行洗脱的操作步骤。
蛋白表达及纯化PPT课件

萃取法
液-液萃取法
利用两种不混溶的溶剂,将目标蛋白 质从一种溶剂中转移到另一种溶剂中 。
反胶团萃取法
利用反胶团形成的微环境,选择性分 离和纯化蛋白质。
色谱法
凝胶色谱法
利用凝胶介质对不同大小的蛋白质颗粒的分离作用,将目标蛋白质与其他杂质 分离开。
离子交换色谱法
利用离子交换剂对不同带电状态的蛋白质的吸附作用,将其与其他蛋白质分离 开。
亲和层析
利用基因工程改造的蛋白质分 子上的特定标签与固相载体上 的配体结合进行分离纯化。常 用的亲和层析包括His-tag亲 和层析、GST亲和层析等。
离子交换层析和凝胶过滤 层析
与抗体药物的纯化类似,离子 交换层析和凝胶过滤层析也可 用于重组蛋白药物的进一步纯 化和精制。
蛋白质相互作用研究中的纯化应用
02
蛋白纯化方法
沉淀法
盐析法
通过加入高浓度的盐溶 液,改变蛋白质的溶解
度使其沉淀下来。
有机溶剂沉淀法
利用有机溶剂降低蛋白 质溶解度的原理,使其
沉淀。
聚合物沉淀法
利用聚合物与蛋白质的 相互作用,使蛋白质沉
淀。
低温沉淀法
在低温条件下,通过改 变蛋白质的溶解度使其
沉淀。
离心法
差速离心法
通过逐渐增加离心力,将 不同大小的蛋白质颗粒分 离开。
扩展床吸附技术
将大颗粒的介质填充于柱中,使流动相能够通过吸附作用去 除杂质,提高纯化效果。
集成化与自动化技术
集成化分离系统
将多个分离步骤集成在一个系统中, 实现连续的分离纯化,提高生产效率。
自动化控制技术
通过自动化控制系统,实现分离纯化 的全流程监控和自动调节,减少人为 误差和操作时间。
蛋白纯化CSM-昆虫细胞中蛋白表达与纯化

14.2 His 标签蛋白表达纯化1、把His 标签蛋白表达质粒,进行转化,转到大肠杆菌的感受态细胞BL21 中,冰上放置30min,加入新鲜的LB 培养基孵育大约1h,然后涂板,于37℃培养箱中进行过夜培养。
2、第二天,小心地挑取培养菌落中的单克隆,加入到10mL 的加有适当抗性的LB液体培养基中,放在37℃的摇床中培养过夜。
3、把过夜培养的菌液倒入800mL 加有适当抗性的LB 液体培养基中,于37℃摇床中摇到大约OD600 值在0.6~0.8 之间,就可以拿出,把菌液放置于4℃冷却菌液20~30min。
4、在超净台中,加入诱导剂IPTG 至终浓度0.1mM,诱导蛋白表达,于18℃摇床中培养6-8h。
5、诱导完成以后,8000 rpm 离心10min,沉淀收集菌体。
6、在沉淀后的细菌菌体中,加入lysis buffer(10 mM 咪唑,300 mM NaCl,50 mMNaH2PO4,用NaOH 调节pH 值至8.0,蛋白酶抑制剂)中重悬,1g 湿重的菌体沉淀用5mL 的裂解缓冲液,进行超声破碎,然后12000rpm 离心20min,离心后取上清。
7、在上清中加入1 mL Ni-NTA beads 到4 mL 裂解液中,于4℃冰箱中轻轻地混合60 min 左右。
8、加入4 mL 的wash buffer(300 mM NaCl,20 mM 咪唑,50 mM NaH2PO4,用NaOH 调节pH 值至8.0,蛋白酶抑制剂),在冰上洗4 次,每次孵育10min。
9、加入elution buffer(250 mM 咪唑,300 mM NaCl,50 mM NaH2PO4,用NaOH调节pH 值至8.0,蛋白酶抑制剂)洗脱重组蛋白4 次,每次用500μl。
14.3 GST 融合蛋白的表达与纯化1、把GST 标签蛋白表达质粒,进行转化,转到大肠杆菌的感受态细胞BL21 中,冰上放置30min,加入新鲜的LB 培养基孵育大约1h,然后涂板,于37℃培养箱中进行过夜培养。
蛋白质表达与纯化技术的研究进展

蛋白质表达与纯化技术的研究进展随着生物技术的发展,蛋白质表达与纯化技术也得到了迅速的发展。
蛋白质是生命物质中至关重要的组成部分,为研究生命的机制及开发生物制药提供了重要的基础和前提。
本文将从蛋白质表达及纯化技术的研究进展入手,介绍相关的前沿技术和方法。
一、蛋白质表达技术的研究进展1.1 原核表达系统原核表达系统是一种常用的蛋白质表达技术,它利用细菌的生物学特性,在大规模表达目标蛋白质的同时,具有快速、高效、经济的优势。
近年来,原核表达系统也得到了不断的改良和优化,例如利用基因工程技术将目标蛋白质表达的速度和表达量得到了显著提高,进一步拓宽了其应用范围。
1.2 酵母表达系统酵母表达系统主要利用酵母菌作为载体表达目标蛋白质,具有高表达量、合成质量好、能够进行翻译后修饰等优点。
在酵母表达系统中,利用选择性培养基的筛选方法可以显著提高目标蛋白质表达的效率和纯度。
1.3 昆虫细胞表达系统昆虫细胞表达系统是一种常用的哺乳动物细胞表达系统,利用昆虫细胞(如Sf9、Sf21细胞等)表达目标蛋白质。
这种系统具有易于维护,表达效率高,重组蛋白质具有天然的哺乳动物的修饰等优点。
目前,昆虫细胞表达系统已经被广泛应用于疫苗、生物药物等领域。
1.4 哺乳动物细胞表达系统哺乳动物细胞表达系统是目前最常用的蛋白质表达技术,通过利用哺乳动物细胞表达目标蛋白质并进行不同程度的修饰,可以得到与天然蛋白质相似的重组蛋白质。
此外,该系统还可应用于细胞培养技术、生物药物研发等领域。
二、蛋白质纯化技术的研究进展2.1 柱层析技术柱层析技术作为蛋白质纯化的核心技术,是一种能根据其化学性质和物理性质特征,利用不同的色谱柱实现组分分离的技术。
随着柱层析技术的发展,液相色谱、气相色谱、毛细管电泳等技术的出现,蛋白质的纯化程度得到了进一步提高。
2.2 薄层凝胶电泳技术薄层凝胶电泳技术是一种以物质的分子量为分离基础,利用电泳原理实现生物大分子分离的技术。
蛋白可溶表达与纯化的详细实验过程

蛋白可溶表达与纯化的详细实验过程2010年02月27日星期六09:02丁香园chrispp作品。
一、证明目的蛋白有表达1、挑取一单菌落接种于含氨苄(Amp,终浓度为100g/mL)的3mL液体LB培养基中37℃,250rpm培养过夜。
2、将过夜培养菌液以1:100转接于含上述抗生素的100mL液体LB中,37℃,250rpm,3.5h (大量培养至OD600=0.6左右)。
3、取出1mL菌液为未诱导的电泳对照,剩余培养液加入诱导物IPTG至终浓度为1mmol/L,继续振荡培养4h。
4、以未诱导菌液与IPTG诱导后菌液作对比,SDS-PAGE检测。
结果如下:图11:pET-32aA3未诱导,2:pET-32aA3未诱导,3:pET-32aA3诱导后,4:pET-32aA3诱导后二、表达的情况,是可溶还是沉淀1、将上述的37℃诱导菌液离心(4℃,12000g,5min),弃上清(LB培养液),沉淀重悬于0.9%NaCl溶液洗菌。
2、将重悬于0.9%NaCl的菌体溶液离心(4℃,12000g,5min),弃上清(0.9%NaCl洗菌液),得菌体,称菌体重量,重悬于8mL PBS(pH7.0)缓冲液,缓冲液用量根据50~100mg/mL的菌液终浓度。
3、超声破菌:冰浴条件下,超声波破碎大肠杆菌,超声设置如下功率:400W,工作:15s,间隔:45s,全程时间:30min(30次左右)以菌液由白变透明,且不粘稠为依据(少量菌液用液氮反复冻融7次以上效果也不错,而且简单,但是DNA 出来后会粘稠,可能加dna酶后会好些,那样好离心,我是用接近最高转速离心的(没加dan酶),不然分不开)4、将超声波破碎的菌液离心(4℃,12000g,15min),分别收集上清和沉淀,各取1mL,加入1mL2×上样缓冲液,沸水浴10min,SDS-PAGE检测。
结果如下:图21:pET-32aA3诱导上清,2:pET-32aA3诱导沉淀三、探索可溶表达条件有上述实验结果可知,目标蛋白在上清也有表达,即存在可溶性的表达,但是量很少,大部分也包涵体的形式存在(沉淀中),而包涵体的存在也证明了蛋白表达量很高,上清可溶蛋白的可操作性等因素都优于对包涵体蛋白的分离纯化,所以可通过探索可溶表达条件,最终来加大上清蛋白的含量,以利于下一步实验的进行。
蛋白表达纯化步骤
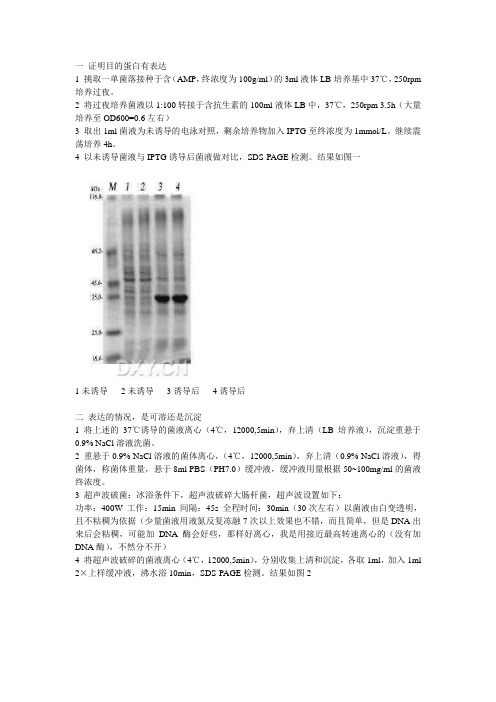
一证明目的蛋白有表达1 挑取一单菌落接种于含(AMP,终浓度为100g/ml)的3ml液体LB培养基中37℃,250rpm 培养过夜。
2 将过夜培养菌液以1:100转接于含抗生素的100ml液体LB中,37℃,250rpm 3.5h(大量培养至OD600=0.6左右)3 取出1ml菌液为未诱导的电泳对照,剩余培养物加入IPTG至终浓度为1mmol/L。
继续震荡培养4h。
4 以未诱导菌液与IPTG诱导后菌液做对比,SDS-PAGE检测。
结果如图一1未诱导2未诱导3诱导后4诱导后二表达的情况,是可溶还是沉淀1 将上述的37℃诱导的菌液离心(4℃,12000,5min),弃上清(LB培养液),沉淀重悬于0.9% NaCl溶液洗菌。
2 重悬于0.9% NaCl溶液的菌体离心,(4℃,12000,5min),弃上清(0.9% NaCl溶液),得菌体,称菌体重量,悬于8ml PBS(PH7.0)缓冲液,缓冲液用量根据50~100mg/ml的菌液终浓度。
3 超声波破菌:冰浴条件下,超声波破碎大肠杆菌,超声波设置如下:功率:400W 工作:15min 间隔:45s 全程时间:30min(30次左右)以菌液由白变透明,且不粘稠为依据(少量菌液用液氮反复冻融7次以上效果也不错,而且简单,但是DNA出来后会粘稠,可能加DNA酶会好些,那样好离心,我是用接近最高转速离心的(没有加DNA酶),不然分不开)4 将超声波破碎的菌液离心(4℃,12000,5min),分别收集上清和沉淀,各取1ml,加入1ml 2×上样缓冲液,沸水浴10min,SDS-PAGE检测。
结果如图2三探索可溶表达条件有上述结果可知,目标蛋白在上清也有表达,即存在可溶性表达,但是量很少,大部分以包涵体的形式存在(沉淀中),而包涵体的存在也证明蛋白的表达量很高,上清可溶性蛋白的可操作性等因素的干扰优于对包涵体蛋白的分离纯化,所以可通过探索可溶性蛋白的表达条件,最终加大上清可溶性蛋白的含量,以便实现下一步实验的进行。
蛋白原核表达纯化原理

蛋白原核表达纯化原理蛋白原核表达纯化是一种常用的生物技术方法,用于大量制备目标蛋白质。
该方法可以在原核细胞中直接表达目标蛋白,然后通过一系列的纯化步骤获得高纯度的蛋白质样品。
以下将详细介绍蛋白原核表达纯化的原理和步骤。
蛋白原核表达纯化的原理主要基于细菌细胞的生物特性。
在表达过程中,目标蛋白的基因会被插入到表达载体中,该载体会被转化到细菌细胞中。
转化后的细菌细胞会利用其自身的代谢机制表达目标蛋白。
蛋白原核表达纯化的步骤主要包括以下几个方面:第一步,构建表达载体。
在这一步骤中,目标蛋白的基因会被插入到表达载体的多克隆位点中。
这个过程可以通过PCR扩增目标基因,然后将其连接到表达载体上。
第二步,转化细菌细胞。
在这一步骤中,经过构建的表达载体会被转化到细菌细胞中。
转化可以使用化学方法或电穿孔等物理方法进行。
第三步,培养表达菌株。
转化后的细菌细胞会被培养在含有适当抗生素的培养基中。
培养的条件包括温度、pH值、搅拌速度等,这些条件可以根据目标蛋白的特性进行优化。
第四步,诱导表达。
在菌株达到一定的生长密度后,可以通过添加适当的诱导剂来诱导目标蛋白的表达。
诱导剂的选择可以根据目标蛋白的特性进行优化。
第五步,收获细胞。
在表达过程中,细菌细胞会合成大量的目标蛋白。
可以通过离心等方法,将细菌细胞从培养基中收获下来。
第六步,裂解细胞。
收获的细菌细胞需要被裂解,以释放目标蛋白。
常用的方法包括超声波、高压等物理方法,以及酶解等化学方法。
第七步,纯化目标蛋白。
裂解后的混合物中含有大量的杂质,需要通过一系列的纯化步骤来获得高纯度的目标蛋白。
常用的纯化方法包括亲和层析、离子交换层析、凝胶过滤等。
经过以上步骤,蛋白原核表达纯化的过程就完成了。
这种方法可以高效地制备目标蛋白,并且可以根据需要进行优化。
蛋白原核表达纯化在生物医药、生物工程等领域有着广泛的应用前景,对于研究目标蛋白的结构和功能具有重要意义。
蛋白质的表达、纯化及检测-分子实验报告

实验目的1.了解外源基因在大肠杆菌细胞中的诱导表达情况2.学会用SDS-PAGE电泳法分离不同分子量的蛋白质3.学习通过亲和层析法纯化目的蛋白4.学会考马斯亮蓝染色法和蛋白质杂交法检测蛋白质实验原理1.外源基因在大肠杆菌细胞中的诱导表达:将外源基因克隆在特殊的表达载体中,让其在E. coli中表达,该表达载体上含有lac操作子的启动子。
在不加诱导剂的条件下培养宿主菌,lacI基因表达的阻遏蛋白LacI与lac操作子结合,使外源基因不能表达;向培养基中加入诱导物IPTG后,LacI阻遏蛋白变构失活,不能与lac操作子结合,外源基因就表达。
2.蛋白质SDS-PAGE电泳分离:SDS-PAGE是最常用的定性分析蛋白质的电泳方式,特别是用于蛋白质纯度检测和测定蛋白质分子量。
其分离原理是根据蛋白质分子量的差异,因为SDS-PAGE的样品处理液及缓冲液的加入破坏了蛋白质的二级、三级、四级等结构,并使SDS与蛋白质充分结合形成SDS-蛋白质复合物,稳定地存在于均一的溶液中,SDS与蛋白质结合后使SDS-蛋白质复合物上带有大量的负电荷,远远超过其原来所带的电荷,从而使蛋白质原来所带的电荷可以忽略不计,消除了不同分子之间原有的电荷差别,其电泳迁移率主要取决于亚基分子质量的大小,这样分离出的谱带也为蛋白质的亚基。
3.考马斯亮蓝法检测蛋白质:考马斯亮蓝是一种蛋白质染料,主要有R-250和G-250两种类型。
考马斯亮蓝可以和蛋白肽链中碱性氨基酸残基或芳香族氨基酸残基(Arg,Trp,Tyr,His,Phe)结合。
考马斯亮蓝R250多用于聚丙烯酰胺凝胶电泳后蛋白质条带的染色;因为考马斯亮蓝R250中的R代表Red,偏红,红蓝色,与蛋白质结合虽然比较缓慢,但是染料可以穿透凝胶,染胶效果好,染色后为蓝色,且与胶的结合可以被洗脱下去,所以可以用来对电泳条带染色。
4.基因融合就是将两个或多个开放读码框按一定顺序连接在一起,融合阅读框架的表达产物是一个杂和蛋白。
蛋白质的表达纯化及结晶

2.3.2.2 蛋白质表达、纯化(1)初始种子培养:挑取阳性克隆到10 mL的含有Amp抗生素的液体LB培养基中,在37℃过夜振荡培养。
(2)扩大培养:将初始种子接入1 L含抗生素的液体培养基中,37℃振荡培养至菌浓OD600为0.6-0.8时,降温至15℃或20℃;一个小时后加入终浓度为0.6 mM的IPTG,并诱导表达过夜。
(3)收集菌体:于4200 rpm,4℃下离心15 min,弃去上清,收获菌体;加入重悬溶液(25 mM Tris-HCl pH8.0,100 mM NaCl),悬浮菌体细胞;细胞破碎前加入终浓度为2 mM的蛋白酶抑制剂PMSF(Phenylmethyl sulfonyl fluoride,苯甲酸磺酰氟)。
(4)超声破碎细胞:400W下,超声3s,间隔6s,工作60次。
(5)超速离心:细胞裂解液于14000 rpm,4℃下离心50 min,收集上清液,进行下一步的分离纯化。
(6)Ni-NTA亲和层析:将上清液体倒入Ni-NTA柱中。
流净后,用wash buffer (25 mM Tris-HCl pH8.0,100 mM NaCl,15 mM imidazole)冲洗10个柱体积,除去杂蛋白;最后使用elution buffer(25 mM Tris-HCl pH8.0,100 mM NaCl,250 mM imidazole)将目的蛋白洗脱下来。
使用SDS-PAGE检测蛋白的可溶性、挂柱效率及蛋白的浓度。
由于本实验中YdiV等蛋白都是连接到pGl01载体中,带有可以用PPase切除的6×His标签。
为了获得更纯净的、不带标签的蛋白,我们在有那个wash buffer冲洗去除杂蛋白以后,每根镍柱中加入5 ml的重悬缓冲液然后加入100-200 μL的PPase,3-5 h后,用5 ml重选冲洗柱子,电泳检测酶切效率。
(7)阴离子交换层析纯化:先平衡离子交换柱。
然后将上一步洗脱下的蛋白用溶液A(25 mM Tris-HCl, pH8.0)稀释4-6倍,上样到离子交换柱Source Q上,使用溶液A与溶液B(25mM Tris-HCl pH8.0,1M NaCl)进行线性梯度洗脱。
蛋白表达及纯化ppt课件

Part 1: 蛋白表达、纯化
蛋白表达流程
1. 目的基因cDNA
2. 表达宿主
3. 载体 4. 设计引物 5. 连接转化 6. 诱导表达 7. 蛋白纯化 8. 鉴定保存
gene
Host
Cellfree Bacteri al
Yeas t
Mammalia n
0.60 0.50 0.40
Protein preparation, extraction, clarification
Cell growth, protein overexpression Cell lysis Removal of cell debris
From: Protein Purification Handbook. Amersham Biosciences. 18-1132-29, Edition AC
转译起始序列
a: mRNA的5末端之独特的结构特征, 是决定mRNA转译起始效率的主 要因素。 b: 在构建外源基因的高效表达载体时 , 需认真选择有效的转录起 始序列。 c: 未鉴定出通用有效的转译起始序列的保守结构 (KOZAK SEQUENCE)
筛选标记: 其编码产物可被快速测定的功能单元。
pET system manual, Novagen, 11th Edition
X蛋白在大肠杆菌中表达条件优化
1:0mmol/L; 2:0.25mmol/L;3:0.5mmol/L; 4:0.75mmol/L;5:1.0mmol/L; 6:1.5mmol/L; 7:2.5mmol/L; 8:3.5mmol/L; 9:5.0mmol/L。 图1.5 X蛋白表达条件:IPTG浓度优化
1:蛋白marker;2~9:分别用IPTG 诱导表达 0、1、 2、3、4、6、8、12 h。
生物化学中的蛋白质表达和纯化

生物化学中的蛋白质表达和纯化蛋白质是细胞中最基本的生物大分子之一,具有重要的结构和功能作用。
在生化实验研究中,常常需要大量的蛋白质作为实验材料。
蛋白质表达和纯化技术是生物化学研究中的关键技术之一。
本文将简要介绍蛋白质表达和纯化的原理和方法。
一、蛋白质表达技术蛋白质表达是将目的基因转录成RNA后再翻译成蛋白质的过程。
蛋白质表达主要有原核细胞和真核细胞两种方法。
原核细胞表达系统主要利用大肠杆菌,真核细胞表达系统则使用哺乳动物细胞,其主要的表达技术有以下几种:(一)重组蛋白质大规模表达重组蛋白质是指人为构建的同源或异源蛋白序列,利用基因工程技术将其导入到表达宿主中进行高效表达的蛋白质。
大肠杆菌是目前最常用的宿主。
一般来说,要将目的基因插入到选择性表达载体中,选用合适的启动子和终止子,将目的蛋白质与标签结合。
表达宿主随后被转化,蛋白质在生长过程中表达出来,随后进行纯化和鉴定。
(二)GST融合蛋白表达GST融合蛋白是利用GST (glutathione S-transferase)标签的蛋白质,将GST和目的蛋白质融合在一起表达,然后通过Glutathione 亲和层析纯化方法纯化目的蛋白质。
GST融合蛋白可以提高目的蛋白质的稳定性和可溶性,使得其在细胞内表达更加稳定。
(三)His标签蛋白表达His标签是一种聚组氨酸标签,可以与Ni2+螯合,因此可采用Ni2+亲和层析的方法纯化。
His标签融合蛋白表达时选择了较少的氨基酸标签,对目标蛋白的生物学性质和功能影响较小。
二、蛋白质纯化技术蛋白质表达和纯化是蛋白质生物化学研究的关键。
通常情况下,表达宿主细胞中的蛋白质必须经过纯化才能得到纯净的蛋白质,获得足够高纯度的蛋白质可用于测定其结构和功能。
(一)离子交换层析法离子交换层析法是利用蛋白质负荷(或正荷)的离子性质与相应的离子交换质团之间进行选择性结合的纯化方法。
离子交换层析法分为阴离子交换层析和阳离子交换层析两种。
cas9 蛋白 生产 工艺

cas9 蛋白生产工艺Cas9蛋白是一种CRISPR-Cas9系统中的关键组成部分,广泛应用于基因组编辑和基因治疗研究中。
本文将介绍Cas9蛋白的生产工艺。
Cas9蛋白的生产工艺可以分为三个主要步骤:基因克隆、蛋白表达和纯化。
基因克隆是Cas9蛋白生产的第一步。
研究人员通常选择含有Cas9基因序列的质粒进行克隆。
通过PCR扩增技术,将Cas9基因从质粒中扩增出来,并进行酶切和连接,将其插入到表达载体中。
然后,将表达载体转化到大肠杆菌等宿主细胞中,通过培养和筛选得到含有Cas9基因的细胞株。
接下来是蛋白表达步骤。
将含有Cas9基因的细胞株培养在含有适当抗生素的培养基中,通过调节培养条件,如温度、pH值、营养物质等,促进Cas9蛋白的表达。
当达到适当的细胞密度后,可以加入诱导剂(如IPTG)来提高Cas9蛋白的表达水平。
经过一定时间的培养后,细胞会产生大量的Cas9蛋白。
对Cas9蛋白进行纯化。
将培养得到的细胞进行离心,分离出细胞上清液。
通过多步酸碱沉淀、离子交换层析、凝胶过滤等技术,可以将Cas9蛋白从细胞上清液中纯化出来。
在纯化过程中,可以使用SDS-PAGE等方法检测Cas9蛋白的纯度和分子量。
最终,通过浓缩和冻干的方式得到纯化后的Cas9蛋白。
需要注意的是,在Cas9蛋白的生产过程中,要严格控制条件,以确保蛋白的活性和稳定性。
同时,要对生产过程进行监测和控制,确保产品符合质量标准。
总结起来,Cas9蛋白的生产工艺包括基因克隆、蛋白表达和纯化三个主要步骤。
通过这些步骤,可以高效地获得纯化后的Cas9蛋白,为基因组编辑和基因治疗研究提供重要的工具和材料。
这对于推动生物医学研究的发展具有重要的意义。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Protein expression
检测该重组质粒在BL21中是否能被诱导表达,并确定在不同IPTG浓度和时间蛋白诱导 效率的差异。 挑单克隆菌落于7ml含有相应抗生素LB培养液的50ml培养管中,37C振荡培养过夜
(8-16h)。 37C,1mM IPTG终浓度诱导蛋白。取700ul过夜摇菌加入7ml含有相应抗生素LB培
外源基因在原核寄主细胞中表达, 它的编码结构 必须是连续的, 不间断的, 处于寄主启动子有效 控制下。
启动子
可使外源基因高水平表达的最佳启动子必须具备以下几个条件 1 ) 强启动子,外源基因的蛋白质的表达量占细胞总蛋白的10% - 30%以上 2 ) 应能呈现出一种低限的基础转录水平 3 ) 应该是诱导型的, 能通过简单的方式,使用廉价的诱导物得以诱导。 常用的大肠杆菌表达载体启动子:
An introduction to liquid chromatography
Protein solution applied to a column
Column = solid porous matrix (stationary phase) + liquid (mobile phase)
β-半乳糖苷酶基因lacZ 荧光素酶基因 (luciferase )基因、 半乳糖激酶基因(galK ) 氯霉素抗性(乙酰转移酶)基因 (cat ) 四环素抗性基因 (tetr ) Tag-
保护碱基 内切酶1 目的基因 内切酶2 保护碱基
X gene
引物的设计:设计一对特异性 引物。上游、下游引物引入酶 切位点
c: 转录终止子还能增强mRNA分子的稳定性, 大大提高蛋白质产物水平。
d:在构建大肠杆菌表达载体时,通常是添加全部终止密码子 , 阻止核糖体跳跃 (skipping )现象。
e:大肠杆菌格外偏爱使用终止密码子UAA, 当其后连上一个U而形成四联核苷酸 的情况下 , 转译终止效率便会得到进一步加强
转译起始序列
Yeast
Mammalian Insect
Expression System Characteristics
Characteristics
doubling time cost of growth medium expression level protein folding
N-linked glycos.
Fractional precipitation of proteins
Precipitate contaminants
Add Precipitant, Centrifugation
Precipitate protein of interest
Discard pellet
Add Precipitant, Centrifugation, Discard supernatant, Resuspend protein
λ噬菌体的PL启动子 大肠杆菌乳糖操纵子lac启动子 色氨酸操纵子trp启动子 pBR322质粒的beta-内酰胺酶启动子
终止子
a: 如果在克隆基因编码区的3末端之后,接上一个有效的转录终止子,便能够 阻止转录通读过位于下游另一个启动子
b: 如果在启动子的上游部位放置一个有效的转录终止子 , 那么由该启动子驱 动的克隆基因的转录便会被限制在最低本底水平。
养液的50ml离心管中,于OD600=0.4-0.6(1:100接种,37C培养时,细菌生长至 OD600=0.4-0.6约需2h)时加入终浓度为1mM的IPTG。 诱导前在37C培养的菌液中取1ml未诱导的对照,按1ml菌液×OD600×100μl的比例 (如1ml OD600为0.4的菌加40ul buffer)加入2×上样缓冲液,混匀,-20℃保存或直接 上样。 根据蛋白分子量大小配置相应浓度的SDS-PAGE胶。(50KD蛋白10%或12%) 诱导3-4h后收集菌液,取1ml样品,测OD600,10000rpm,1min离心去上清后加入 相应体积的2×上样缓冲液,vortex。 将收集的菌液在水中煮5min,上样10μl ,120V电压走浓缩胶,加电压至150V进入 分离胶电泳,直到染料刚好走到胶底。 取下胶,考马斯亮蓝染色至少30min(染液若是新配的,染20min就够了),脱色液 脱色。若急需看到结果,可以将胶在500ml蒸馏水中煮沸两次即可看到条带。
IPTG induction
1. Even in the absence of IPTG, there is some expression of T7 RNA polymerase from the lacUV5 promoter.
2. The degree of toxicity will vary from protein to protein.
Protein isolation, concentration, and stabilization
Reversible precipitation with salt or
organic molecules
From: Protein Purification Handbook. Amersham Biosciences. 18-1132-29, Edition AC
大肠杆菌表达外源基因的劣势
缺乏对真核生物蛋白质的复性功能 缺乏对真核生物蛋白质的修饰加工系统 内源性蛋白酶降解空间构象不正确的异源蛋白 细胞周质内含有种类繁多的内毒素(endotoxin)
Vector
二 大肠杆菌表达载体的基本组成
一个良好的大肠杆菌表达载体: 复制起点(ori) 多克隆位点。 筛选标记(抗菌素抗性基因、Tag、筛选标记) 控制和调节转录与翻译的必不可少的原件
X基因
内切酶1 目的基因
+
内切酶2 保护碱基
R1
R2
Your destination vector
Your linearized vector
1:DNA marker; 2:重组质粒pET28-X双酶切; 3:空质粒pET28a双酶切
图图1.14.X3 基重但因组为发质同生粒义点pE突突T变2变8(-(XAS双GerU酶)→切A鉴G定C),
pET system manual, Novagen, 11th Edition
X蛋白在大肠杆菌中表达条件优化
1:0mmol/L; 2:0.25mmol/L;3:0.5mmol/L; 4:0.75mmol/L;5:1.0mmol/L; 6:1.5mmol/L; 7:2.5mmol/L; 8:3.5mmol/L; 9:5.0mmol/L。
Protein preparation, extraction, clarification
Cell growth, protein overexpression
Cell lysis Removal of cell debris
From: Protein Purification Handbook. Amersham Biosciences. 18-1132-29, Edition AC
a: mRNA的5末端之独特的结构特征, 是决定mRNA转译起始效率的主 要因素。 b: 在构建外源基因的高效表达载体时 , 需认真选择有效的转录起 始序列。 c: 未鉴定出通用有效的转译起始序列的保守结构 (KOZAK SEQUENCE)
筛选标记: 其编码产物可被快速测定的功能单元。 追踪某些特定的DNA结构 (重组质粒 )是否已经导 入寄主细胞 同任何一种目的启动子连接 , 其表达活性可作为 检测启动子功能的依据。
O-linked glycos. phosphorylation acetylation acylation g-carboxylation project cost
E. coli
rapid (30 min) low
high refolding may be
required no
no no no no no low
Yeast
rapid (90 min) low
low – high refolding may be required high mannose
yes yes yes yes no low
Insect
slow (18-24 hrs) high
low – high proper folding
simple, no sialic acid yes yes yes yes no
图1.5 X蛋白表达条件:IPTG浓度优化
1:蛋白marker;2~9:分别用IPTG 诱导表达 0、1、 2、3、4、6、8、12 h。
图1.6 X蛋白表达条件:诱导时间优化
Protein Purification
Characterize function, activity, structure Use in assays Raise antibodies many other reasons ...
蛋白表达、纯化
抗体制备应用及ELISA间接法
卢士强 2010-11-18
Part 1: 蛋白表达、纯化
蛋白表达流程
1. 目的基因cDNA 2. 表达宿主 3. 载体 4. 设计引物 5. 连接转化 6. 诱导表达 7. 蛋白纯化 8. 鉴定保存
gene
Host
Cell-free
Bacterial
Adapted from: Protein Purification Handbook. Amersham Biosciences. 18-1132-29, Edition AC
Basic scheme of protein purification
From: Protein Purification Handbook. Amersham Biosciences. 18-1132-29, Edition AC