实验十一食品中黄曲霉毒素B1的检测
实验十一-食品中黄曲霉毒素B1的检测

4.植物油:
用小烧杯称取4.0g样品,用20.0mL石油醚,将样品移 于125mL分液漏斗中,用20.0mL 甲醇水(55 +45)溶 液分次洗烧杯,溶液一并移人分液漏斗中。(精炼油 4.0g样为4.575mL,可直接用移液器加入分液漏斗再 加溶剂后振摇)振摇2min,静置分层后,放出下层甲 醇水溶液于750mL蒸发皿中。再用5.0mL甲醇水溶液 重复振摇提取一次,提取液一并加入蒸发皿中,以下 按自“65℃水浴通风挥干”起,依法操作。
二、实验原理:
本试剂盒采用间接竞争ELISA方法,样品中的黄曲霉 毒素B1经提取、脱脂、浓缩后与定量特异性抗体反应, 多余的游离抗体则与酶标板内的包被抗原结合,加入 酶标记物和底物后显色,与标准曲线比较测定含量。
三、仪器与试剂
(一)试剂盒组成 1、 AFB1抗原包被16孔×6条形带框酶标板; 2、抗体; 3、酶标二抗; 4、空白对照液; 5、底物(1mg×4); 6、底物液A; 7、底物液B; 8、终止液; 9、PBS一T洗液; 10、一次性手套。 (二)仪器 1、酶标仪; 2、离心机。
5.其它食品:可按照G8 5009有关章节提供的方法操 作,最终提取物(相当于4.0g样 品)应收集于2.0m1 甲醇一PBS(20+80)中。
(二)样品测定
1、将下列试剂稀释后备用 PBS一T洗液:390mL蒸榴水稀释(1:40); 底物液A: 9mI蒸溜水稀释(1:9); 抗体: 7.0mL洗液稀释(1:140); 酶标二抗:10mL洗液稀释(1:200); 阴性对照液:200μl抗体十200μl空白对照液在玻
3.花生(脂肪含量15.0-45.0%):
样品去壳去皮粉碎后称取20.0g,加入250mL具塞三角瓶中,准确 加人100.0mL甲醇水(55 +45)溶液和30mL石油醚,盖塞后滴水封 严,150rpm振荡30min,静置15min后用快速定性滤纸过滤于 125mL分液漏斗中,待分层后,放出下层甲醇水溶液于100mL烧 杯内,从中取20.0 mL(相当于4.0g样品)置于另一125mL分液漏 斗中,加入20.0mL三氯甲烷,振摇2min,静置分层(如有乳化现 象可滴加甲醇促使分层).放出三氯甲烷于75mL蒸发皿中,再加 5.0mL三氯甲烷于分液漏斗中重复振摇提取后,放出三氯甲烷层一 并于蒸发皿中,以下按上述“65℃水浴通风挥干”起,依法操作。
对食品中黄曲霉毒素B1的检测方法及分析

2010 NO.23S c i enc e a nd T ech n ol o gy I nno v at i on Her a l d研 究 报 告1 黄曲霉毒素B t测定原理与步骤方法在食品中黄曲霉毒素B1检测方法很多,有些样品如薯类及部分酱料、油类样品受样品杂质干扰的影响,因而使方法的灵敏度降低。
目前通常采用双向展开法,可以提高了检测方法的灵敏度,如测定了一份薯类样品,按灵敏度由原法的25ppb提高到5ppb,同时测定了含杂质较多的花生油、酱油及曲种等样品,情况亦是如此。
而原法灵敏度为5ppb的样品(如花生及部分花生油样品等)用双向展开法展开,其灵敏度亦可以进一步提高,有的可提高到1.25ppb。
今将双向展开法的原理及操作步骤介绍如下。
1.1原理因无水乙醚不能推动黄曲霉毒素B-(以下简称B。
)。
在用乙醚将薄层板横展时,可以把干扰的杂质推到样品点的旁边,而不像用乙醚纵展时,有时在样品的过道中留有部分杂质的底色。
在乙醚横展后再用丙酮:氯仿纵展,可以使样品在B1标准点相应处的杂质底色大量的减少.从而提高了原有方法的灵敏度。
1.2试剂(补充的)无水乙醚,分析纯,在无水乙醚中加入少许无水硫酸钠贮存。
1.3操作步骤主要补充双向展开法的操作步骤,其它参照B1测定法。
1.3.1滴加方法由于在具体测定条件下,用无水乙醚展开后Bt仍有很少的移动(因无水乙醚中含微量的水或醇以及空气中的湿度太大等均可以使B,移动),因此在滴加时必需同时滴加两块层析板。
在一块5×20cm的薄层板上距下端3cm的基线上滴加以下两个点:第1个点离左边边距0.8~1cm处滴加B。
标准液Ⅲ(0.04微克/毫升)10微升;第2个点离第一个点的间隔距离约1.8~2.0cm处滴加样液20微升。
在另一块上的滴加样式完全与第一块相同,祗在第二个样品点上再滴加B1标准液Ⅲ0.04(微克/毫升)10毫升。
1.3.2展开方法①横展:在展开槽内的长边放置玻璃支架一个,再加无水乙醚10毫升。
食品中黄曲霉毒素B1的测定

实验六食品中黄曲霉毒素B1的测定一、实验目的与要求学习薄层层析法测定粮食中黄曲霉毒素B1原理及步骤。
二、实验原理样品中AFT B1经有机溶剂提取、净化、浓缩并经薄层色谱分离后,在波长365nm紫外光下产生荧光,根据其在薄层板上显示荧光的强度与标准品AFT B1最低检出量产生的荧光强度比较来测定样品中AFT B1的含量。
三、实验仪器与试剂仪器:玻璃板:5×20cm;涂布器;色谱展开槽(25×6×4cm);紫外光灯: 100-125W,带有波长365nm滤光片;微量注射器:20uL,10uL各一支试剂:三氯甲烷(AR);正己烷(AR 沸程30-60)或石油醚(沸程60-90);甲醇(AR);苯(AR);乙腈(AR);无水乙醚(AR);丙酮(AR)。
(以上试剂应重蒸,并应经检验对薄层色谱测定无干扰)苯-乙腈(98:2)混合溶液;甲醇-水(55:45)混合溶液;三氟乙酸(AR);氯化钠(AR);无水硫酸钠(AR);硅胶G(薄层色谱用)。
5%次氯酸钠溶液:称取100g漂白精,加入500mL水,搅匀。
另将80g工业用碳酸钠(Na2CO3.10H2O)溶于500mL温水中,倒入上述溶液中,搅匀,澄清过滤后贮存于带橡皮塞的玻璃瓶中,用作消毒剂。
黄曲霉毒素B1标准贮备液:用百万分之一的微量分析天平精密称取1-1.2mgAFT B1标准品,先加入2mL乙腈溶解后,再用苯稀释至100mL,避光置于4℃冰箱中保存。
使用前用紫外分光光度计测定其浓度,再用苯-乙腈混合溶液调整其浓度为10ug/mL。
(在350nm测定AFT B1的苯-乙腈溶液的吸光度,其摩尔消光系数为19800,可根据朗伯-比尔定律求出其浓度)。
黄曲霉毒素B1标准应用液I(1ug/mL):吸取1.0mL10ug/mL的黄曲霉毒素B1标准贮备液于10mL容量瓶中,加苯-乙腈混合溶液定容。
黄曲霉毒素B1标准应用液II(0.2ug/mL):吸取1.0mL 1ug/mL的黄曲霉毒素B1标准应用液I于5mL容量瓶中,加苯-乙腈混合溶液定容。
食品中黄曲霉毒素B1 检测方法研究
仪,主要包含了传感和自动化识别两大系统。由于综合应用 质也可能会产生影响,降低或增加待测信号,形成基质效
了数学、物理、材料、神经及生理等诸多理论,能迅速完成 应。原因主要是生物样品中内源性成分的存在或融入样品
复杂气味和气体的识别,通过气味数学模型的构建及应用, 前处理中的外源性成分的影响。所以在样品本身、基质及
关键词:食品;黄曲霉毒素 B1;检测方法
当前,人们高度重视食品安全问题,在探索和研究各类 障人们的身体健康。
食品安全检测方法方面已取得了瞩目成果。黄曲霉毒素 B1 的毒性和致癌性极强,通过污染粮食作物及其制品从而对人
畜健康构成威胁,需高度重视其检测工作的开展。基于此,
本文介绍了几种用于检测食品中黄曲霉毒素 B1 的方法。 1 食品中黄曲霉毒素 B1 检测重要性
优势
劣势
电子鼻法
响应时间短、检测速度快,测定评估范围广,能避免人为误差, 无法识别分子水平上单个可挥发物标志物
重复性好
高效液相色谱 - 质谱联用法
重现性好、灵敏度高
程序烦琐、检测周期长、成本较高
时间分辨荧光免疫分析法
操作流程短、有效期长、灵敏度高、检测便捷、 试剂盒昂贵
食品科技
食品中黄曲霉毒素 B1 检测方法研究
王 媛
(连云港市赣榆区综合检验检测中心,江苏连云港 222100)
金标免疫层析法检测黄曲霉毒素b1的方法

金标免疫层析法检测黄曲霉毒素B1的方法1.引言黄曲霉毒素B1是一种广泛存在于食品、饲料中的毒素,对人体和动物健康造成严重威胁。
对黄曲霉毒素B1进行有效的检测至关重要。
金标免疫层析法作为一种快速、准确并且易于使用的检测方法,被广泛应用于食品安全和质量控制领域。
本文将就金标免疫层析法检测黄曲霉毒素B1的方法进行探讨,并提供深度和广度兼具的分析,以期帮助您更全面地了解这一检测技术。
2.金标免疫层析法的原理金标免疫层析法是一种基于抗体和抗原之间特异性识别和结合原理的检测方法。
简单来说,它利用了抗体对特定分子的高度特异性识别能力,通过在复杂样品中将目标分子与标记有金纳米颗粒的抗体结合,从而实现对目标分子的定量和定性检测。
金标免疫层析法具有操作简便、快速灵敏、不需要仪器设备等特点,因此在食品安全领域得到了广泛的应用。
3.黄曲霉毒素B1的特性和对人体健康的危害黄曲霉毒素B1是一种热稳定性很强的毒素,它容易在食品和饲料中积累。
长期摄入受污染的食品或饲料会导致人类和动物出现急性或慢性中毒,表现为呕吐、腹泻、免疫抑制等严重症状,甚至可能导致肝癌等疾病的发生。
对黄曲霉毒素B1的检测至关重要,金标免疫层析法作为一种快速、敏感的检测方法,能够有效地帮助人们及时发现并控制食品中的黄曲霉毒素B1污染。
4.金标免疫层析法检测黄曲霉毒素B1的步骤金标免疫层析法检测黄曲霉毒素B1的步骤通常包括样品制备、抗体标记金纳米颗粒、检测纸条制备和数据读取等。
将样品进行处理,提取其中的黄曲霉毒素B1,并将其与标记有金纳米颗粒的抗体结合。
将混合物加载到检测纸条上,金标免疫层析法会在纸条上形成不同程度的色带,根据色带浓度的变化可以判断出样品中黄曲霉毒素B1的含量。
通过专门的读取设备或者肉眼观察检测纸条上色带的变化来获得测试结果。
5.金标免疫层析法的优势金标免疫层析法作为一种快速、准确、便捷的检测方法,具有以下几点优势。
它无需昂贵的仪器设备,操作简便,使用者无需专业的技术背景即可进行检测。
高效液相色谱法检测农产品中黄曲霉毒素B1的含量

分析检测高效液相色谱法检测农产品中黄曲霉毒素B1的含量韩 宇(四川省甘孜藏族自治州食品药品检验所,四川康定 626000)摘 要:目的:建立高效液相色谱法联合免疫磁固相萃取法测定农产品食品中黄曲霉毒素B1(Aflatoxin B1,AFB1)含量的分析方法。
方法:采用70%乙腈溶液提取花生、玉米、大豆、小麦、豌豆和绿豆样品中的AFB1,然后利用免疫磁珠净化、萃取和富集提取液中的AFB1,采用高效液相色谱法进行定量分析。
结果:方法的检出限为0.03~0.92 μg·kg-1,回收率为79.52%~97.53%。
利用该方法对市场上购买的20份花生、玉米、大豆、小麦、豌豆、绿豆样品中的AFB1进行检测,除了玉米和绿豆中未检测出AFB1外,其余实验样品中均检测出一定含量的AFB1。
结论:该方法简便、快速、准确,适用于复杂样品中黄曲霉毒素B1的测定。
关键词:高效液相色谱法;免疫磁固相萃取;黄曲霉毒素B1Determination of Aspergillus Flavus B1 in Agricultural Products by High-Performance Liquid ChromatographyHAN Yu(Ganzi Tibetan Autonomous Prefecture Institute for Food and Drug Control, Kangding 626000, China)Abstract: Objective: To establish a method for the determination of Aflatoxin B1 (AFB1) in agricultural products by high performance liquid chromatography combined with immunomagnetic solid phase extraction. Method: AFB1 was extracted from samples of peanuts, corn, soybeans, wheat, peas, and mung beans using a 70% acetonitrile solution, and then AFB1 was purified, extracted and enriched by immunomagnetic beads, and quantitative analysis was performed by HPLC. Result: The limits of detection were 0.03~0.92 μg·kg-1, and the recoveries were 79.52%~97.53%. AFB1 in 20 samples of peanut, corn, soybean, wheat, pea and mung bean purchased from the market was detected by this method. AFB1 content was detected in all the other samples except corn and mung bean. Conclusion: The method is simple, rapid, accurate and suitable for the determination of aflatoxin B1 in complex samples.Keywords: high-performance liquid chromatography; immunomagnetic solid-phase extraction; aspergillus flavus B1黄曲霉毒素是一种由黄曲霉菌产生的有毒代谢产物,在自然界中分布广泛,在粮食和饲料等农产品中广泛存在。
生鲜乳中黄曲霉毒素B1、B2、G1、G2、M1、M2的测定 液相色谱-柱后光化学衍生法山东标准2020版

生鲜乳中黄曲霉毒素B1、B2、G1、G2、M1、M2的测定液相色谱-柱后光化学衍生法警示:整个分析操作过程应在指定区域内进行。
该区域应避光(阳光),具备相对独立的操作台和废弃物存放装置。
在整个实验过程中,操作者应按照接触剧毒物的要求采取相应的保护措施。
1 范围本标准规定了测定生鲜乳中黄曲霉毒素B1、B2、G1、G2、M1、M2含量的液相色谱-柱后光化学衍生测定方法。
本标准适用于生鲜乳中黄曲霉毒素B1、B2、G1、G2、M1、M2的定量测定。
本标准方法的检出限:黄曲霉毒素B1为0.002 μg/kg,黄曲霉毒素B2为0.002 μg/kg,黄曲霉毒素G2为0.002 μg/kg,黄曲霉毒素G1为0.002 μg/kg,黄曲霉毒素M1为0.005 μg/kg,黄曲霉毒素M2为0.002 μg/kg;本标准方法的定量限:黄曲霉毒素B1为0.006 μg/kg,黄曲霉毒素B2为0.005 μg/kg,黄曲霉毒素G2为0.005 μg/kg,黄曲霉毒素G1为0.006 μg/kg,黄曲霉毒素M1为0.015 μg/kg,黄曲霉毒素M2为0.006 μg/kg。
2 规范性引用文件下列文件对于本文件的应用是必不可少的。
凡是注日期的引用文件,仅所注日期的版本适用于本文件。
凡是不注日期的引用文件,其最新版本(包括所有的修改单)适用于本文件。
GB/T 6682 分析实验室用水规格和试验方法3 原理试样中的黄曲霉毒素B1、B2、G1、G2、M1、M2用甲醇溶液提取,提取液经黄曲霉素毒素免疫亲和柱净化、富集,用高效液相色谱分离后经柱后光化学衍生,荧光检测器检测,外标法定量。
4 试剂和材料除非另有说明,在分析中仅使用确认为分析纯的试剂,水为GB/T 6682中规定的一级水。
4.1 甲醇:色谱纯。
4.2 乙腈:色谱纯。
4.3 甲醇水溶液:取50 mL甲醇,加入50 mL纯水,混合均匀。
4.4 流动相:乙腈+水=20+80。
ELISA法检测油菜籽中的黄曲霉毒素B1

分析检测ELISA法检测油菜籽中的黄曲霉毒素B1杨 权1,孙婷琳2,王 燕3,余 佳3,李利霞1,熊 英1*(1.孝感市公共检验检测中心,湖北孝感 432000;2.湖北省粮油食品质量监督检测中心,湖北武汉 43000;3.三峡公共检验检测中心,湖北宜昌 443000)摘 要:为了建立油菜籽中黄曲霉毒素B1(Aflatoxin B1,AFB1)的快速检测方法,本文采用酶联免疫吸附法,选取不同浓度的提取液、不同种类、不同比例的稀释液检测样本的加标回收率,并进行了准确度、精密度及重复性的评价。
结果发现用60%甲醇1︰5、超纯水1︰4稀释检测油菜籽样本中黄曲霉毒素B1的加标回收率为99%~106%,相对标准偏差小于5%。
结果表明,酶联免疫吸附法测定油菜籽中黄曲霉毒素B1含量的准确性,重现性,稳定性好,可广泛用于油菜籽中黄曲霉毒素B1的检测。
关键词:AFB1;酶联免疫吸附法;油菜籽黄曲霉毒素是一组真菌(霉菌)毒素,是黄曲霉和寄生曲霉的代谢产物[1],广泛存在于花生、大豆等农产品中,是影响花生、大豆、芝麻等农产品油料质量安全的重要因素。
黄曲霉毒素具有毒性和强致癌性,毒性远远高于氰化物、砷化物和有机农药的毒性[2]。
其中黄曲霉毒素B1是目前已知致癌性最强的天然毒素,对人及动物肝脏组织有破坏作用,人、畜摄入了黄曲霉毒素超标的食物或饲料,可发生急性中毒,出现急性肝炎、出血性坏死、肝细胞脂肪变性和胆管增生。
当微量持续摄入,可造成慢性中毒,出现肝实质细胞变性、肝硬化等,动物生长发育迟缓,体重减轻,母畜不孕或产仔少等系列症状,可诱发肝癌、胃癌等多种部位的癌症。
目前黄曲霉毒素的常规检测方法有薄层色谱法、高效液相色谱法、高效液相色谱-串联质谱法及酶联免疫吸附法等[3-4]。
油菜是我国传统的油料作物,也是我国主要的油料作物。
菜籽油一直是我国居民的传统生活用油,在我国植物油供给结构中,菜籽油居第二位[5]。
在菜籽油实际生产中,往往会由油菜籽中引入黄曲霉毒素B1,导致成品菜籽油中可能会含有微量黄曲霉毒素B1。
食品中黄曲霉毒素B1的测定

Science &Technology Vision 科技视界操作次序加入量孔号123456789150微升50微升A 00.10.250.512……DCCC C C CCC混匀23孔号234567891.890 1.548 1.2410.9190.6700.509 1.586 1.5400引言黄曲霉毒素B 1是一种高毒性、高致癌性的物质,除可引发肝癌外,也可引起其他部位如肾、胃、支气管、腺体和皮下组织的癌肿,是食品卫生部门常规的检测项目之一。
之前的方法都不能更好的检测出粮食作物、饲料和发酵食品中黄曲霉毒素的含量,今用酶联免疫吸附法能更灵敏准确的检出。
该法是把抗原抗体免疫反应和酶的高效催化作用原理有机地结合起来的一种新型检测技术,其主要依据有三点:第一,抗原(抗体)能结合到固相载体的表面并仍保持其免疫活性;第二,抗体(抗原)与酶结合所形成的结合物仍保持免疫活性和酶的活性;第三,结合物与相应的抗原(抗体)反应后,结合的酶仍能催化底物生成有色物质,而颜色的深浅可定量抗体(抗原)的含量。
酶联免疫吸附分析法主要有三种测定方法:即间接法、抗体夹心法和竞争法。
前两种主要用于测定抗体和大分子抗原,适用于临床诊断上;而竞争法是测定小分子抗原的方法,因而尤其适用于食品卫生分析,该法主要有四步:1)将人工抗原吸附于固相载体,温育后清洗;2)加入含有抗原的待测液和抗体,温育后洗涤;3)加入酶标抗体,温育后清洗;4)加酶底物,温育后显色,并进行检测和结果判断。
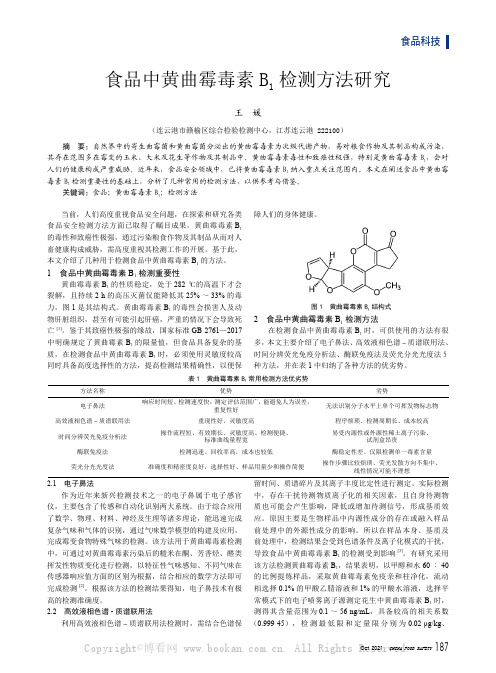
图1黄曲霉毒素化学结构1实验部分1.1酶联免疫吸附法的原理利用固相酶联免疫吸附原理,将AFB 1特异性抗体包被与聚苯乙烯微量反应板的空穴中,再加入样品提取液(未加抗原)及酶标AFB 1抗原(已知抗原),使两者与抗体之间进行免疫竞争及应,然后加酶底物显色,颜色的深浅取决于抗体和酶标AFB 1抗原结合的量,即样品中AFB 1多,则被抗体结合酶标AFB 1抗原少,颜色浅,反之则深。
谈食品中黄曲霉毒素B1的检测

谈食品中黄曲霉毒素B1的检测作者:山瑛来源:《中国科技纵横》2016年第01期【摘要】食品中黄曲霉毒素的测定方法是使用GB/T18979-2003《食品中黄曲霉毒素的测定免疫亲和层析净化高效液相色谱法和荧光光度法》中免疫亲和层析法高效液相色谱法进行的测定,但是在实际测定中发现许多细节问题在测定中容易忽视,而会影响测定的结果,本文针对笔者长期的实验发现的问题分别对样品的处理、标准溶液及衍生液的处理、免疫亲和柱等方面做了归纳分析。
【关键词】黄曲霉毒素B1的测定注意事项免疫亲和层析法高效液相色谱法黄曲霉毒素B1(Aflatoxin B1简写为AFB1)是二氢呋喃氧杂萘邻酮的衍生物,含有一个双呋喃环和一个氧杂萘邻酮(香豆素)。
黄曲霉毒素B1是已知的化学物质中致癌性最强的一种。
黄曲霉毒素B1对包括人和若干动物具有强烈的毒性,其毒性作用主要是对肝脏的损害。
B1是最危险的致癌物,经常在玉米,花生,棉花种子,一些干果中常能检测到。
它们在紫外线照射下能产生荧光,根据荧光颜色不同,将其分为B族和G族两大类及其衍生物。
AFT目前已发现20余种。
AFT主要污染粮油食品、动植物食品等;如花生、玉米,大米、小麦、豆类、坚果类、肉类、乳及乳制品、水产品等均有黄曲霉毒素污染。
其中以花生和玉米污染最严重。
家庭自制发酵食品也能检出黄曲霉毒素,尤其是高温高湿地区的粮油及制品种检出率更高。
黄曲霉毒素耐热,280℃才可裂解,故一般烹调加工温度下难以破坏。
本文对GB/T18979-2003《食品中黄曲霉毒素的测定免疫亲和层析净化高效液相色谱法和荧光光度法》中免疫亲和层析法高效液相色谱法测定食品中黄曲霉毒素B1进行探讨:原理试样经甲醇-水提取,提取液经过滤、稀释后,滤液经过含有黄曲霉毒素特异抗体的免疫亲和柱净化,此抗体对黄曲霉毒素B1具有专一性,黄曲霉毒素交联在层析介质中的抗体上。
用水或吐温-20/PBS将免疫亲和柱上杂质除去,以甲醇通过免疫亲和层析柱洗脱,洗脱液通过带荧光检测器的高效液相色谱仪柱后碘液衍生测定黄曲霉毒素B1的含量。
食品中黄曲霉毒素B族和G族的测定

食品安全国家标准食品中黄曲霉毒素B族和G族的测定1范围本标准规定了食品中黄曲霉毒素B1㊁黄曲霉毒素B2㊁黄曲霉毒素G1㊁黄曲霉毒素G2(以下简称A F TB1㊁A F TB2㊁A F T G1和A F T G2)的测定方法㊂本标准第一法为同位素稀释液相色谱-串联质谱法,适用于谷物及其制品㊁豆类及其制品㊁坚果及籽类㊁油脂及其制品㊁调味品㊁婴幼儿配方食品和婴幼儿辅助食品中A F TB1㊁A F TB2㊁A F TG1和A F TG2的测定㊂本标准第二法为高效液相色谱-柱前衍生法,适用于谷物及其制品㊁豆类及其制品㊁坚果及籽类㊁油脂及其制品㊁调味品㊁婴幼儿配方食品和婴幼儿辅助食品中A F T B1㊁A F T B2㊁A F T G1和A F T G2的测定㊂本标准第三法为高效液相色谱-柱后衍生法,适用于谷物及其制品㊁豆类及其制品㊁坚果及籽类㊁油脂及其制品㊁调味品㊁婴幼儿配方食品和婴幼儿辅助食品中A F T B1㊁A F T B2㊁A F T G1和A F T G2的测定㊂本标准第四法为酶联免疫吸附筛查法,适用于谷物及其制品㊁豆类及其制品㊁坚果及籽类㊁油脂及其制品㊁调味品㊁婴幼儿配方食品和婴幼儿辅助食品中A F TB1的测定㊂本标准第五法为薄层色谱法,适用于谷物及其制品㊁豆类及其制品㊁坚果及籽类㊁油脂及其制品㊁调味品中A F TB1的测定㊂第一法同位素稀释液相色谱-串联质谱法2原理试样中的黄曲霉毒素B1㊁黄曲霉毒素B2㊁黄曲霉毒素G1㊁黄曲霉毒素G2,用乙腈-水溶液或甲醇-水溶液提取,提取液用含1%T r i t o nX-100(或吐温-20)的磷酸盐缓冲溶液稀释后(必要时经黄曲霉毒素固相净化柱初步净化),通过免疫亲和柱净化和富集,净化液浓缩㊁定容和过滤后经液相色谱分离,串联质谱检测,同位素内标法定量㊂3试剂和材料除非另有说明,本方法所用试剂均为分析纯,水为G B/T6682规定的一级水㊂3.1试剂3.1.1乙腈(C H3C N):色谱纯㊂3.1.2甲醇(C H3O H):色谱纯㊂3.1.3乙酸铵(C H3C O O N H4):色谱纯㊂3.1.4氯化钠(N a C l)㊂3.1.5磷酸氢二钠(N a2H P O4)㊂3.1.6磷酸二氢钾(K H2P O4)㊂3.1.7氯化钾(K C l)㊂3.1.8盐酸(H C l)㊂3.1.9 T r i t o nX-100[C14H22O(C2H4O)n](或吐温-20,C58H114O26)㊂3.2试剂配制3.2.1乙酸铵溶液(5mm o l/L):称取0.39g乙酸铵,用水溶解后稀释至1000m L,混匀㊂3.2.2乙腈-水溶液(84+16):取840m L乙腈加入160m L水,混匀㊂3.2.3甲醇-水溶液(70+30):取700m L甲醇加入300m L水,混匀㊂3.2.4乙腈-水溶液(50+50):取50m L乙腈加入50m L水,混匀㊂3.2.5乙腈-甲醇溶液(50+50):取50m L乙腈加入50m L甲醇,混匀㊂3.2.610%盐酸溶液:取1m L盐酸,用纯水稀释至10m L,混匀㊂3.2.7磷酸盐缓冲溶液(以下简称P B S):称取8.00g氯化钠㊁1.20g磷酸氢二钠(或2.92g十二水磷酸氢二钠)㊁0.20g磷酸二氢钾㊁0.20g氯化钾,用900m L水溶解,用盐酸调节p H至7.4ʃ0.1,加水稀释至1000m L㊂3.2.81%T r i t o nX-100(或吐温-20)的P B S:取10m LT r i t o nX-100(或吐温-20),用P B S稀释至1000m L㊂3.3标准品3.3.1 A F TB1标准品(C17H12O6,C A S:1162-65-8):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂3.3.2 A F TB2标准品(C17H14O6,C A S:7220-81-7):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂3.3.3 A F T G1标准品(C17H12O7,C A S:1165-39-5):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂3.3.4 A F T G2标准品(C17H14O7,C A S:7241-98-7):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂3.3.5同位素内标13C17-A F TB1(C17H12O6,C A S:157449-45-0):纯度ȡ98%,浓度为0.5μg/m L㊂3.3.6同位素内标13C17-A F TB2(C17H14O6,C A S:157470-98-8):纯度ȡ98%,浓度为0.5μg/m L㊂3.3.7同位素内标13C17-A F T G1(C17H12O7,C A S:157444-07-9):纯度ȡ98%,浓度为0.5μg/m L㊂3.3.8同位素内标13C17-A F T G2(C17H14O7,C A S:157462-49-7):纯度ȡ98%,浓度为0.5μg/m L㊂注:标准物质可以使用满足溯源要求的商品化标准溶液㊂3.4标准溶液配制3.4.1标准储备溶液(10μg/m L):分别称取A F TB1㊁A F TB2㊁A F TG1和A F TG21m g(精确至0.01m g),用乙腈溶解并定容至100m L㊂此溶液浓度约为10μg/m L㊂溶液转移至试剂瓶中后,在-20ħ下避光保存,备用㊂临用前进行浓度校准(校准方法参见附录A)㊂3.4.2混合标准工作液(100n g/m L):准确移取混合标准储备溶液(1.0μg/m L)1.00m L至100m L容量瓶中,乙腈定容㊂此溶液密封后避光-20ħ下保存,三个月有效㊂3.4.3混合同位素内标工作液(100n g/m L):准确移取0.5μg/m L13C17-A F TB1㊁13C17-A F TB2㊁13C17-A F T G1和13C17-A F T G2各2.00m L,用乙腈定容至10m L㊂在-20ħ下避光保存,备用㊂3.4.4标准系列工作溶液:准确移取混合标准工作液(100n g/m L)10μL㊁50μL㊁100μL㊁200μL㊁500μL㊁800μL㊁1000μL至10m L容量瓶中,加入200μL100n g/m L的同位素内标工作液,用初始流动相定容至刻度,配制浓度点为0.1n g/m L㊁0.5n g/m L㊁1.0n g/m L㊁2.0n g/m L㊁5.0n g/m L㊁8.0n g/m L㊁10.0n g/m L的系列标准溶液㊂4仪器和设备4.1匀浆机㊂4.2高速粉碎机㊂4.3组织捣碎机㊂4.4超声波/涡旋振荡器或摇床㊂4.5天平:感量0.01g和0.00001g㊂4.6涡旋混合器㊂4.7高速均质器:转速6500r/m i n~24000r/m i n㊂4.8离心机:转速ȡ6000r/m i n㊂4.9玻璃纤维滤纸:快速㊁高载量㊁液体中颗粒保留1.6μm㊂4.10固相萃取装置(带真空泵)㊂4.11氮吹仪㊂4.12液相色谱-串联质谱仪:带电喷雾离子源㊂4.13液相色谱柱㊂4.14免疫亲和柱:A F TB1柱容量ȡ200n g,A F TB1柱回收率ȡ80%,A F TG2的交叉反应率ȡ80%(验证方法参见附录B)㊂注:对于不同批次的亲和柱在使用前需进行质量验证㊂4.15黄曲霉毒素专用型固相萃取净化柱或功能相当的固相萃取柱(以下简称净化柱):对复杂基质样品测定时使用㊂4.16微孔滤头:带0.22μm微孔滤膜(所选用滤膜应采用标准溶液检验确认无吸附现象,方可使用)㊂4.17筛网:1mm~2mm试验筛孔径㊂4.18p H计㊂5分析步骤使用不同厂商的免疫亲和柱,在样品上样㊁淋洗和洗脱的操作方面可能会略有不同,应该按照供应商所提供的操作说明书要求进行操作㊂警示:整个分析操作过程应在指定区域内进行㊂该区域应避光(直射阳光)㊁具备相对独立的操作台和废弃物存放装置㊂在整个实验过程中,操作者应按照接触剧毒物的要求采取相应的保护措施㊂5.1样品制备5.1.1液体样品(植物油㊁酱油㊁醋等)采样量需大于1L,对于袋装㊁瓶装等包装样品需至少采集3个包装(同一批次或号),将所有液体样品在一个容器中用匀浆机混匀后,其中任意的100g(m L)样品进行检测㊂5.1.2固体样品(谷物及其制品㊁坚果及籽类㊁婴幼儿谷类辅助食品等)采样量需大于1k g,用高速粉碎机将其粉碎,过筛,使其粒径小于2mm孔径试验筛,混合均匀后缩分至100g,储存于样品瓶中,密封保存,供检测用㊂5.1.3半流体(腐乳㊁豆豉等)采样量需大于1k g(L),对于袋装㊁瓶装等包装样品需至少采集3个包装(同一批次或号),用组织捣碎机捣碎混匀后,储存于样品瓶中,密封保存,供检测用㊂5.2样品提取5.2.1液体样品5.2.1.1植物油脂称取5g试样(精确至0.01g)于50m L离心管中,加入100μL同位素内标工作液(3.4.3)振荡混合后静置30m i n㊂加入20m L乙腈-水溶液(84+16)或甲醇-水溶液(70+30),涡旋混匀,置于超声波/涡旋振荡器或摇床中振荡20m i n(或用均质器均质3m i n),在6000r/m i n下离心10m i n,取上清液备用㊂5.2.1.2酱油㊁醋称取5g试样(精确至0.01g)于50m L离心管中,加入125μL同位素内标工作液振荡混合后静置30m i n㊂用乙腈或甲醇定容至25m L(精确至0.1m L),涡旋混匀,置于超声波/涡旋振荡器或摇床中振荡20m i n(或用均质器均质3m i n),在6000r/m i n下离心10m i n(或均质后玻璃纤维滤纸过滤),取上清液备用㊂5.2.2固体样品5.2.2.1一般固体样品称取5g试样(精确至0.01g)于50m L离心管中,加入100μL同位素内标工作液振荡混合后静置30m i n㊂加入20.0m L乙腈-水溶液(84+16)或甲醇-水溶液(70+30),涡旋混匀,置于超声波/涡旋振荡器或摇床中振荡20m i n(或用均质器均质3m i n),在6000r/m i n下离心10m i n(或均质后玻璃纤维滤纸过滤),取上清液备用㊂5.2.2.2婴幼儿配方食品和婴幼儿辅助食品称取5g试样(精确至0.01g)于50m L离心管中,加入100μL同位素内标工作液振荡混合后静置30m i n㊂加入20.0m L乙腈-水溶液(50+50)或甲醇-水溶液(70+30),涡旋混匀,置于超声波/涡旋振荡器或摇床中振荡20m i n(或用均质器均质3m i n),在6000r/m i n下离心10m i n(或均质后玻璃纤维滤纸过滤),取上清液备用㊂5.2.3半流体样品称取5g试样(精确至0.01g)于50m L离心管中,加入100μL同位素内标工作液振荡混合后静置30m i n㊂加入20.0m L乙腈-水溶液(84+16)或甲醇-水溶液(70+30),置于超声波/涡旋振荡器或摇床中振荡20m i n(或用均质器均质3m i n),在6000r/m i n下离心10m i n(或均质后玻璃纤维滤纸过滤),取上清液备用㊂5.3样品净化5.3.1免疫亲和柱净化5.3.1.1上样液的准备准确移取4m L上清液,加入46m L1%T r i t i o nX-100(或吐温-20)的P B S(使用甲醇-水溶液提取时可减半加入),混匀㊂5.3.1.2免疫亲和柱的准备将低温下保存的免疫亲和柱恢复至室温㊂5.3.1.3试样的净化待免疫亲和柱内原有液体流尽后,将上述样液移至50m L注射器筒中,调节下滴速度,控制样液以1m L/m i n~3m L/m i n的速度稳定下滴㊂待样液滴完后,往注射器筒内加入2ˑ10m L水,以稳定流速淋洗免疫亲和柱㊂待水滴完后,用真空泵抽干亲和柱㊂脱离真空系统,在亲和柱下部放置10m L刻度试管,取下50m L的注射器筒,加入2ˑ1m L甲醇洗脱亲和柱,控制1m L/m i n~3m L/m i n的速度下滴,再用真空泵抽干亲和柱,收集全部洗脱液至试管中㊂在50ħ下用氮气缓缓地将洗脱液吹至近干,加入1.0m L初始流动相,涡旋30s溶解残留物,0.22μm滤膜过滤,收集滤液于进样瓶中以备进样㊂5.3.2黄曲霉毒素固相净化柱和免疫亲和柱同时使用(对花椒㊁胡椒和辣椒等复杂基质)5.3.2.1净化柱净化移取适量上清液,按净化柱操作说明进行净化,收集全部净化液㊂5.3.2.2免疫亲和柱净化用刻度移液管准确吸取上述净化液4m L,加入46m L1%T r i t i o n X-100(或吐温-20)的P B S[使用甲醇-水溶液提取时,加入23m L1%T r i t i o n X-100(或吐温-20)的P B S],混匀㊂按5.3.1.2和5.3.1.3处理㊂注:全自动(在线)或半自动(离线)的固相萃取仪器可优化操作参数后使用㊂5.4液相色谱参考条件液相色谱参考条件列出如下:a)流动相:A相:5mm o l/L乙酸铵溶液;B相:乙腈-甲醇溶液(50+50);b)梯度洗脱:32%B(0m i n~0.5m i n),45%B(3m i n~4m i n),100%B(4.2m i n~4.8m i n),32%B(5.0m i n~7.0m i n);c)色谱柱:C18柱(柱长100mm,柱内径2.1mm;填料粒径1.7μm),或相当者;d)流速:0.3m L/m i n;e)柱温:40ħ;f)进样体积:10μL㊂5.5质谱参考条件质谱参考条件列出如下:a)检测方式:多离子反应监测(MR M);b)离子源控制条件:参见表1;c)离子选择参数:参见表2;d)子离子扫描图:参见图C.1~图C.8;e)液相色谱-质谱图:见图C.9㊂表1离子源控制条件电离方式E S I+毛细管电压/k V3.5锥孔电压/V30射频透镜1电压/V14.9射频透镜2电压/V15.1表1(续)离子源温度/ħ150锥孔反吹气流量/(L/h)50脱溶剂气温度/ħ500脱溶剂气流量/(L/h)800电子倍增电压/V650表2离子选择参数表化合物名称母离子(m/z)定量离子(m/z)碰撞能量e V定性离子(m/z)碰撞能量e V离子化方式A F TB13132852224138E S I+13C17-A F TB13302552330135E S I+A F TB23152872525928E S I+13C17-A F TB23323032527328E S I+A F T G13292432528325E S I+13C17-A F T G13462572529925E S I+A F T G23312453028527E S I+13C17-A F T G23482593030127E S I+5.6定性测定试样中目标化合物色谱峰的保留时间与相应标准色谱峰的保留时间相比较,变化范围应在ʃ2.5%之内㊂每种化合物的质谱定性离子必须出现,至少应包括一个母离子和两个子离子,而且同一检测批次,对同一化合物,样品中目标化合物的两个子离子的相对丰度比与浓度相当的标准溶液相比,其允许偏差不超过表3规定的范围㊂表3定性时相对离子丰度的最大允许偏差相对离子丰度/%>5020~5010~20ɤ10允许相对偏差/%ʃ20ʃ25ʃ30ʃ505.7标准曲线的制作在5.4㊁5.5的液相色谱串联质谱仪分析条件下,将标准系列溶液由低到高浓度进样检测,以A F TB1㊁A F TB2㊁A F TG1和A F TG2色谱峰与各对应内标色谱峰的峰面积比值-浓度作图,得到标准曲线回归方程,其线性相关系数应大于0.99㊂5.8试样溶液的测定取5.3处理得到的待测溶液进样,内标法计算待测液中目标物质的质量浓度,按第6章计算样品中待测物的含量㊂待测样液中的响应值应在标准曲线线性范围内,超过线性范围则应适当减少取样量重新测定㊂5.9空白试验不称取试样,按5.2和5.3的步骤做空白实验㊂应确认不含有干扰待测组分的物质㊂6分析结果的表述试样中A F TB1㊁A F TB2㊁A F T G1和A F T G2的残留量按式(1)计算:X=ρˑV1ˑV3ˑ1000V2ˑmˑ1000 (1)式中:X 试样中A F TB1㊁A F TB2㊁A F T G1或A F T G2的含量,单位为微克每千克(μg/k g);ρ 进样溶液中A F TB1㊁A F TB2㊁A F T G1或A F T G2按照内标法在标准曲线中对应的浓度,单位为纳克每毫升(n g/m L);V1 试样提取液体积(植物油脂㊁固体㊁半固体按加入的提取液体积;酱油㊁醋按定容总体积),单位为毫升(m L);V3 样品经净化洗脱后的最终定容体积,单位为毫升(m L);1000 换算系数;V2 用于净化分取的样品体积,单位为毫升(m L);m 试样的称样量,单位为克(g)㊂计算结果保留三位有效数字㊂7精密度在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的20%㊂8其他当称取样品5g时,A F TB1的检出限为:0.03μg/k g,A F TB2的检出限为0.03μg/k g,A F TG1的检出限为0.03μg/k g,A F T G2的检出限为0.03μg/k g;A F TB1的定量限为0.1μg/k g,A F TB2的定量限为0.1μg/k g,A F T G1的定量限为0.1μg/k g,A F T G2的定量限为0.1μg/k g㊂第二法高效液相色谱-柱前衍生法9原理试样中的黄曲霉毒素B1㊁黄曲霉毒素B2㊁黄曲霉毒素G1㊁黄曲霉毒素G2,用乙腈-水溶液或甲醇-水溶液的混合溶液提取,提取液经黄曲霉毒素固相净化柱净化去除脂肪㊁蛋白质㊁色素及碳水化合物等干扰物质,净化液用三氟乙酸柱前衍生,液相色谱分离,荧光检测器检测,外标法定量㊂10试剂和材料除非另有说明,本方法所用试剂均为分析纯,水为G B/T6682规定的一级水㊂10.1试剂10.1.1甲醇(C H3O H):色谱纯㊂10.1.2乙腈(C H3C N):色谱纯㊂10.1.3正己烷(C6H14):色谱纯㊂10.1.4三氟乙酸(C F3C O O H)㊂10.2试剂配制10.2.1乙腈-水溶液(84+16):取840m L乙腈加入160m L水㊂10.2.2甲醇-水溶液(70+30):取700m L甲醇加入300m L水㊂10.2.3乙腈-水溶液(50+50):取500m L乙腈加入500m L水㊂10.2.4乙腈-甲醇溶液(50+50):取500m L乙腈加入500m L甲醇㊂10.3标准品10.3.1 A F TB1标准品(C17H12O6,C A S号:1162-65-8):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂10.3.2 A F TB2标准品(C17H14O6,C A S号:7220-81-7):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂10.3.3 A F T G1标准品(C17H12O7,C A S号:1165-39-5):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂10.3.4 A F T G2标准品(C17H14O7,C A S号:7241-98-7):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂注:标准物质可以使用满足溯源要求的商品化标准溶液㊂10.4标准溶液配制10.4.1标准储备溶液(10μg/m L):分别称取A F T B1㊁A F T B2㊁A F T G1和A F T G21m g(精确至0.01m g),用乙腈溶解并定容至100m L㊂此溶液浓度约为10μg/m L㊂溶液转移至试剂瓶中后,在-20ħ下避光保存,备用㊂临用前进行浓度校准(校准方法参见附录A)㊂10.4.2混合标准工作液(A F TB1和A F T G1:100n g/m L,A F TB2和A F T G2:30n g/m L):准确移取A F TB1和A F T G1标准储备溶液各1m L,A F TB2和A F T G2标准储备溶液各300μL至100m L容量瓶中,乙腈定容㊂密封后避光-20ħ下保存,三个月内有效㊂10.4.3标准系列工作溶液:分别准确移取混合标准工作液10μL㊁50μL㊁200μL㊁500μL㊁1000μL㊁2000μL㊁4000μL至10m L容量瓶中,用初始流动相定容至刻度(含A F T B1和A F T G1浓度为0.1n g/m L㊁0.5n g/m L㊁2.0n g/m L㊁5.0n g/m L㊁10.0n g/m L㊁20.0n g/m L㊁40.0n g/m L,A F T B2和A F T G2浓度为0.03n g/m L㊁0.15n g/m L㊁0.6n g/m L㊁1.5n g/m L㊁3.0n g/m L㊁6.0n g/m L㊁12n g/m L 的系列标准溶液)㊂11仪器和设备11.1匀浆机㊂11.2高速粉碎机㊂11.3组织捣碎机㊂11.4超声波/涡旋振荡器或摇床㊂11.5天平:感量0.01g和0.00001g㊂11.6涡旋混合器㊂11.7高速均质器:转速6500r/m i n~24000r/m i n㊂11.8离心机:转速ȡ6000r/m i n㊂11.9玻璃纤维滤纸:快速㊁高载量㊁液体中颗粒保留1.6μm㊂11.10氮吹仪㊂11.11液相色谱仪:配荧光检测器㊂11.12色谱分离柱㊂11.13黄曲霉毒素专用型固相萃取净化柱(以下简称净化柱),或相当者㊂11.14一次性微孔滤头:带0.22μm微孔滤膜(所选用滤膜应采用标准溶液检验确认无吸附现象,方可使用)㊂11.15筛网:1mm~2mm试验筛孔径㊂11.16恒温箱㊂11.17p H计㊂12分析步骤12.1样品制备12.1.1液体样品(植物油㊁酱油㊁醋等)采样量需大于1L,对于袋装㊁瓶装等包装样品需至少采集3个包装(同一批次或号),将所有液体样品在一个容器中用匀浆机混匀后,其中任意的100g(m L)样品进行检测㊂12.1.2固体样品(谷物及其制品㊁坚果及籽类㊁婴幼儿谷类辅助食品等)采样量需大于1k g,用高速粉碎机将其粉碎,过筛,使其粒径小于2mm孔径试验筛,混合均匀后缩分至100g,储存于样品瓶中,密封保存,供检测用㊂12.1.3半流体(腐乳㊁豆豉等)采样量需大于1k g(L),对于袋装㊁瓶装等包装样品需至少采集3个包装(同一批次或号),用组织捣碎机捣碎混匀后,储存于样品瓶中,密封保存,供检测用㊂12.2样品提取12.2.1液体样品12.2.1.1植物油脂称取5g试样(精确至0.01g)于50m L离心管中,加入20m L乙腈-水溶液(84+16)或甲醇-水溶液(70+30),涡旋混匀,置于超声波/涡旋振荡器或摇床中振荡20m i n(或用均质器均质3m i n),在6000r/m i n下离心10m i n,取上清液备用㊂12.2.1.2酱油㊁醋称取5g试样(精确至0.01g)于50m L离心管中,用乙腈或甲醇定容至25m L(精确至0.1m L),涡旋混匀,置于超声波/涡旋振荡器或摇床中振荡20m i n(或用均质器均质3m i n),在6000r/m i n下离心10m i n(或均质后玻璃纤维滤纸过滤),取上清液备用㊂12.2.2固体样品12.2.2.1一般固体样品称取5g试样(精确至0.01g)于50m L离心管中,加入20.0m L乙腈-水溶液(84+16)或甲醇-水溶液(70+30),涡旋混匀,置于超声波/涡旋振荡器或摇床中振荡20m i n(或用均质器均质3m i n),在6000r/m i n下离心10m i n(或均质后玻璃纤维滤纸过滤),取上清液备用㊂12.2.2.2婴幼儿配方食品和婴幼儿辅助食品称取5g试样(精确至0.01g)于50m L离心管中,加入20.0m L乙腈-水溶液(50+50)或甲醇-水溶液(70+30),涡旋混匀,置于超声波/涡旋振荡器或摇床中振荡20m i n(或用均质器均质3m i n),在6000r/m i n下离心10m i n(或均质后玻璃纤维滤纸过滤),取上清液备用㊂12.2.3半流体样品称取5g试样(精确至0.01g)于50m L离心管中,加入20.0m L乙腈-水溶液(84+16)或甲醇-水溶液(70+30),置于超声波/涡旋振荡器或摇床中振荡20m i n(或用均质器均质3m i n),在6000r/m i n下离心10m i n(或均质后玻璃纤维滤纸过滤),取上清液备用㊂12.3样品黄曲霉毒素固相净化柱净化移取适量上清液,按净化柱操作说明进行净化,收集全部净化液㊂12.4衍生用移液管准确吸取4.0m L净化液于10m L离心管后在50ħ下用氮气缓缓地吹至近干,分别加入200μL正己烷和100μL三氟乙酸,涡旋30s,在40ħʃ1ħ的恒温箱中衍生15m i n,衍生结束后,在50ħ下用氮气缓缓地将衍生液吹至近干,用初始流动相定容至1.0m L,涡旋30s溶解残留物,过0.22μm滤膜,收集滤液于进样瓶中以备进样㊂12.5色谱参考条件色谱参考条件列出如下:a)流动相:A相:水,B相:乙腈-甲醇溶液(50+50);b)梯度洗脱:24%B(0m i n~6m i n),35%B(8.0m i n~10.0m i n),100%B(10.2m i n~11.2m i n),24%B(11.5m i n~13.0m i n);c)色谱柱:C18柱(柱长150mm或250mm,柱内径4.6mm,填料粒径5.0μm),或相当者;d)流速:1.0m L/m i n;e)柱温:40ħ;f)进样体积:50μL;g)检测波长:激发波长360n m;发射波长440n m;h)液相色谱图:参见图D.1㊂12.6样品测定12.6.1标准曲线的制作系列标准工作溶液由低到高浓度依次进样检测,以峰面积为纵坐标-浓度为横坐标作图,得到标准曲线回归方程㊂12.6.2试样溶液的测定待测样液中待测化合物的响应值应在标准曲线线性范围内,浓度超过线性范围的样品则应稀释后重新进样分析㊂12.6.3空白试验不称取试样,按12.2㊁12.3和12.4的步骤做空白实验㊂应确认不含有干扰待测组分的物质㊂13分析结果的表述试样中A F TB1㊁A F TB2㊁A F T G1和A F T G2的残留量按式(2)计算:X=ρˑV1ˑV3ˑ1000V2ˑmˑ1000 (2)式中:X 试样中A F TB1㊁A F TB2㊁A F T G1或A F T G2的含量,单位为微克每千克(μg/k g);ρ 进样溶液中A F TB1㊁A F TB2㊁A F T G1或A F T G2按照外标法在标准曲线中对应的浓度,单位为纳克每毫升(n g/m L);V1 试样提取液体积(植物油脂㊁固体㊁半固体按加入的提取液体积;酱油㊁醋按定容总体积),单位为毫升(m L);V3 净化液的最终定容体积,单位为毫升(m L);1000 换算系数;V2 净化柱净化后的取样液体积,单位为毫升(m L);m 试样的称样量,单位为克(g)㊂计算结果保留三位有效数字㊂14精密度在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的20%㊂15其他当称取样品5g时,柱前衍生法的A F TB1的检出限为0.03μg/k g,A F TB2的检出限为0.03μg/k g, A F TG1的检出限为0.03μg/k g,A F TG2的检出限为0.03μg/k g;柱前衍生法的A F TB1的定量限为0.1μg/ k g,A F TB2的定量限为0.1μg/k g,A F TG1的定量限为0.1μg/k g,A F TG2的定量限为0.1μg/k g㊂第三法高效液相色谱-柱后衍生法导语:下述方法的仪器检测部分,包括碘或溴试剂衍生㊁光化学衍生㊁电化学衍生等柱后衍生方法,可根据实际情况,选择其中一种方法即可㊂16原理试样中的黄曲霉毒素B1㊁黄曲霉毒素B2㊁黄曲霉毒素G1㊁黄曲霉毒素G2,用乙腈-水溶液或甲醇-水溶液的混合溶液提取,提取液经免疫亲和柱净化和富集,净化液浓缩㊁定容和过滤后经液相色谱分离,柱后衍生(碘或溴试剂衍生㊁光化学衍生㊁电化学衍生等),经荧光检测器检测,外标法定量㊂17试剂和材料除非另有说明,本方法所用试剂均为分析纯,水为G B/T6682规定的一级水㊂17.1试剂17.1.1甲醇(C H3O H):色谱纯㊂17.1.2乙腈(C H3C N):色谱纯㊂17.1.3氯化钠(N a C l)㊂17.1.4磷酸氢二钠(N a2H P O4)㊂17.1.5磷酸二氢钾(K H2P O4)㊂17.1.6氯化钾(K C l)㊂17.1.7盐酸(H C l)㊂17.1.8 T r i t o nX-100[C14H22O(C2H4O)n](或吐温-20,C58H114O26)㊂17.1.9碘衍生使用试剂:碘(I2)㊂17.1.10溴衍生使用试剂:三溴化吡啶(C5H6B r3N2)㊂17.1.11电化学衍生使用试剂:溴化钾(K B r)㊁浓硝酸(HN O3)㊂17.2试剂配制17.2.1乙腈-水溶液(84+16):取840m L乙腈加入160m L水㊂17.2.2甲醇-水溶液(70+30):取700m L甲醇加入300m L水㊂17.2.3乙腈-水溶液(50+50):取500m L乙腈加入500m L水㊂17.2.4乙腈-水溶液(10+90):取100m L乙腈加入900m L水㊂17.2.5乙腈-甲醇溶液(50+50):取500m L乙腈加入500m L甲醇㊂17.2.6磷酸盐缓冲溶液(以下简称P B S):称取8.00g氯化钠㊁1.20g磷酸氢二钠(或2.92g十二水磷酸氢二钠)㊁0.20g磷酸二氢钾㊁0.20g氯化钾,用900m L水溶解,用盐酸调节p H至7.4,用水定容至1000m L㊂17.2.71%T r i t o nX-100(或吐温-20)的P B S:取10m LT r i t o nX-100,用P B S定容至1000m L㊂17.2.80.05%碘溶液:称取0.1g碘,用20m L甲醇溶解,加水定容至200m L,用0.45μm的滤膜过滤,现配现用(仅碘柱后衍生法使用)㊂17.2.95m g/L三溴化吡啶水溶液:称取5m g三溴化吡啶溶于1L水中,用0.45μm的滤膜过滤,现配现用(仅溴柱后衍生法使用)㊂17.3标准品17.3.1 A F TB1标准品(C17H12O6,C A S号:1162-65-8):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂17.3.2 A F TB2标准品(C17H14O6,C A S号:7220-81-7):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂17.3.3 A F T G1标准品(C17H12O7,C A S号:1165-39-5):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂17.3.4 A F T G2标准品C17H14O7,C A S号:7241-98-7):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂注:标准物质可以使用满足溯源要求的商品化标准溶液㊂17.4标准溶液配制17.4.1标准储备溶液(10μg/m L):分别称取A F T B1㊁A F T B2㊁A F T G1和A F T G21m g(精确至0.01m g),用乙腈溶解并定容至100m L㊂此溶液浓度约为10μg/m L㊂溶液转移至试剂瓶中后,在-20ħ下避光保存,备用㊂临用前进行浓度校准(校准方法参见附录A)㊂17.4.2混合标准工作液(A F TB1和A F T G1:100n g/m L,A F TB2和A F T G2:30n g/m L):准确移取A F TB1和A F T G1标准储备溶液各1m L,A F TB2和A F T G2标准储备溶液各300μL至100m L容量瓶中,乙腈定容㊂密封后避光-20ħ下保存,三个月内有效㊂17.4.3标准系列工作溶液:分别准确移取混合标准工作液10μL㊁50μL㊁200μL㊁500μL㊁1000μL㊁2000μL㊁4000μL至10m L容量瓶中,用初始流动相定容至刻度(含A F T B1和A F T G1浓度为0.1n g/m L㊁0.5n g/m L㊁2.0n g/m L㊁5.0n g/m L㊁10.0n g/m L㊁20.0n g/m L㊁40.0n g/m L,A F T B2和A F T G2浓度为0.03n g/m L㊁0.15n g/m L㊁0.6n g/m L㊁1.5n g/m L㊁3.0n g/m L㊁6.0n g/m L㊁12n g/m L 的系列标准溶液)㊂18仪器和设备18.1匀浆机㊂18.2高速粉碎机㊂18.3组织捣碎机㊂18.4超声波/涡旋振荡器或摇床㊂18.5天平:感量0.01g和0.00001g㊂18.6涡旋混合器㊂18.7高速均质器:转速6500r/m i n~24000r/m i n㊂18.8离心机:转速ȡ6000r/m i n㊂18.9玻璃纤维滤纸:快速㊁高载量㊁液体中颗粒保留1.6μm㊂18.10固相萃取装置(带真空泵)㊂18.11氮吹仪㊂18.12液相色谱仪:配荧光检测器(带一般体积流动池或者大体积流通池)㊂注:当带大体积流通池时不需要再使用任何型号或任何方式的柱后衍生器㊂18.13液相色谱柱㊂18.14光化学柱后衍生器(适用于光化学柱后衍生法)㊂18.15溶剂柱后衍生装置(适用于碘或溴试剂衍生法)㊂18.16电化学柱后衍生器(适用于电化学柱后衍生法)㊂18.17免疫亲和柱:A F TB1柱容量ȡ200n g,A F TB1柱回收率ȡ80%,A F T G2的交叉反应率ȡ80% (验证方法参见附录B)㊂注:对于每个批次的亲和柱使用前需质量验证㊂18.18黄曲霉毒素固相净化柱或功能相当的固相萃取柱(以下简称净化柱):对复杂基质样品测定时使用㊂18.19一次性微孔滤头:带0.22μm微孔滤膜(所选用滤膜应采用标准溶液检验确认无吸附现象,方可使用)㊂18.20筛网:1mm~2mm试验筛孔径㊂19分析步骤使用不同厂商的免疫亲和柱,在样品的上样㊁淋洗和洗脱的操作方面可能略有不同,应该按照供应商所提供的操作说明书要求进行操作㊂警示:整个分析操作过程应在指定区域内进行㊂该区域应避光(直射阳光)㊁具备相对独立的操作台和废弃物存放装置㊂在整个实验过程中,操作者应按照接触剧毒物的要求采取相应的保护措施㊂19.1样品制备同12.1㊂19.2样品提取同12.2㊂19.3样品净化19.3.1免疫亲和柱净化19.3.1.1上样液的准备准确移取4m L上述上清液,加入46m L1%T r i t o nX-100(或吐温-20)的P B S(使用甲醇-水溶液提取时可减半加入),混匀㊂19.3.1.2免疫亲和柱的准备将低温下保存的免疫亲和柱恢复至室温㊂19.3.1.3试样的净化免疫亲和柱内的液体放弃后,将上述样液移至50m L注射器筒中,调节下滴速度,控制样液以1m L/m i n~3m L/m i n的速度稳定下滴㊂待样液滴完后,往注射器筒内加入2ˑ10m L水,以稳定流速淋洗免疫亲和柱㊂待水滴完后,用真空泵抽干亲和柱㊂脱离真空系统,在亲和柱下部放置10m L刻度试管,取下50m L的注射器筒,2ˑ1m L甲醇洗脱亲和柱,控制1m L/m i n~3m L/m i n的速度下滴,再用真空泵抽干亲和柱,收集全部洗脱液至试管中㊂在50ħ下用氮气缓缓地将洗脱液吹至近干,用初始流动相定容至1.0m L,涡旋30s溶解残留物,0.22μm滤膜过滤,收集滤液于进样瓶中以备进样㊂19.3.2黄曲霉毒素固相净化柱和免疫亲和柱同时使用(对花椒㊁胡椒和辣椒等复杂基质)19.3.2.1净化柱净化移取适量上清液,按净化柱操作说明进行净化,收集全部净化液㊂19.3.2.2免疫亲和柱净化用刻度移液管准确吸取上部净化液4m L,加入46m L1%T r i t o nX-100(或吐温-20)的P B S(使用甲醇-水溶液提取时可减半加入),混匀㊂按19.4.1.3处理㊂注:全自动(在线)或半自动(离线)的固相萃取仪器可优化操作参数后使用㊂19.4液相色谱参考条件19.4.1无衍生器法(大流通池直接检测)液相色谱参考条件列出如下:a)流动相:A相,水;B相,乙腈-甲醇(50+50);b)等梯度洗脱条件:A,65%;B,35%;。
黄曲霉毒素B1检测方法的分析

黄曲霉毒素B1检测方法的分析叶雪珠 王小骊 赵燕申 董秀金(浙江省农科院农产品质量标准研究所,杭州,310021)摘 要 黄曲霉毒素B1是一种致癌物质,许多国家已将其限量标准提高,黄曲霉毒素B1检测方法的检出限已因其毒性而提高,并已成为国际贸易中新的技术壁垒。
文中介绍了黄曲霉毒素B1的几种常见检测方法,并对其特点及检出限进行了概括性的分析。
关键词 黄曲霉毒素B1,检测方法,第一作者:硕士,助理研究员。
收稿时间:2003-07-11黄曲霉毒素(AFT)是公认的天然强致癌物质之一,其中以黄曲霉毒素B1(AFTB1)毒性最大,它们常存在于各种动物饲料和人类食品中。
用污染的饲料饲养畜禽,会导致肉、乳及其制品的污染,人类食用被污染的食品会导致急性中毒,引起肝脏坏死出血,慢性中毒可引起肝癌。
同时用被污染的饲料饲养畜禽会使畜禽生产率降低,增重减慢,造成重大经济损失[1,2]。
迄今为止,急性AFT 中毒病例在世界上许多国家,尤其在发展中国家报道较多。
因此各国对食品及饲料中AFTB1的限量进行了规定[3],欧盟等国在2002年对粮食、花生及其产品中的AFT B1的限量进行了修订,再次提高上述农产品中的AFTB1的限量标准,如供人类直接食用的花生AFTB1限量为 2 g/kg,作为食品原料进口的花生AFTB1限量为 8 g/kg 。
我国也先后于1982、1992和1998年制定并实施了食品和饲料中AFTB1允许含量的强制性国家标准,但限量要求仍低于国外。
目前在出口食品中AFTB1已经成为限制出口的技术壁垒,为保障食用者的健康,应对技术壁垒,在全球贸易竞争中立于不败之地,必须加强AFT B1的监测工作,提高AFTB1的检测技术已是势在必行。
近年来,关于AFTB1的检测方法研究较多,大致可分为薄层层析法(TLC )、液相色谱法(H PLC)和酶联免疫法(ELISA)。
本文根据相关报道,总结分析了AFT B1的几种检测方法,并对其特点及检出限进行了比较。
食品中黄曲霉毒素B族和G族的测定
食品安全国家标准食品中黄曲霉毒素B族和G族的测定1范围本标准规定了食品中黄曲霉毒素B1㊁黄曲霉毒素B2㊁黄曲霉毒素G1㊁黄曲霉毒素G2(以下简称A F TB1㊁A F TB2㊁A F T G1和A F T G2)的测定方法㊂本标准第一法为同位素稀释液相色谱-串联质谱法,适用于谷物及其制品㊁豆类及其制品㊁坚果及籽类㊁油脂及其制品㊁调味品㊁婴幼儿配方食品和婴幼儿辅助食品中A F TB1㊁A F TB2㊁A F TG1和A F TG2的测定㊂本标准第二法为高效液相色谱-柱前衍生法,适用于谷物及其制品㊁豆类及其制品㊁坚果及籽类㊁油脂及其制品㊁调味品㊁婴幼儿配方食品和婴幼儿辅助食品中A F T B1㊁A F T B2㊁A F T G1和A F T G2的测定㊂本标准第三法为高效液相色谱-柱后衍生法,适用于谷物及其制品㊁豆类及其制品㊁坚果及籽类㊁油脂及其制品㊁调味品㊁婴幼儿配方食品和婴幼儿辅助食品中A F T B1㊁A F T B2㊁A F T G1和A F T G2的测定㊂本标准第四法为酶联免疫吸附筛查法,适用于谷物及其制品㊁豆类及其制品㊁坚果及籽类㊁油脂及其制品㊁调味品㊁婴幼儿配方食品和婴幼儿辅助食品中A F TB1的测定㊂本标准第五法为薄层色谱法,适用于谷物及其制品㊁豆类及其制品㊁坚果及籽类㊁油脂及其制品㊁调味品中A F TB1的测定㊂第一法同位素稀释液相色谱-串联质谱法2原理试样中的黄曲霉毒素B1㊁黄曲霉毒素B2㊁黄曲霉毒素G1㊁黄曲霉毒素G2,用乙腈-水溶液或甲醇-水溶液提取,提取液用含1%T r i t o nX-100(或吐温-20)的磷酸盐缓冲溶液稀释后(必要时经黄曲霉毒素固相净化柱初步净化),通过免疫亲和柱净化和富集,净化液浓缩㊁定容和过滤后经液相色谱分离,串联质谱检测,同位素内标法定量㊂3试剂和材料除非另有说明,本方法所用试剂均为分析纯,水为G B/T6682规定的一级水㊂3.1试剂3.1.1乙腈(C H3C N):色谱纯㊂3.1.2甲醇(C H3O H):色谱纯㊂3.1.3乙酸铵(C H3C O O N H4):色谱纯㊂3.1.4氯化钠(N a C l)㊂3.1.5磷酸氢二钠(N a2H P O4)㊂3.1.6磷酸二氢钾(K H2P O4)㊂3.1.7氯化钾(K C l)㊂3.1.8盐酸(H C l)㊂3.1.9 T r i t o nX-100[C14H22O(C2H4O)n](或吐温-20,C58H114O26)㊂3.2试剂配制3.2.1乙酸铵溶液(5mm o l/L):称取0.39g乙酸铵,用水溶解后稀释至1000m L,混匀㊂3.2.2乙腈-水溶液(84+16):取840m L乙腈加入160m L水,混匀㊂3.2.3甲醇-水溶液(70+30):取700m L甲醇加入300m L水,混匀㊂3.2.4乙腈-水溶液(50+50):取50m L乙腈加入50m L水,混匀㊂3.2.5乙腈-甲醇溶液(50+50):取50m L乙腈加入50m L甲醇,混匀㊂3.2.610%盐酸溶液:取1m L盐酸,用纯水稀释至10m L,混匀㊂3.2.7磷酸盐缓冲溶液(以下简称P B S):称取8.00g氯化钠㊁1.20g磷酸氢二钠(或2.92g十二水磷酸氢二钠)㊁0.20g磷酸二氢钾㊁0.20g氯化钾,用900m L水溶解,用盐酸调节p H至7.4ʃ0.1,加水稀释至1000m L㊂3.2.81%T r i t o nX-100(或吐温-20)的P B S:取10m LT r i t o nX-100(或吐温-20),用P B S稀释至1000m L㊂3.3标准品3.3.1 A F TB1标准品(C17H12O6,C A S:1162-65-8):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂3.3.2 A F TB2标准品(C17H14O6,C A S:7220-81-7):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂3.3.3 A F T G1标准品(C17H12O7,C A S:1165-39-5):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂3.3.4 A F T G2标准品(C17H14O7,C A S:7241-98-7):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂3.3.5同位素内标13C17-A F TB1(C17H12O6,C A S:157449-45-0):纯度ȡ98%,浓度为0.5μg/m L㊂3.3.6同位素内标13C17-A F TB2(C17H14O6,C A S:157470-98-8):纯度ȡ98%,浓度为0.5μg/m L㊂3.3.7同位素内标13C17-A F T G1(C17H12O7,C A S:157444-07-9):纯度ȡ98%,浓度为0.5μg/m L㊂3.3.8同位素内标13C17-A F T G2(C17H14O7,C A S:157462-49-7):纯度ȡ98%,浓度为0.5μg/m L㊂注:标准物质可以使用满足溯源要求的商品化标准溶液㊂3.4标准溶液配制3.4.1标准储备溶液(10μg/m L):分别称取A F TB1㊁A F TB2㊁A F TG1和A F TG21m g(精确至0.01m g),用乙腈溶解并定容至100m L㊂此溶液浓度约为10μg/m L㊂溶液转移至试剂瓶中后,在-20ħ下避光保存,备用㊂临用前进行浓度校准(校准方法参见附录A)㊂3.4.2混合标准工作液(100n g/m L):准确移取混合标准储备溶液(1.0μg/m L)1.00m L至100m L容量瓶中,乙腈定容㊂此溶液密封后避光-20ħ下保存,三个月有效㊂3.4.3混合同位素内标工作液(100n g/m L):准确移取0.5μg/m L13C17-A F TB1㊁13C17-A F TB2㊁13C17-A F T G1和13C17-A F T G2各2.00m L,用乙腈定容至10m L㊂在-20ħ下避光保存,备用㊂3.4.4标准系列工作溶液:准确移取混合标准工作液(100n g/m L)10μL㊁50μL㊁100μL㊁200μL㊁500μL㊁800μL㊁1000μL至10m L容量瓶中,加入200μL100n g/m L的同位素内标工作液,用初始流动相定容至刻度,配制浓度点为0.1n g/m L㊁0.5n g/m L㊁1.0n g/m L㊁2.0n g/m L㊁5.0n g/m L㊁8.0n g/m L㊁10.0n g/m L的系列标准溶液㊂4仪器和设备4.1匀浆机㊂4.2高速粉碎机㊂4.3组织捣碎机㊂4.4超声波/涡旋振荡器或摇床㊂4.5天平:感量0.01g和0.00001g㊂4.6涡旋混合器㊂4.7高速均质器:转速6500r/m i n~24000r/m i n㊂4.8离心机:转速ȡ6000r/m i n㊂4.9玻璃纤维滤纸:快速㊁高载量㊁液体中颗粒保留1.6μm㊂4.10固相萃取装置(带真空泵)㊂4.11氮吹仪㊂4.12液相色谱-串联质谱仪:带电喷雾离子源㊂4.13液相色谱柱㊂4.14免疫亲和柱:A F TB1柱容量ȡ200n g,A F TB1柱回收率ȡ80%,A F TG2的交叉反应率ȡ80%(验证方法参见附录B)㊂注:对于不同批次的亲和柱在使用前需进行质量验证㊂4.15黄曲霉毒素专用型固相萃取净化柱或功能相当的固相萃取柱(以下简称净化柱):对复杂基质样品测定时使用㊂4.16微孔滤头:带0.22μm微孔滤膜(所选用滤膜应采用标准溶液检验确认无吸附现象,方可使用)㊂4.17筛网:1mm~2mm试验筛孔径㊂4.18p H计㊂5分析步骤使用不同厂商的免疫亲和柱,在样品上样㊁淋洗和洗脱的操作方面可能会略有不同,应该按照供应商所提供的操作说明书要求进行操作㊂警示:整个分析操作过程应在指定区域内进行㊂该区域应避光(直射阳光)㊁具备相对独立的操作台和废弃物存放装置㊂在整个实验过程中,操作者应按照接触剧毒物的要求采取相应的保护措施㊂5.1样品制备5.1.1液体样品(植物油㊁酱油㊁醋等)采样量需大于1L,对于袋装㊁瓶装等包装样品需至少采集3个包装(同一批次或号),将所有液体样品在一个容器中用匀浆机混匀后,其中任意的100g(m L)样品进行检测㊂5.1.2固体样品(谷物及其制品㊁坚果及籽类㊁婴幼儿谷类辅助食品等)采样量需大于1k g,用高速粉碎机将其粉碎,过筛,使其粒径小于2mm孔径试验筛,混合均匀后缩分至100g,储存于样品瓶中,密封保存,供检测用㊂5.1.3半流体(腐乳㊁豆豉等)采样量需大于1k g(L),对于袋装㊁瓶装等包装样品需至少采集3个包装(同一批次或号),用组织捣碎机捣碎混匀后,储存于样品瓶中,密封保存,供检测用㊂5.2样品提取5.2.1液体样品5.2.1.1植物油脂称取5g试样(精确至0.01g)于50m L离心管中,加入100μL同位素内标工作液(3.4.3)振荡混合后静置30m i n㊂加入20m L乙腈-水溶液(84+16)或甲醇-水溶液(70+30),涡旋混匀,置于超声波/涡旋振荡器或摇床中振荡20m i n(或用均质器均质3m i n),在6000r/m i n下离心10m i n,取上清液备用㊂5.2.1.2酱油㊁醋称取5g试样(精确至0.01g)于50m L离心管中,加入125μL同位素内标工作液振荡混合后静置30m i n㊂用乙腈或甲醇定容至25m L(精确至0.1m L),涡旋混匀,置于超声波/涡旋振荡器或摇床中振荡20m i n(或用均质器均质3m i n),在6000r/m i n下离心10m i n(或均质后玻璃纤维滤纸过滤),取上清液备用㊂5.2.2固体样品5.2.2.1一般固体样品称取5g试样(精确至0.01g)于50m L离心管中,加入100μL同位素内标工作液振荡混合后静置30m i n㊂加入20.0m L乙腈-水溶液(84+16)或甲醇-水溶液(70+30),涡旋混匀,置于超声波/涡旋振荡器或摇床中振荡20m i n(或用均质器均质3m i n),在6000r/m i n下离心10m i n(或均质后玻璃纤维滤纸过滤),取上清液备用㊂5.2.2.2婴幼儿配方食品和婴幼儿辅助食品称取5g试样(精确至0.01g)于50m L离心管中,加入100μL同位素内标工作液振荡混合后静置30m i n㊂加入20.0m L乙腈-水溶液(50+50)或甲醇-水溶液(70+30),涡旋混匀,置于超声波/涡旋振荡器或摇床中振荡20m i n(或用均质器均质3m i n),在6000r/m i n下离心10m i n(或均质后玻璃纤维滤纸过滤),取上清液备用㊂5.2.3半流体样品称取5g试样(精确至0.01g)于50m L离心管中,加入100μL同位素内标工作液振荡混合后静置30m i n㊂加入20.0m L乙腈-水溶液(84+16)或甲醇-水溶液(70+30),置于超声波/涡旋振荡器或摇床中振荡20m i n(或用均质器均质3m i n),在6000r/m i n下离心10m i n(或均质后玻璃纤维滤纸过滤),取上清液备用㊂5.3样品净化5.3.1免疫亲和柱净化5.3.1.1上样液的准备准确移取4m L上清液,加入46m L1%T r i t i o nX-100(或吐温-20)的P B S(使用甲醇-水溶液提取时可减半加入),混匀㊂5.3.1.2免疫亲和柱的准备将低温下保存的免疫亲和柱恢复至室温㊂5.3.1.3试样的净化待免疫亲和柱内原有液体流尽后,将上述样液移至50m L注射器筒中,调节下滴速度,控制样液以1m L/m i n~3m L/m i n的速度稳定下滴㊂待样液滴完后,往注射器筒内加入2ˑ10m L水,以稳定流速淋洗免疫亲和柱㊂待水滴完后,用真空泵抽干亲和柱㊂脱离真空系统,在亲和柱下部放置10m L刻度试管,取下50m L的注射器筒,加入2ˑ1m L甲醇洗脱亲和柱,控制1m L/m i n~3m L/m i n的速度下滴,再用真空泵抽干亲和柱,收集全部洗脱液至试管中㊂在50ħ下用氮气缓缓地将洗脱液吹至近干,加入1.0m L初始流动相,涡旋30s溶解残留物,0.22μm滤膜过滤,收集滤液于进样瓶中以备进样㊂5.3.2黄曲霉毒素固相净化柱和免疫亲和柱同时使用(对花椒㊁胡椒和辣椒等复杂基质)5.3.2.1净化柱净化移取适量上清液,按净化柱操作说明进行净化,收集全部净化液㊂5.3.2.2免疫亲和柱净化用刻度移液管准确吸取上述净化液4m L,加入46m L1%T r i t i o n X-100(或吐温-20)的P B S[使用甲醇-水溶液提取时,加入23m L1%T r i t i o n X-100(或吐温-20)的P B S],混匀㊂按5.3.1.2和5.3.1.3处理㊂注:全自动(在线)或半自动(离线)的固相萃取仪器可优化操作参数后使用㊂5.4液相色谱参考条件液相色谱参考条件列出如下:a)流动相:A相:5mm o l/L乙酸铵溶液;B相:乙腈-甲醇溶液(50+50);b)梯度洗脱:32%B(0m i n~0.5m i n),45%B(3m i n~4m i n),100%B(4.2m i n~4.8m i n),32%B(5.0m i n~7.0m i n);c)色谱柱:C18柱(柱长100mm,柱内径2.1mm;填料粒径1.7μm),或相当者;d)流速:0.3m L/m i n;e)柱温:40ħ;f)进样体积:10μL㊂5.5质谱参考条件质谱参考条件列出如下:a)检测方式:多离子反应监测(MR M);b)离子源控制条件:参见表1;c)离子选择参数:参见表2;d)子离子扫描图:参见图C.1~图C.8;e)液相色谱-质谱图:见图C.9㊂表1离子源控制条件电离方式E S I+毛细管电压/k V3.5锥孔电压/V30射频透镜1电压/V14.9射频透镜2电压/V15.1表1(续)离子源温度/ħ150锥孔反吹气流量/(L/h)50脱溶剂气温度/ħ500脱溶剂气流量/(L/h)800电子倍增电压/V650表2离子选择参数表化合物名称母离子(m/z)定量离子(m/z)碰撞能量e V定性离子(m/z)碰撞能量e V离子化方式A F TB13132852224138E S I+13C17-A F TB13302552330135E S I+A F TB23152872525928E S I+13C17-A F TB23323032527328E S I+A F T G13292432528325E S I+13C17-A F T G13462572529925E S I+A F T G23312453028527E S I+13C17-A F T G23482593030127E S I+5.6定性测定试样中目标化合物色谱峰的保留时间与相应标准色谱峰的保留时间相比较,变化范围应在ʃ2.5%之内㊂每种化合物的质谱定性离子必须出现,至少应包括一个母离子和两个子离子,而且同一检测批次,对同一化合物,样品中目标化合物的两个子离子的相对丰度比与浓度相当的标准溶液相比,其允许偏差不超过表3规定的范围㊂表3定性时相对离子丰度的最大允许偏差相对离子丰度/%>5020~5010~20ɤ10允许相对偏差/%ʃ20ʃ25ʃ30ʃ505.7标准曲线的制作在5.4㊁5.5的液相色谱串联质谱仪分析条件下,将标准系列溶液由低到高浓度进样检测,以A F TB1㊁A F TB2㊁A F TG1和A F TG2色谱峰与各对应内标色谱峰的峰面积比值-浓度作图,得到标准曲线回归方程,其线性相关系数应大于0.99㊂5.8试样溶液的测定取5.3处理得到的待测溶液进样,内标法计算待测液中目标物质的质量浓度,按第6章计算样品中待测物的含量㊂待测样液中的响应值应在标准曲线线性范围内,超过线性范围则应适当减少取样量重新测定㊂5.9空白试验不称取试样,按5.2和5.3的步骤做空白实验㊂应确认不含有干扰待测组分的物质㊂6分析结果的表述试样中A F TB1㊁A F TB2㊁A F T G1和A F T G2的残留量按式(1)计算:X=ρˑV1ˑV3ˑ1000V2ˑmˑ1000 (1)式中:X 试样中A F TB1㊁A F TB2㊁A F T G1或A F T G2的含量,单位为微克每千克(μg/k g);ρ 进样溶液中A F TB1㊁A F TB2㊁A F T G1或A F T G2按照内标法在标准曲线中对应的浓度,单位为纳克每毫升(n g/m L);V1 试样提取液体积(植物油脂㊁固体㊁半固体按加入的提取液体积;酱油㊁醋按定容总体积),单位为毫升(m L);V3 样品经净化洗脱后的最终定容体积,单位为毫升(m L);1000 换算系数;V2 用于净化分取的样品体积,单位为毫升(m L);m 试样的称样量,单位为克(g)㊂计算结果保留三位有效数字㊂7精密度在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的20%㊂8其他当称取样品5g时,A F TB1的检出限为:0.03μg/k g,A F TB2的检出限为0.03μg/k g,A F TG1的检出限为0.03μg/k g,A F T G2的检出限为0.03μg/k g;A F TB1的定量限为0.1μg/k g,A F TB2的定量限为0.1μg/k g,A F T G1的定量限为0.1μg/k g,A F T G2的定量限为0.1μg/k g㊂第二法高效液相色谱-柱前衍生法9原理试样中的黄曲霉毒素B1㊁黄曲霉毒素B2㊁黄曲霉毒素G1㊁黄曲霉毒素G2,用乙腈-水溶液或甲醇-水溶液的混合溶液提取,提取液经黄曲霉毒素固相净化柱净化去除脂肪㊁蛋白质㊁色素及碳水化合物等干扰物质,净化液用三氟乙酸柱前衍生,液相色谱分离,荧光检测器检测,外标法定量㊂10试剂和材料除非另有说明,本方法所用试剂均为分析纯,水为G B/T6682规定的一级水㊂10.1试剂10.1.1甲醇(C H3O H):色谱纯㊂10.1.2乙腈(C H3C N):色谱纯㊂10.1.3正己烷(C6H14):色谱纯㊂10.1.4三氟乙酸(C F3C O O H)㊂10.2试剂配制10.2.1乙腈-水溶液(84+16):取840m L乙腈加入160m L水㊂10.2.2甲醇-水溶液(70+30):取700m L甲醇加入300m L水㊂10.2.3乙腈-水溶液(50+50):取500m L乙腈加入500m L水㊂10.2.4乙腈-甲醇溶液(50+50):取500m L乙腈加入500m L甲醇㊂10.3标准品10.3.1 A F TB1标准品(C17H12O6,C A S号:1162-65-8):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂10.3.2 A F TB2标准品(C17H14O6,C A S号:7220-81-7):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂10.3.3 A F T G1标准品(C17H12O7,C A S号:1165-39-5):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂10.3.4 A F T G2标准品(C17H14O7,C A S号:7241-98-7):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂注:标准物质可以使用满足溯源要求的商品化标准溶液㊂10.4标准溶液配制10.4.1标准储备溶液(10μg/m L):分别称取A F T B1㊁A F T B2㊁A F T G1和A F T G21m g(精确至0.01m g),用乙腈溶解并定容至100m L㊂此溶液浓度约为10μg/m L㊂溶液转移至试剂瓶中后,在-20ħ下避光保存,备用㊂临用前进行浓度校准(校准方法参见附录A)㊂10.4.2混合标准工作液(A F TB1和A F T G1:100n g/m L,A F TB2和A F T G2:30n g/m L):准确移取A F TB1和A F T G1标准储备溶液各1m L,A F TB2和A F T G2标准储备溶液各300μL至100m L容量瓶中,乙腈定容㊂密封后避光-20ħ下保存,三个月内有效㊂10.4.3标准系列工作溶液:分别准确移取混合标准工作液10μL㊁50μL㊁200μL㊁500μL㊁1000μL㊁2000μL㊁4000μL至10m L容量瓶中,用初始流动相定容至刻度(含A F T B1和A F T G1浓度为0.1n g/m L㊁0.5n g/m L㊁2.0n g/m L㊁5.0n g/m L㊁10.0n g/m L㊁20.0n g/m L㊁40.0n g/m L,A F T B2和A F T G2浓度为0.03n g/m L㊁0.15n g/m L㊁0.6n g/m L㊁1.5n g/m L㊁3.0n g/m L㊁6.0n g/m L㊁12n g/m L 的系列标准溶液)㊂11仪器和设备11.1匀浆机㊂11.2高速粉碎机㊂11.3组织捣碎机㊂11.4超声波/涡旋振荡器或摇床㊂11.5天平:感量0.01g和0.00001g㊂11.6涡旋混合器㊂11.7高速均质器:转速6500r/m i n~24000r/m i n㊂11.8离心机:转速ȡ6000r/m i n㊂11.9玻璃纤维滤纸:快速㊁高载量㊁液体中颗粒保留1.6μm㊂11.10氮吹仪㊂11.11液相色谱仪:配荧光检测器㊂11.12色谱分离柱㊂11.13黄曲霉毒素专用型固相萃取净化柱(以下简称净化柱),或相当者㊂11.14一次性微孔滤头:带0.22μm微孔滤膜(所选用滤膜应采用标准溶液检验确认无吸附现象,方可使用)㊂11.15筛网:1mm~2mm试验筛孔径㊂11.16恒温箱㊂11.17p H计㊂12分析步骤12.1样品制备12.1.1液体样品(植物油㊁酱油㊁醋等)采样量需大于1L,对于袋装㊁瓶装等包装样品需至少采集3个包装(同一批次或号),将所有液体样品在一个容器中用匀浆机混匀后,其中任意的100g(m L)样品进行检测㊂12.1.2固体样品(谷物及其制品㊁坚果及籽类㊁婴幼儿谷类辅助食品等)采样量需大于1k g,用高速粉碎机将其粉碎,过筛,使其粒径小于2mm孔径试验筛,混合均匀后缩分至100g,储存于样品瓶中,密封保存,供检测用㊂12.1.3半流体(腐乳㊁豆豉等)采样量需大于1k g(L),对于袋装㊁瓶装等包装样品需至少采集3个包装(同一批次或号),用组织捣碎机捣碎混匀后,储存于样品瓶中,密封保存,供检测用㊂12.2样品提取12.2.1液体样品12.2.1.1植物油脂称取5g试样(精确至0.01g)于50m L离心管中,加入20m L乙腈-水溶液(84+16)或甲醇-水溶液(70+30),涡旋混匀,置于超声波/涡旋振荡器或摇床中振荡20m i n(或用均质器均质3m i n),在6000r/m i n下离心10m i n,取上清液备用㊂12.2.1.2酱油㊁醋称取5g试样(精确至0.01g)于50m L离心管中,用乙腈或甲醇定容至25m L(精确至0.1m L),涡旋混匀,置于超声波/涡旋振荡器或摇床中振荡20m i n(或用均质器均质3m i n),在6000r/m i n下离心10m i n(或均质后玻璃纤维滤纸过滤),取上清液备用㊂12.2.2固体样品12.2.2.1一般固体样品称取5g试样(精确至0.01g)于50m L离心管中,加入20.0m L乙腈-水溶液(84+16)或甲醇-水溶液(70+30),涡旋混匀,置于超声波/涡旋振荡器或摇床中振荡20m i n(或用均质器均质3m i n),在6000r/m i n下离心10m i n(或均质后玻璃纤维滤纸过滤),取上清液备用㊂12.2.2.2婴幼儿配方食品和婴幼儿辅助食品称取5g试样(精确至0.01g)于50m L离心管中,加入20.0m L乙腈-水溶液(50+50)或甲醇-水溶液(70+30),涡旋混匀,置于超声波/涡旋振荡器或摇床中振荡20m i n(或用均质器均质3m i n),在6000r/m i n下离心10m i n(或均质后玻璃纤维滤纸过滤),取上清液备用㊂12.2.3半流体样品称取5g试样(精确至0.01g)于50m L离心管中,加入20.0m L乙腈-水溶液(84+16)或甲醇-水溶液(70+30),置于超声波/涡旋振荡器或摇床中振荡20m i n(或用均质器均质3m i n),在6000r/m i n下离心10m i n(或均质后玻璃纤维滤纸过滤),取上清液备用㊂12.3样品黄曲霉毒素固相净化柱净化移取适量上清液,按净化柱操作说明进行净化,收集全部净化液㊂12.4衍生用移液管准确吸取4.0m L净化液于10m L离心管后在50ħ下用氮气缓缓地吹至近干,分别加入200μL正己烷和100μL三氟乙酸,涡旋30s,在40ħʃ1ħ的恒温箱中衍生15m i n,衍生结束后,在50ħ下用氮气缓缓地将衍生液吹至近干,用初始流动相定容至1.0m L,涡旋30s溶解残留物,过0.22μm滤膜,收集滤液于进样瓶中以备进样㊂12.5色谱参考条件色谱参考条件列出如下:a)流动相:A相:水,B相:乙腈-甲醇溶液(50+50);b)梯度洗脱:24%B(0m i n~6m i n),35%B(8.0m i n~10.0m i n),100%B(10.2m i n~11.2m i n),24%B(11.5m i n~13.0m i n);c)色谱柱:C18柱(柱长150mm或250mm,柱内径4.6mm,填料粒径5.0μm),或相当者;d)流速:1.0m L/m i n;e)柱温:40ħ;f)进样体积:50μL;g)检测波长:激发波长360n m;发射波长440n m;h)液相色谱图:参见图D.1㊂12.6样品测定12.6.1标准曲线的制作系列标准工作溶液由低到高浓度依次进样检测,以峰面积为纵坐标-浓度为横坐标作图,得到标准曲线回归方程㊂12.6.2试样溶液的测定待测样液中待测化合物的响应值应在标准曲线线性范围内,浓度超过线性范围的样品则应稀释后重新进样分析㊂12.6.3空白试验不称取试样,按12.2㊁12.3和12.4的步骤做空白实验㊂应确认不含有干扰待测组分的物质㊂13分析结果的表述试样中A F TB1㊁A F TB2㊁A F T G1和A F T G2的残留量按式(2)计算:X=ρˑV1ˑV3ˑ1000V2ˑmˑ1000 (2)式中:X 试样中A F TB1㊁A F TB2㊁A F T G1或A F T G2的含量,单位为微克每千克(μg/k g);ρ 进样溶液中A F TB1㊁A F TB2㊁A F T G1或A F T G2按照外标法在标准曲线中对应的浓度,单位为纳克每毫升(n g/m L);V1 试样提取液体积(植物油脂㊁固体㊁半固体按加入的提取液体积;酱油㊁醋按定容总体积),单位为毫升(m L);V3 净化液的最终定容体积,单位为毫升(m L);1000 换算系数;V2 净化柱净化后的取样液体积,单位为毫升(m L);m 试样的称样量,单位为克(g)㊂计算结果保留三位有效数字㊂14精密度在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的20%㊂15其他当称取样品5g时,柱前衍生法的A F TB1的检出限为0.03μg/k g,A F TB2的检出限为0.03μg/k g, A F TG1的检出限为0.03μg/k g,A F TG2的检出限为0.03μg/k g;柱前衍生法的A F TB1的定量限为0.1μg/ k g,A F TB2的定量限为0.1μg/k g,A F TG1的定量限为0.1μg/k g,A F TG2的定量限为0.1μg/k g㊂第三法高效液相色谱-柱后衍生法导语:下述方法的仪器检测部分,包括碘或溴试剂衍生㊁光化学衍生㊁电化学衍生等柱后衍生方法,可根据实际情况,选择其中一种方法即可㊂16原理试样中的黄曲霉毒素B1㊁黄曲霉毒素B2㊁黄曲霉毒素G1㊁黄曲霉毒素G2,用乙腈-水溶液或甲醇-水溶液的混合溶液提取,提取液经免疫亲和柱净化和富集,净化液浓缩㊁定容和过滤后经液相色谱分离,柱后衍生(碘或溴试剂衍生㊁光化学衍生㊁电化学衍生等),经荧光检测器检测,外标法定量㊂17试剂和材料除非另有说明,本方法所用试剂均为分析纯,水为G B/T6682规定的一级水㊂17.1试剂17.1.1甲醇(C H3O H):色谱纯㊂17.1.2乙腈(C H3C N):色谱纯㊂17.1.3氯化钠(N a C l)㊂17.1.4磷酸氢二钠(N a2H P O4)㊂17.1.5磷酸二氢钾(K H2P O4)㊂17.1.6氯化钾(K C l)㊂17.1.7盐酸(H C l)㊂17.1.8 T r i t o nX-100[C14H22O(C2H4O)n](或吐温-20,C58H114O26)㊂17.1.9碘衍生使用试剂:碘(I2)㊂17.1.10溴衍生使用试剂:三溴化吡啶(C5H6B r3N2)㊂17.1.11电化学衍生使用试剂:溴化钾(K B r)㊁浓硝酸(HN O3)㊂17.2试剂配制17.2.1乙腈-水溶液(84+16):取840m L乙腈加入160m L水㊂17.2.2甲醇-水溶液(70+30):取700m L甲醇加入300m L水㊂17.2.3乙腈-水溶液(50+50):取500m L乙腈加入500m L水㊂17.2.4乙腈-水溶液(10+90):取100m L乙腈加入900m L水㊂17.2.5乙腈-甲醇溶液(50+50):取500m L乙腈加入500m L甲醇㊂17.2.6磷酸盐缓冲溶液(以下简称P B S):称取8.00g氯化钠㊁1.20g磷酸氢二钠(或2.92g十二水磷酸氢二钠)㊁0.20g磷酸二氢钾㊁0.20g氯化钾,用900m L水溶解,用盐酸调节p H至7.4,用水定容至1000m L㊂17.2.71%T r i t o nX-100(或吐温-20)的P B S:取10m LT r i t o nX-100,用P B S定容至1000m L㊂17.2.80.05%碘溶液:称取0.1g碘,用20m L甲醇溶解,加水定容至200m L,用0.45μm的滤膜过滤,现配现用(仅碘柱后衍生法使用)㊂17.2.95m g/L三溴化吡啶水溶液:称取5m g三溴化吡啶溶于1L水中,用0.45μm的滤膜过滤,现配现用(仅溴柱后衍生法使用)㊂17.3标准品17.3.1 A F TB1标准品(C17H12O6,C A S号:1162-65-8):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂17.3.2 A F TB2标准品(C17H14O6,C A S号:7220-81-7):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂17.3.3 A F T G1标准品(C17H12O7,C A S号:1165-39-5):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂17.3.4 A F T G2标准品C17H14O7,C A S号:7241-98-7):纯度ȡ98%,或经国家认证并授予标准物质证书的标准物质㊂注:标准物质可以使用满足溯源要求的商品化标准溶液㊂17.4标准溶液配制17.4.1标准储备溶液(10μg/m L):分别称取A F T B1㊁A F T B2㊁A F T G1和A F T G21m g(精确至0.01m g),用乙腈溶解并定容至100m L㊂此溶液浓度约为10μg/m L㊂溶液转移至试剂瓶中后,在-20ħ下避光保存,备用㊂临用前进行浓度校准(校准方法参见附录A)㊂17.4.2混合标准工作液(A F TB1和A F T G1:100n g/m L,A F TB2和A F T G2:30n g/m L):准确移取A F TB1和A F T G1标准储备溶液各1m L,A F TB2和A F T G2标准储备溶液各300μL至100m L容量瓶中,乙腈定容㊂密封后避光-20ħ下保存,三个月内有效㊂17.4.3标准系列工作溶液:分别准确移取混合标准工作液10μL㊁50μL㊁200μL㊁500μL㊁1000μL㊁2000μL㊁4000μL至10m L容量瓶中,用初始流动相定容至刻度(含A F T B1和A F T G1浓度为0.1n g/m L㊁0.5n g/m L㊁2.0n g/m L㊁5.0n g/m L㊁10.0n g/m L㊁20.0n g/m L㊁40.0n g/m L,A F T B2和A F T G2浓度为0.03n g/m L㊁0.15n g/m L㊁0.6n g/m L㊁1.5n g/m L㊁3.0n g/m L㊁6.0n g/m L㊁12n g/m L 的系列标准溶液)㊂18仪器和设备18.1匀浆机㊂18.2高速粉碎机㊂18.3组织捣碎机㊂18.4超声波/涡旋振荡器或摇床㊂18.5天平:感量0.01g和0.00001g㊂18.6涡旋混合器㊂18.7高速均质器:转速6500r/m i n~24000r/m i n㊂18.8离心机:转速ȡ6000r/m i n㊂18.9玻璃纤维滤纸:快速㊁高载量㊁液体中颗粒保留1.6μm㊂18.10固相萃取装置(带真空泵)㊂18.11氮吹仪㊂18.12液相色谱仪:配荧光检测器(带一般体积流动池或者大体积流通池)㊂注:当带大体积流通池时不需要再使用任何型号或任何方式的柱后衍生器㊂18.13液相色谱柱㊂18.14光化学柱后衍生器(适用于光化学柱后衍生法)㊂18.15溶剂柱后衍生装置(适用于碘或溴试剂衍生法)㊂18.16电化学柱后衍生器(适用于电化学柱后衍生法)㊂18.17免疫亲和柱:A F TB1柱容量ȡ200n g,A F TB1柱回收率ȡ80%,A F T G2的交叉反应率ȡ80% (验证方法参见附录B)㊂注:对于每个批次的亲和柱使用前需质量验证㊂18.18黄曲霉毒素固相净化柱或功能相当的固相萃取柱(以下简称净化柱):对复杂基质样品测定时使用㊂18.19一次性微孔滤头:带0.22μm微孔滤膜(所选用滤膜应采用标准溶液检验确认无吸附现象,方可使用)㊂18.20筛网:1mm~2mm试验筛孔径㊂19分析步骤使用不同厂商的免疫亲和柱,在样品的上样㊁淋洗和洗脱的操作方面可能略有不同,应该按照供应商所提供的操作说明书要求进行操作㊂警示:整个分析操作过程应在指定区域内进行㊂该区域应避光(直射阳光)㊁具备相对独立的操作台和废弃物存放装置㊂在整个实验过程中,操作者应按照接触剧毒物的要求采取相应的保护措施㊂19.1样品制备同12.1㊂19.2样品提取同12.2㊂19.3样品净化19.3.1免疫亲和柱净化19.3.1.1上样液的准备准确移取4m L上述上清液,加入46m L1%T r i t o nX-100(或吐温-20)的P B S(使用甲醇-水溶液提取时可减半加入),混匀㊂19.3.1.2免疫亲和柱的准备将低温下保存的免疫亲和柱恢复至室温㊂19.3.1.3试样的净化免疫亲和柱内的液体放弃后,将上述样液移至50m L注射器筒中,调节下滴速度,控制样液以1m L/m i n~3m L/m i n的速度稳定下滴㊂待样液滴完后,往注射器筒内加入2ˑ10m L水,以稳定流速淋洗免疫亲和柱㊂待水滴完后,用真空泵抽干亲和柱㊂脱离真空系统,在亲和柱下部放置10m L刻度试管,取下50m L的注射器筒,2ˑ1m L甲醇洗脱亲和柱,控制1m L/m i n~3m L/m i n的速度下滴,再用真空泵抽干亲和柱,收集全部洗脱液至试管中㊂在50ħ下用氮气缓缓地将洗脱液吹至近干,用初始流动相定容至1.0m L,涡旋30s溶解残留物,0.22μm滤膜过滤,收集滤液于进样瓶中以备进样㊂19.3.2黄曲霉毒素固相净化柱和免疫亲和柱同时使用(对花椒㊁胡椒和辣椒等复杂基质)19.3.2.1净化柱净化移取适量上清液,按净化柱操作说明进行净化,收集全部净化液㊂19.3.2.2免疫亲和柱净化用刻度移液管准确吸取上部净化液4m L,加入46m L1%T r i t o nX-100(或吐温-20)的P B S(使用甲醇-水溶液提取时可减半加入),混匀㊂按19.4.1.3处理㊂注:全自动(在线)或半自动(离线)的固相萃取仪器可优化操作参数后使用㊂19.4液相色谱参考条件19.4.1无衍生器法(大流通池直接检测)液相色谱参考条件列出如下:a)流动相:A相,水;B相,乙腈-甲醇(50+50);b)等梯度洗脱条件:A,65%;B,35%;。
黄曲霉毒素B1的检测方法

饲料研究2018年第1期黄曲霉毒素B i的轤测方法徐荣1张佩华2*(1.湖南农业大学动物科学技术学院,湖南长沙410128;2.湖南畜禽安全生产协同创新中心,湖南长沙410128)摘要:目前研究进展显示,黄曲霉毒素Bi(Aflatoxin B i简写为AFBi)是化学物质中致癌性最强的一种,对人和动物都会造成危害,本文旨在对黄曲霉毒素B i的毒性和检测方法进行综述。
关键词:黄曲霉毒素B1毒性检测方法黄曲霉毒素B,主要是由黄曲霉和寄生曲霉 产生的次级代谢产物,是谷物污染中常见的具有 强致癌性、致毒性、致畸性的真菌毒素,在生产中 容易造成经济损失、影响人畜健康,所以黄曲霉毒 素B,的检测方法在饲料生产和人类生活中具有 重要意义。
1黄曲霉毒素Bi简介AFB1是含有一个双呋喃环和一个氧杂萘邻 酮(香豆素)的二氢呋喃氧杂萘邻酮的衍生物,是 目前已知化学致癌性最强的物质之一,可以通过 储存、加工等各种途径污染粮油等农产品、食品和 饲料等,直接或间接危害人类和动物的健康。
2黄曲霉毒素Bi的毒性黄曲霉毒素B,的毒性要比呕吐毒素的毒性 强30倍,比玉米赤霉烯酮的毒性强20倍。
黄曲霉 毒素B i的急性毒性是础霜的68倍,氰化钾的10 倍,慢性毒性可诱发癌变,致癌能力为二甲基亚硝 胺的75倍,比二甲基偶氨苯高900倍,人的原发性肝癌也很可能与黄曲霉毒素有关。
当人类大量摄入AFB,时,可发生急性中毒,出现急性肝炎、出血性坏死、肝细胞脂肪变性和胆 管增生;微量持续摄人,可造成慢性中毒,生长障 碍,引起纤维性病变,致使纤维组织增生。
AFB,具 有致癌性,可导致人类肝癌和食道癌。
当动物家禽摄入AFB1时,采食量,体增重和 饲料转化率均会降低;还会导致不孕、流产、肺水 肿、疫病易感性增加等现象发生。
3黄曲霉毒素Bi的检测方法世界各国及国际组织都制定了黄曲霉毒素B,的限量标准。
GB 5009.22-2016《食品安全国家标 准食品中黄曲霉毒素B族和G族的测定》中规定 当称取样品5g时,柱前衍生法AFB,的检出限为 0.03滋g/kg。
食品中黄曲霉毒素B1检测方法研究

Iustry科技文苑行业70 食品安全导刊 2019年1~2月2.2 补充食品检测细则食品检测监管的法律法规或行业规范是指导具体食品检测行为的重要内容,必须确保其与现实需求接轨,且具备充足的可操作性。
因此,有关部门必须从检测市场发展现状出发,有针对性的补充食品检测细则,还需在广泛调查的基础上进行有针对性的调整与更新,以保持实施细则的时效性,确保其应有的作用得以发挥。
2.3 构建第三方检测制度根据上述分析可知,目前我国食品质量安全检测资源大多掌握在政府及有关部门手中,权责不清的问题仍然存在,检测覆盖面亦不足,故应加强第三方检测主体的建设,以提高食品安全检测制度的灵活性和客观性。
第三方检测机构独立于监管部门和食品经营企业之外,充分利用社会资源,具有重要的补充作用,能够达到优化食品检测效果。
2.4 加强基层检测能力建设我国各级检测的实力差距大,应加大基层检测机构的建设,弥补省级检测机构检测能力的不足。
国家应建立地方食品监督检验机构,与省级食品检测机构形成合力,共同做好管辖区域内的食品质量安全检测工作。
同时还应加大基层检测的资金投入,确保其达到应有的检测能力。
3 结语综上所述,随着人们食品安全意识的不断提高,社会对于食品质量要求愈加严格。
各级政府及相关部门应加强食品检测,通过加大技术研究及应用、补充检测细则、建立第三方检测制度以及加大基层检测建设等方法,提高我国食品检测水平,营造出健康而安全的食品生产与消费氛围。
参考文献:[1] 薛庆根.美国食品安全体系及对我国的启示[J ].经济纵横,2006(2):62.[2] 张旭霞,林博,李小宁.食品安全检测技术存在的问题与对策[J ].现代农业科技,2012,(15).[3] 薛茂云,刘大纶.论食品安全问题及其解决途径[J ].江苏商论,2009,(07).食品中黄曲霉毒素B 1检测方法研究□ 张宏博 靳志敏 高智慧 郑玉山(通讯作者) 内蒙古自治区食品检验检测中心摘 要:黄曲霉毒素(AF)是广泛存在于自然界中的真菌毒素,而在所有的真菌毒素中,AFB 1的毒性、致癌性较大,其能阻止蛋白、酶和凝血因子合成,又能抑制葡萄糖、脂肪酸、代谢产物的合成,从而引起免疫抑制、脂肪退化和DNA 损伤。
花生及其制品中黄曲霉毒素B1的检测试验分析

花生及其制品中黄曲霉毒素B1的检测试验分析
马玉红
【期刊名称】《中国食品工业》
【年(卷),期】2024()3
【摘要】为实现对花生及其制品中黄曲霉毒素B1进行定性和定量分析,本研究通过同位素内标法和低温高速离心净化法建立了一种高效液相色谱-质谱法的测定方法。
该方法检出限为0.003μg/kg,定量限为0.010μg/kg,精密度和准确率良好;通过该方法对市面上的18种花生及其制品的商品进行抽检,合格率为100%;生物可给性分析结果表明,花生奶的生物可给性较高;胃肠消化阶段的黄曲霉毒素B1生物可给性显著高于胃消化阶段。
【总页数】3页(P75-77)
【作者】马玉红
【作者单位】乌鲁木齐市天山区疾病预防控制中心
【正文语种】中文
【中图分类】TS2
【相关文献】
1.花生油中黄曲霉毒素B1(AFB1)的检测及分析
2.花生及其制品中黄曲霉毒素B1免疫亲和柱净化-荧光快速检测技术
3.间接竞争酶联免疫分析方法检测花生中黄曲霉毒素B1
4.花生及其制品中黄曲霉毒素B1胶体金快速检测方法的建立
因版权原因,仅展示原文概要,查看原文内容请购买。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
样品去壳去皮粉碎后称取20.0g,加入250mL具塞三角瓶中,准确 加人100.0mL甲醇水(55 +45)溶液和30mL石油醚,盖塞后滴水封 严,150rpm振荡30min,静置15min后用快速定性滤纸过滤于 125mL分液漏斗中,待分层后,放出下层甲醇水溶液于100mL烧 杯内,从中取20.0 mL(相当于4.0g样品)置于另一125mL分液漏 斗中,加入20.0mL三氯甲烷,振摇2min,静置分层(如有乳化现 象可滴加甲醇促使分层).放出三氯甲烷于75mL蒸发皿中,再加 5.0mL三氯甲烷于分液漏斗中重复振摇提取后,放出三氯甲烷层一 并于蒸发皿中,以下按上述“65℃水浴通风挥干”起,依法操作。
3、显色反应:加入绿色试剂100μl,摇匀,室温反应 10min,加入红色试剂100μl,终止反应。
4、结果检测:在450nm处用酶标仪检测。
七、注意事项及说明
2. 玉米(脂肪含量3.0~5.0%):
样品粉碎后过20目筛,称取20.0g,加入250mL具塞锥形瓶中。准 确加入50.0mL甲醇一水(80+20)溶液和15.0mL石油醚,盖塞后 滴水封严,150rpm振荡30min。用快速定性滤纸过滤于125mL分 液漏斗中,待分层后,放出下层甲醇水溶液于50mL烧杯内,从中取 10.0mL (相当于4.0g样品)于75mL蒸发皿中,以下按上述“65℃ 水浴通风挥干”起,依法操作。
实验二十、食品中黄曲霉毒素B1的检测
标准依据: GB/T5009.22-2003食品中黄曲霉毒素B1的测 定
一、实验目的与要求:
1、学习酶联免疫(ELISA)方法测定 法
2、了解酶联免疫(ELISA)的工作原理,正确熟练使用 酶标仪。
5.其它食品:可按照G8 5009有关章节提供的方法操 作,最终提取物(相当于4.0g样 品)应收集于2.0m1 甲醇一PBS(20+80)中。
(二)样品测定
1、将下列试剂稀释后备用 PBS一T洗液:390mL蒸榴水稀释(1:40); 底物液A: 9mI蒸溜水稀释(1:9); 抗体: 7.0mL洗液稀释(1:140); 酶标二抗:10mL洗液稀释(1:200); 阴性对照液:200μl抗体十200μl空白对照液在玻
五、数据处理及计算结果
1、求出各孔的0.D校正值:0.D校正值=O.D 实测值一空白对照孔0.D值(均值)。
2、求出待测样的0.D%值:0.D%=[O.D校正 值/阴性对照孔0.D校正值(均值)] × 100%。
3、求出待测样AFB1含量(ng/g):在标准 竞争抑制曲线上找到待测样0.D%值(座标Y 值)的对应点,该点座标X值的反对数(10x) 即为样品中AFB1的含量(ng/g)
璃试管内混合振荡后静置。
2、抗体抗原反应 将抗体与等量样品提取液(溶液为甲醇一PBS)在玻
璃试管内混合振荡后室温 静置15分钟。启封酶标板, 用洗液2×3分钟洗板后在吸水纸上拍干,分别在适当 孔位加入此抗体抗原反应液及空白对照液(3孔)和阴 性对照液(3孔),130μl/孔,37℃湿盒中孵育(或加 盖以保持相对湿度),2小时后,倒掉反应液并拍干, 用洗液3×3分钟洗板,拍干。 3、酶标记反应 加入酶标二抗,100μl/孔,37℃盒中孵育1小时后, 用洗液5×3分钟洗板,拍干。 4、显色反应 lmg底物十2.5m1底物液A +42μl底物液B,待底物充 分溶解后加入酶标板,100μl/孔,37℃15分钟后加 终止液40μl/孔。 5、测定 酶标仪490nm测定各孔0.D值。
四、实验步骤
(一)样品中AFB1的提取
1. 大米和小麦(脂肪含量<3.0%):
样品粉碎后过20目筛,称取20.0g,加入250m1具塞 锥形瓶中。准确加人60mL三氯甲烷,盖塞后滴水封 严,150rpm振荡30min。静置后,用快速定性滤纸过 滤于50mL烧杯中,立即取12mL滤液(相当4.0g样品) 于75mL蒸发皿中。65℃水浴通风挥干。用2.0mL甲 醇一PBS (20+80)分三次(0.8mL、0.7mL、0.5mL) 溶解并彻底冲洗蒸发皿中凝结物,移至小试管加盖振 荡后静置待测。此液每毫升相当2.0g样品。
二、实验原理:
本试剂盒采用间接竞争ELISA方法,样品中的黄曲霉 毒素B1经提取、脱脂、浓缩后与定量特异性抗体反应, 多余的游离抗体则与酶标板内的包被抗原结合,加入 酶标记物和底物后显色,与标准曲线比较测定含量。
三、仪器与试剂
(一)试剂盒组成 1、 AFB1抗原包被16孔×6条形带框酶标板; 2、抗体; 3、酶标二抗; 4、空白对照液; 5、底物(1mg×4); 6、底物液A; 7、底物液B; 8、终止液; 9、PBS一T洗液; 10、一次性手套。 (二)仪器 1、酶标仪; 2、离心机。
六、使用进口试剂盒检测的主要实验步骤
1、样品提取:取5.0g样品与锥形瓶中,加入70%甲醇 25ml,振摇3min,过滤于分液漏斗中,静置分层,放 出下层的甲醇水提取液,即为样品提取液。(亦可根 据样品中黄曲霉毒素含量进行适当稀释为待测样液)
2、免疫反应:红色微孔中加入蓝色试剂100μl,分别加 入标准品和样品各100μl,三次混匀,移100μl到白色 抗体孔,摇匀,室温反应10min,弃去液体,用去离 子水洗5次,用吸水纸扣干。
4.植物油:
用小烧杯称取4.0g样品,用20.0mL石油醚,将样品移 于125mL分液漏斗中,用20.0mL 甲醇水(55 +45) 溶液分次洗烧杯,溶液一并移人分液漏斗中。(精炼 油4.0g样为4.575mL,可直接用移液器加入分液漏斗 再加溶剂后振摇)振摇2min,静置分层后,放出下层 甲醇水溶液于750mL蒸发皿中。再用5.0mL甲醇水溶 液重复振摇提取一次,提取液一并加入蒸发皿中,以 下按自“65℃水浴通风挥干”起,依法操作。