常染色体显性遗传多囊肾研究进展
胎儿常染色体显性遗传性多囊肾病的研究进展

《中国产前诊断杂志(电子版)》 2020年第12卷第1期·综述· 胎儿常染色体显性遗传性多囊肾病的研究进展朱雨露 综述 徐岚 审校(汕头大学医学院第一附属医院,广东汕头 515041)【摘要】 常染色体显性多囊肾病(autosomal dominantpolycystickidneydisease,ADPKD)是最常见的遗传性肾病之一。
通常,ADPKD在早期无明显自觉症状,多在成年后发病,临床上该病以双肾多发性囊肿为主要特征,并常伴发肝、胰、脾等器官囊性病变及心脑血管病变。
ADPKD亦可在儿童期甚至在胎儿期发病,一旦发病,其程度较成年患者更加严重,疾病进展也更加迅猛。
诊断为患有ADPKD的胎儿的母亲在随后的妊娠中,胎儿有50%的复发机会。
胎儿超声检查在胎儿常染色体显性多囊肾病的病因学研究和预后评估中是必不可少的,在症状出现前,产前即进行胎儿基因检测并尽早干预有助于避免具有致命缺陷的胎儿出生,目前已经有多种基因测试方法可用于检测ADPKD。
本文旨在对胎儿常染色体显性遗传性多囊肾病在发病机制、临床特点、产前检查、产前诊断与治疗方面的最新研究进展进行综合评述。
【关键词】 遗传咨询;先天畸形;产前诊断;产前超声;常染色体显性遗传性多囊肾病;遗传学【中图分类号】 R714.55 【文献标识码】 A犇犗犐:10.13470/j.cnki.cjpd.2020.01.012 通信作者:徐岚,E mail:xulandoctor@163.com 常染色体显性多囊肾病(autosomal dominantpolycystickidneydisease,ADPKD)是一种以常染色体显性遗传方式遗传的疾病,是一种累及多系统、多脏器的全身性疾病。
在活产婴儿中发病率约为1/500~1/1000[1],大约8%的透析或接受肾移植的患者患有ADPKD[2]。
ADPKD的特征在于双肾多发性囊肿,该疾病的标志是在儿童早期肾脏中数百个微观充满液体的囊肿的发展,它们以不同的速率甚至以指数倍的方式连续生长(每年增加2%~10%),逐渐导致正常肾组织的损失[3],最终将导致终末期肾病(end stagekidneydisease,ESKD)。
常染色体显性遗传多囊肾治疗的研究进展

“ 二次打击” 模式 , 该模 式认 为首先存在有 多数胚层 P D1 P D K 和 K 2基 因 突变 的 内在 特 性 , 次 是 后 天 因 其 素 作用 下 肾和 肝 囊 肿 的 发 生 、 展 … 。P D等 位 基 发 K
因的 体细 胞 突变 不 受胚 胎 突 变 的影 响是 否是 唯 一或 最 主要 的引 起 A P D囊 肿 形成 的机 制 尚未 确定 , DK 如 果 A P D是 细 胞水 平 的 隐性 疾病 这 一假 说 被证 实 , DK 基 因替代 疗 法 在 理 论 上 是 可 能 的 。 OkRde国 家 a i g 实验 室多 囊 肾模 型 (rk模 型 ) 过 野 生 型 op o p 通 rk基 因作 为转 移 基 因 的表 达 , 得 其 肾脏 表 型得 到 挽救 , 使
因对 正 常 组 织 是 否 安 全 , 以及 P D1 度 表 达 也 可 K 过 声生囊肿表型的事实等 , 使人们对基 因治疗 A P D DK 的可 行性 存 在疑 虑 。
12 口连环 蛋 白 . 一 8连环 蛋 白 ( -a n ) 一 种 双 功 能 蛋 白 , 一  ̄ct i 是 3 en 在
如果 体细 胞 突变 对 于 A P D的 囊 肿 发 展重 要 DK
的话 , 么 减 少 体 细 胞 突 变 率 , 延 缓 疾 病 的 发 展 , 那 可 这种 观 点 日益 为人 们所 接 受 。人 们 设 想使 用 能够 防
止 内源和外源性 突变原活性 的药 物 , 1 如 相酶 的抑 制剂 p S , - Z 或使用 能够加强 去毒作用 的药 物 , 2 X 如 相酶诱导剂 o ia, h r 但它们都还未尝试应用在 A P pz D一
1 针对 基 因 突变 的治 疗
多囊肾的诊断及治疗新进展

多囊肾的诊断及治疗新进展
李林;梅长林
【期刊名称】《中国医师进修杂志》
【年(卷),期】2006(029)016
【摘要】@@ 人类对于常染色体显性多囊肾病(autosomal dominant polycystic kidney disease,ADPKD)的研究经历了漫长的过程.公元前460年,希波克拉底描述了一种酷似多囊肾病的疾病.1856年Bristowe 在伦敦病理学会上首次报道了1例囊性肾病合并囊性肝病的病例.直到近几十年,随着遗传学、分子生物学等的发展,人们才逐渐对多囊肾病有了科学的认识.
【总页数】3页(P4-6)
【作者】李林;梅长林
【作者单位】200003,上海,第二军医大学长征医院肾内科,解放军肾脏病研究
所;200003,上海,第二军医大学长征医院肾内科,解放军肾脏病研究所
【正文语种】中文
【中图分类】R6
【相关文献】
1.多囊肾的诊断及治疗新进展 [J], 李林;梅长林
2.第四届重庆介入放射学学术年会暨国家级继续医学教育项目肝癌影像诊断及介入治疗新进展和非血管介入治疗新进展征文通知 [J],
3.2007年脑卒中诊断治疗新进展高级培训班(原名“脑卒中治疗及康复新进展高级研修班”)通知 [J],
4.第四届重庆介入放射学学术年会暨国家级继续医学教育项目征文通知(肝癌影像诊断及介入治疗新进展和非血管介入治疗新进展) [J],
5.第四届重庆介入放射学学术年会暨国家级继续医学教育项目征文通知(肝癌影像诊断及介入治疗新进展和非血管介入治疗新进展) [J],
因版权原因,仅展示原文概要,查看原文内容请购买。
常染色体显性遗传性多囊肾病
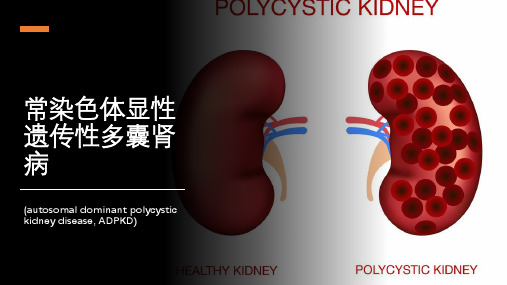
流行病学ห้องสมุดไป่ตู้
•
所有种族都可发生ADPKD,报道的患病率为1:1000。在美国每年
开始透析的患者中,大约有5%的肾病由ADPKD引起。
•
ADPKD主要由两种基因突变引起:16号染色体上的PKD1(编码多
囊蛋白-1)或4号染色体上的PKD2(编码多囊蛋白-2)。大多数患者可维持
正常肾功能到30岁左右。一旦GFR开始下降,其每年可平均降低4.4-
➢ 多囊肝 多数不需治疗。肝脏明显增大可引起腹胀、呼吸困难、胃食管反流、门静脉高压等。可根据病情选 择肝囊肿穿刺硬化、去顶减压术、肝部分切除术或肝移植术
➢ 颅内动脉瘤 对于有动脉瘤和蛛网膜下腔出血家族史的病人,推荐MRI血管造影检查确诊。直径大于10mm的动 脉瘤应采取介入或手术治疗
治疗
肾脏替代治疗 ➢ 包括血液透析、腹膜透析和肾移植。目前认为ADPKD病人腹膜透析与血液透析的并发症和 长期存活率无明显差异。移植后肾存活率、并发症与其他肾移植人群相似
治疗
对症治疗 ➢ 疼痛
急性疼痛针对病因进行治疗。慢性疼痛,程度轻者或一过性疼痛卧床休息并观察,如疼痛持续或较 重按止痛阶梯序贯药物治疗,仍不能缓解可考虑囊肿穿刺硬化、囊肿去顶减压术及多囊肾切除术
➢ 出血 多为囊肿出血所致,呈自限性,轻者绝对卧床休息、止痛、多饮水。出血量大、保守疗法效果差可 行选择性血管栓塞或出血侧肾脏切除
泌尿道和囊肿感染,引起ADPKD病人发热的首要病因,主要表现为膀胱炎、肾盂肾炎、囊肿感 染和肾周脓肿,逆行性感染为主要途径
临床表现
肾外表现 ➢ 囊性病变
可累及肝脏、胰腺、脾脏、卵巢及蛛网膜等器官。其中肝囊肿最常见,大多数病人无症状,少数 可表现为疼痛、囊肿感染和出血
肾病内科成人多囊肾病的遗传学与治疗

肾病内科成人多囊肾病的遗传学与治疗肾病内科成人多囊肾病是一种常见的常染色体显性遗传疾病,通常由PKD1和PKD2基因突变引起。
本文将探讨成人多囊肾病的遗传学以及目前的治疗方案。
一、多囊肾病的遗传学成人多囊肾病是一种遗传疾病,主要由PKD1和PKD2基因的突变引起。
PKD1基因突变是最常见的致病基因,在成人多囊肾病患者中约占85%的比例,而PKD2基因突变约占15%。
这两个基因编码蛋白质分别是多囊肾病1蛋白(polycystin-1)和多囊肾病2蛋白(polycystin-2)。
多囊肾病的遗传方式为常染色体显性遗传,也就是说,只需要一个受影响的父亲或母亲传递该基因就可以使子女患病。
如果父母都携带PKD1或PKD2基因的突变,则子女患病的风险为50%。
在遗传学研究中发现,多囊肾病的表型和基因型之间存在一定的相关性。
PKD1基因突变导致的多囊肾病表现为更严重的疾病,病情进展更快,导致肾功能衰竭的风险更高。
而PKD2基因突变引起的多囊肾病病情较为温和,病程进展较为缓慢。
二、多囊肾病的治疗目前,对于成人多囊肾病的治疗主要还是以缓解疾病症状、控制并发症、维持肾功能为主要目标。
以下是一些常用的治疗措施:1. 药物治疗:对于多囊肾病的治疗,目前尚无特效药物。
然而,一些研究表明,例如利尿剂、抗高血压药物等可以控制高血压、减轻水肿等症状,从而改善患者的生活质量。
2. 饮食调控:合理的饮食调控对于多囊肾病患者的治疗和康复很重要。
建议减少高盐、高脂肪的饮食,增加膳食纤维的摄入以及适量饮水,有助于缓解肾功能损害。
3. 进行手术:在极少数病例中,如果囊肿较大、引起严重的疼痛或影响其他脏器的功能,可能需要进行手术治疗。
手术方式包括囊肿穿刺引流、囊肿去顶术等。
4. 肾脏移植:对于终末期肾脏病患者,肾脏移植是最有效的治疗方法之一。
肾脏移植可以帮助患者恢复正常的肾功能,并提高生活质量。
总结成人多囊肾病是一种常见的遗传疾病,由PKD1和PKD2基因突变引起。
多囊肾病的发病机制及诊治进展1‘

梅长林 解放军肾脏病研究所 第二军医大学长征医院肾内科
主要内容
❖ 多囊肾病概述 ❖ 多囊肾病的发病机制 ❖ 多囊肾病的治疗研究
国外研究进展 我们的研究结果
多囊肾病
〔polycystic kidney disease,
PKD〕
常染色体显性多囊
肾病〔autosomal
dominant
❖ 预期结果:ACEI/ARB联合治疗与ACEI单独治疗相比将更加 显著地延缓多囊肾病的进展,而且严格的血压控制也比常规 血压控制要更加显著地延缓多囊肾病进展
❖
研究 B组
❖ 试验人群:GFR 30-60 mL/min/1.73m2 且合并有高血压的 ADPKD患者
❖ 试验目的:将血压控制在≤130/80mmHg的情况下,比较 ACEI/ARB联合治疗与ACEI单独治疗对延缓ADPKD患者到 达研究终点〔GFR下降至基线的50%、ESRD或死亡〕影响
RAS存在于囊肿衬里上皮细胞
Renin
Am J Physiol Renal Physiol 287: F775–F788, 2004.
Angiotensinogen
ANG Ⅱ
ACE
ANG Ⅱ
AT1 receptors
Mahmoud Loghman-Adham et al. The intrarenal renin-angiotensin system in autosomal
❖ Study B:moderately advanced disease〔GFR 30-60
❖
mL/min/1.73 m2〕
❖
研究A组
❖ 试验人群:GFR >60 mL/min/1.73m2 且BP>130/80 mmHg的 ADPKD患者
肾病内科多囊肾的遗传与治疗进展

肾病内科多囊肾的遗传与治疗进展多囊肾是一种常见的遗传性疾病,主要表现为肾脏内多个囊肿的形成。
本文将从遗传机制和治疗进展两个方面进行探讨。
一、多囊肾的遗传机制多囊肾可分为两种类型,一种为常染色体显性遗传型,另一种为常染色体隐性遗传型。
1. 常染色体显性遗传型常染色体显性遗传型多囊肾(ADPKD)是指由PKD1和PKD2基因突变引起的多囊肾。
大多数情况下,ADPKD是由PKD1基因突变所导致。
PKD1和PKD2基因编码的蛋白质分别称为多囊肾蛋白1和多囊肾蛋白2,它们参与了细胞内钙离子转运的调节。
当PKD1或PKD2基因发生突变时,细胞内钙离子转运受到影响,导致多囊肾的形成。
2. 常染色体隐性遗传型常染色体隐性遗传型多囊肾(ARPKD)是由PKHD1基因突变引起的。
PKHD1基因编码的蛋白质称为多囊肾蛋白,它参与了肾小管发育的调节。
PKHD1基因突变导致多囊肾蛋白功能异常,进而影响肾小管的正常发育。
二、多囊肾的治疗进展目前,多囊肾的治疗主要以缓解症状、延缓病情进展和减轻并发症为目标。
以下介绍一些常见的治疗方法及其进展情况。
1. 药物治疗目前,针对多囊肾的药物治疗主要集中在控制相关症状和延缓病情进展方面。
对于ADPKD患者,托拉塞米德是一种常用的药物,可以减缓囊肿的生长速度。
另外,一些研究还发现利用mTOR抑制剂和vasopressin V2受体拮抗剂能够达到减缓多囊肾生长的效果。
2. 血液透析和肾移植对于晚期多囊肾患者,血液透析和肾移植是常见的治疗手段。
血液透析通过机器代替肾脏进行部分或全部的血液过滤,帮助排除体内垃圾和余液。
肾移植则是将供体的健康肾脏移植到患者体内,以取代功能受损的肾脏。
3. 基因治疗随着基因编辑技术的发展,基因治疗逐渐成为多囊肾治疗的新方向。
通过修复或替换患者体内异常基因,可以恢复肾脏的正常功能。
目前,基因治疗仍处于实验室研究阶段,但它有望为多囊肾的治疗带来革命性的突破。
三、结语多囊肾是一种遗传性疾病,其遗传机制和治疗进展备受关注。
mTOR抑制剂治疗常染色体显性多囊肾病的研究进展

知有 3种 基 因的突变导致 了 A P D,其 抑制细胞凋 亡 的发 生 。因此 ,有 学 者 轻 、血压下降 、囊肿出血风险降低等 ,甚 DK 中2种最 为常见 :为 P D ( 占 8% ) 提出使用 m O K 1 约 5 T R抑制剂 ,通过抑制 m O 至可能对 A P D的相关并发症如脑 动脉 TR DK 和 P D ( 占 1% ) K2 约 5 ;第 3种 是 P D , 信号通路 的异 常表 达 ,延缓 囊壁 细胞 增 瘤 、左心 室 肥 厚 也有 良好 的治 疗作 用 。 K 3
莫司靶分 子 ( a m l ntgt f aa y g0 at ,V G m m a a re o pm — r卅h f o i a r c r E F) 的 过 度 表 达 有 曙光。 目前 ,有 4个较为详细的临床试验 c ,m O ) 激 活 参 与 其 中 。 i n TR 关“ 。A P D后 期 发 生 间质 纤 维 化 ,更 观察 了 m O 抑 制剂 治 疗 A P D的 效 DK TR DK 1 mO T R参与 A P D囊肿形成 DK 加剧了肾小球滤过率 (lm rl lao 果 ,其 中 3个采 用 肾脏 囊肿 体积改 变 go e a ft t n u r ir i A P D复杂 的 病 理 生 理 过 程 早 已 被 r e F DK a ,G R) 的下 降。而 m O t T R抑制 剂对 作 为 首要 观 察 终 点 。这 些 试 验 的 区别 只 是
。 常 染 色 体 显 性 多 囊 肾 病 ( u sm l 积 ‘ J at o a o
d m ia t oy y tc i e die s o n n p lc si kdn y s a e, ADP —
比,囊肿体积增长率更适合用来代表疾病
常染色体显性遗传病之多囊肾病的研究和治疗

常染色体显性遗传病之多囊肾病的研究和治疗摘要:控制一种遗传性状的显性基因位于常染色体上,其遗传方式称为常染色体显性遗传(AD),由这种致病基因引起的疾病称为常染色体显性遗传病。
常染色体显性遗传型多囊肾病(autosomal Dominant polycystic kidney disease,ADPKD)是一种最常见的单基因遗传性肾病,发病率约为1/400~1/1000。
ADPKD多在3()岁~50岁之间发病,囚此过去常称为“成人型多囊肾病”,实际上ADPKD可在任何年龄发病,包括妊娠时的胎儿,故“成人型”这一术涪并不准确,现已废用。
ADPKD临床表现为双侧肾脏皮、髓质有多个液性囊肿形成和增大,晚期伴肾功能损害。
可累及多个系统。
正文:一多囊肾病的概述1.什么叫多囊肾多囊肾是肾脏的皮质和髓质出现多个囊肿的一种遗传性肾脏疾病。
2.还患者发病原因90%异常基因位于16号染色体的短臂。
另有10%不到患者的异常基因位于4号染色体的短臂。
患者的基因型在常染色体显性遗传病中,假定用A表示显性致病基因,a表示相对应的隐性正常基因,则患者的基因型有两种,显性纯合体(AA)和杂合体(Aa),基因型aa的个体正常,但临床上所见到的患者大多数为杂合体。
二.系谱特点1、男女发病几率相等;2、父母有一方患病,子女有50%获得囊肿基因而发病,如父母均患此病,子女发病率增加到75%;3、不患病的子女不携带囊肿基因,其下代(孙代)也不会发病,即不会隔代遗传。
真正不经父母遗传而由基因突变而发病的情况极少见。
三.多囊肾的症状1、肾肿大两侧肾病变进展不对称,大小有差异,至晚期两肾可占满整个腹腔,肾表面布有很多囊肿,使肾形不规则,凹凸不平,质地较硬。
2、肾区疼痛为其重要症状常为腰背部压迫感或钝痛,也有剧痛,有时为腹痛。
3、血尿约半数病人呈镜下血尿,可有发作性肉眼血尿,此系囊肿壁血管破裂所致。
4、高血压。
5、肾功能不全本病迟早要发生肾功能不全。
自噬在常染色体显性遗传性多囊肾病中的作用研究进展

自噬在常染色体显性遗传性多囊肾病中的作用研究进展
何彬;高艺源;施声淦;蒋培都
【期刊名称】《中国药理学通报》
【年(卷),期】2022(38)3
【摘要】自噬是一种细胞内成分经溶酶体途径被降解的过程,对维持细胞内环境稳态及细胞成分自我更新具有重要作用。
研究表明自噬与常染色体显性遗传性多囊肾病(autosomal dominant polycystic kidney disease,ADPKD)的囊肿生成密切相关。
进一步研究发现,ADPKD疾病模型中存在自噬受损和自噬增强两种现象,自噬
失调影响了ADPKD的发生发展。
因此,自噬的调节可能成为ADPKD治疗新策略。
经mTOR依赖性和mTOR非依赖性途径调节自噬的药物也显示出了缓解ADPKD 症状的作用。
本文就自噬在ADPKD中的作用研究进展进行综述,为进一步研究自
噬与ADPKD及其药物调控提供参考。
【总页数】5页(P321-325)
【作者】何彬;高艺源;施声淦;蒋培都
【作者单位】电子科技大学医学院;四川省医学科学院/电子科技大学附属医院药学部个体化药物治疗四川省重点实验室
【正文语种】中文
【中图分类】R322.61;R392
【相关文献】
1.体细胞突变在常染色体显性多囊肾发病中作用的研究进展
2.胰岛素样生长因子在常染色体显性遗传性多囊肾病发病中的作用
3.胎儿常染色体显性遗传性多囊肾病的研究进展
4.常染色体显性遗传性多囊肾病中的能量代谢改变
5.常染色体显性遗传性多囊肾病的研究进展
因版权原因,仅展示原文概要,查看原文内容请购买。
美国多囊肾最新研究报告

美国多囊肾最新研究报告美国多囊肾最新研究报告引言多囊肾是一种常见的肾脏遗传性疾病,其特征是肾脏内出现多囊状扩张。
这种疾病通常会导致慢性肾功能衰竭,对患者的生活质量和寿命都有严重影响。
近年来,美国的研究人员在多囊肾的治疗和预防方面做出了重要的突破,本文将对美国多囊肾最新的研究进展进行综述。
1. 多囊肾的病因和遗传机制多囊肾是一种常染色体显性遗传的疾病,主要由PKD1和PKD2基因突变引起。
这两个基因编码了多囊肾蛋白1和多囊肾蛋白2,这两种蛋白在肾小管上皮细胞中起着重要的调控作用。
突变导致了多囊肾蛋白的功能失调,进而导致肾小管细胞功能障碍和增生。
2. 分子机制研究的进展近年来,美国的研究人员通过对多囊肾基因的研究,揭示了多囊肾发病的分子机制。
他们发现,多囊肾蛋白在细胞内形成复合物,并与其他蛋白相互作用,调控细胞增殖和分化。
同时,他们还发现了多囊肾蛋白对一些细胞信号通路的调节作用。
这些研究结果为多囊肾的治疗提供了新的靶点和方向。
3. 研究进展近年来,美国的研究人员在多囊肾的治疗和预防方面做出了重要的突破。
他们发现了一些潜在的药物和干预方法,可以延缓疾病的进展,并改善患者的生活质量。
以下是一些相关的研究进展:•药物治疗–研究人员发现一些已经上市的药物对多囊肾有一定的治疗作用,例如利拉鲁肽。
–研究人员还通过筛选化合物库发现了一种新的药物,具有降低多囊肾蛋白表达的作用。
•细胞治疗–美国的研究人员利用干细胞技术,成功地将修复后的肾小管细胞移植到多囊肾患者的肾脏中,取得了显著的治疗效果。
–另外,研究人员还通过基因编辑技术,成功地修复了多囊肾基因突变,为多囊肾的基因治疗提供了新的思路。
•遗传咨询和干预–研究人员提出了遗传咨询和干预的重要性,通过遗传咨询,患者和家族成员可以了解遗传风险,并采取相应的预防措施。
–研究人员还通过基因编辑技术,成功地修复了多囊肾基因突变,为多囊肾的基因治疗提供了新的思路。
4. 未来的研究方向美国的多囊肾研究在治疗和预防方面取得了重要的进展,但仍有许多问题需要解决。
多囊肾的发病机制及其治疗的研究进展

肿块等 , 这些 临床表 现的出现常常是 肾实质 已遭到严重 破坏 的 及其 直径 大小 的作 用。基 因发 生突 变后 导致纤 毛正 常结 构 和 信号 , 更严重 的是这些损 害将不断加重 。尽管 多数患者 在 中年 功能发生障碍 , 由正常情况下尿流速率传递 的终 止信号不能传 导 致 肾小管 进 行性 扩 张, 最 后产 生 囊肿 J 。 以后发 病 , 但 目前很 多证据 表 明 A D P K D最早 在胎 儿期 即可被 递至肾小 管细胞 ,
诊断, 且病情 比成 年发病 者更 重 。S e l i s t r e等 对 5 2例儿 童 此外 , 已被证实 内皮细胞 上 的纤 毛长度 直接 的深入研究 , 证 实肾脏 功能正 常的 期发病 的 A D P K D患者 [ 平 均年 龄 ( 1 0± 4 ) 岁; 年龄分 布 1岁 一 调控 J l 7岁] 的研究证实 了肾脏部 位存在确切损害 , 且其 中 7 7 % 出现 A D P K D患者 , 其增 高的局部 多 巴胺 水平 导致 了 内皮 依赖 性 血 了高血 压 、 蛋 白尿以及 肾功能 损害 。因此 , 在诊 断 A D P K D时 , 患者 , 并进行及 时干预 , 延缓疾病进 展 , 提高其生存质量 。
且参 与 P C 1的定位 J , 主要表 达于 多囊肾 ( P K D) 是 一种 常见的常染色体遗传 病 , 为单基 因遗 透的非选择性阳离子通道 , 传, 遗传学上将 其分为显性 和 隐性两 大类 , 两 种类 型均 造成 双 内质网 、 细胞膜底外侧及 初级 纤毛 。P K D 2突变 所致 疾病相 对
2 分子遗传学
管舒 张的减少。这一多巴胺 通路 的发 现将会成 为 A D P K D潜 在
3 . 2 “ 二次打击 ” 学说 目前 大多数研究者认 为 P K D 1 及
Ⅰ型常染色体显性遗传型多囊肾病常用的基因诊断技术研究进展

活动 , 有利 于 改善 患 肢 的血 液 循 环 , 防止 肌 肉萎 缩 及 关 节
僵直 。
3 6 预防关节挛缩 : . 卧床期间要保持 适当 的床 上运 动锻炼 , 预防肢体废用性挛 缩及关 节挛 缩 ; 外 , 注意保 持各 关节 此 要
功能位置 , 特别 是患肢应 始终 处在功 能状 态下 , 样不 致于 这 骨折愈后站立不起来 。
4 指导功能锻炼
神志等体征变化 , 发现 问题及 时处 理。
维普资讯
・
9 ・ 6
中国现代药物应用 2 0 0 8年 4月第 2卷第 7期
C i dD hnJM0 mgAo1A r 0 8 V 12。o 7 o. o 0 . o. N . 2
3 1 警惕心脑血管 的并发症 : . 进入老年 期 , 环系统发生 明 循
3 3 预防呼吸道并 发症 : . 老年人 由于呼吸相对 减弱 , 有慢 并 性 支气 管炎 , 肺源性 心脏病 史 , 以长 期 卧床及术后 病人 易 所
4 1 老 年人健康 时其 活动就 已减退 , . 一旦 生病或手 术后 活
动量更是 减少 , 不利于骨愈合 。因此 , 卧床 期间 , 根据不 同情 况指导病人在 床上进行 活动。
3 护 理人 员应 耐心宣 传 、 帮助 、 督促 和指 导老年人 进行练 习 。要采取循序渐 进 的原则 , 才能起 到 防止关 节粘 连、 肉 肌 萎缩 , 以促进 骨折愈合 , 最终达到康复 的预期 目标 。
体细胞突变在常染色体显性多囊肾发病中作用的研究进展

体细胞突变在常染色体显性多囊肾发病中作用的研究进展王琛;孔祥阳;徐剑;李嵘;冯超【期刊名称】《重庆医学》【年(卷),期】2016(045)028【总页数】3页(P4014-4016)【关键词】多囊肾,常染色体显性;PKD1基因;PKD2基因;三链DNA【作者】王琛;孔祥阳;徐剑;李嵘;冯超【作者单位】昆明理工大学附属医院肾内科 650032;昆明理工大学医学院疾病与药物遗传实验室 650500;昆明理工大学医学院疾病与药物遗传实验室 650500;昆明理工大学附属医院肾内科 650032;昆明理工大学附属医院肾内科 650032;昆明理工大学附属医院肾内科 650032【正文语种】中文【中图分类】R692.1常染色体显性多囊肾(autosomal dominant polycystic kidney disease,ADPKD)是一种遗传性单基因疾病,由多囊肾基因1(polycyseic kidneydisease gene,PKD1)、PKD2基因突变所致,称Ⅰ型ADPKD,Ⅱ型ADPKD。
其发病率很高,1/1 000~1/400,为常见肾脏遗传病[1-2] 。
一般成年发病,又称成人型多囊肾,临床上也见不同年龄患者。
PKD2突变50%的患者在60 岁左右可发展为终末期肾病(end-stage renal disease,ESRD),约占晚期肾病的10%(Ⅰ型ADPKD比Ⅱ型ADPKD发病早20年,Ⅰ型ADPKD恶化为终末肾病的平均年龄为54.3岁,而Ⅱ型ADPKD为74.0岁)[1,3] 。
除了损伤双侧肾脏以外,ADPKD基因还累及全身多个器官,例如多囊肝、颅内动脉血管瘤、二尖瓣脱垂综合征、腹股沟疝、大肠息室、主动脉夹层、胆囊增生,严重危害人类健康[4]。
目前,已报道克隆ADPKD的致病基因有3个:PKD1、PKD2、PKD3。
其中约85%的患者为ⅠADPKD,由PKD1基因突变所致;约15%的患者为ⅡADPKD,由PKD2基因突变所致。
常染色体显性多囊肾基因突变研究进展

[基金项目]山东省自然科学基金资助项目(Y 2002C 12)。
・综述与讲座・常染色体显性多囊肾基因突变研究进展刘 征,丁克家(山东省立医院,山东济南250021)[关键词] 遗传性疾病;多囊肾疾病;基因突变[中图分类号] R 69211 [文献标识码] A [文章编号] 10022266X (2006)2120090202 常染色体显性多囊肾病(AD PKD ),又称成人型多囊肾病。
据国外文献报道,到60岁时大约有50%的患者出现终末期肾衰竭。
该病为单基因遗传病,存在遗传异质性。
目前已知的引起该病的突变基因可能有3个,分别为PKD 1基因、PKD 2基因、PKD 3基因。
其中85%患者由PKD 1基因突变引起[1],约15%患者由PKD 2基因突变引起。
PKD 3基因由于少见,研究不多,故至今尚未定位克隆。
1 PKD 1基因PKD 1基因定位在第16号染色体短臂1区3带3亚带(16p 1313),总跨度约53kb ,由46个外显子组成,其转录的mRNA 长度1411kb ,编码的多囊蛋白产物称为PC 1。
PKD 1基因的近端区域(16p 1311)含有三个同源基因位点(H G 2A 、H G 2B 和H G 2C ),与PKD 1基因同源性极高。
PKD 1基因的35~46外显子为单拷贝区,1~34外显子由于存在同源基因出现3次核苷酸重复,这给基因测序和突变检测带来了极大困难。
为此,不同实验室采用了多种不同方法,包括蛋白截断实验(PT T )、变性梯度凝胶电泳(D GGE )、单链构象多态性分析(SSCP )、高压液相色谱法(DH PL C )及直接测序法。
每种方法各有利弊,PT T 无法检测没有造成蛋白异常终止的错义突变;D GGE 由于需要在电泳前在产物两次加上一对GC “夹子”,增加了操作难度;SSCP 虽然简便,但存在5%~30%的漏检率;DH PL C 需要专门的仪器设备;直接测序虽然检出率较高,但花费较大。
常染色体显性遗传多囊肾病一家系报告

剂( AC E I ) 类 降压 药 、 大 黄 及 虫 草制 剂 等 保 肾治 疗 , 彩 超提 示多囊 肝 、 多囊 肾 、 肾结石 。此 次入 院病情 较 重, 出 现持续 性 肉眼血 尿 , 肌酐 达 1 2 9 0 . 1 0 , u mo l / L , 结合 病 史 、 体 征及 辅助 检查 , 诊断 为 常染色 体显性 遗 传 多囊 肾病 、 囊肿 出血 、 慢性 肾脏 病 5期 、 囊 肿感染 、 肾结 石 、 继发 性 甲状旁 腺功 能亢 进 。 病例 2 : 先证 者 之外祖 母 ( I 1 ) , 据 先证 者 回忆 , 其外 祖母 约 5 3岁 时 因反复 双下肢 浮肿 伴血 尿去世 , 因当时 医疗条 件所 限 , 未 明确 诊 断 , 结合 AD P KD遗
腹胀 痛加 重 , 合并 持续 性 肉眼血 尿 , 伴发热 3 8 . 5 ℃。 人 院后查 体 , 体温 3 6 . 5 ℃, 血压 l l 1 / 9 0 mmHg , 肾病
[ 作者单 位] 南 京 医 科 大 学 第 二 附属 医 院 肾脏 病 中 心, 南 京
2 1 0 0 0 3 ; 通 讯 作者 , E — ma i l : j wy a n g @n j mu . e d u . c a 本文2 0 1 3 —0 5 —1 7收 到 , 2 0 1 3 —0 6 —1 9修 回 , 2 0 1 3 —0 7 —0 2接 受
左右, 未 系统治 疗 , 未 进行 血液 透 析 , 5 1岁 因尿 毒症
去世 。
6 0 t _ t mo l / L, 尿蛋 白( 2 +) , 尿 隐血 ( +) 。
病例 1 1 : 先证者 之子 ( I V 1 ) , 2 9岁 , 无 高血压 、 血 尿、 肾功能 不全 病 史 , 无任何不适 , 1 O余 年 未 体 检 ,
常染色体显性遗传性多囊性肾病发病机制的研究进展及治疗前景

其氨基酸组成与电压激活的钙离子通道 a 亚单位 1
及P KD1 似 。多 囊 蛋 白一 多囊 蛋 白一 过 存 相 1和 2通
在 于细胞 外 区 的 末 端 而 相 互 作 用 。破 坏 P D1 K
和 2 存在多嘧啶区 , 中在 2 2 其 1内含子 中的多嘧啶
区是迄今 为止 在人 类 基 因组 中发现 的最 长 的 区域 , 长 25k , . b推测 多 嘧 啶区 的存 在 可能 引 起 三螺 旋 体
的形 成 , 而使 得突 变容 易发生 。 从
P 1基 因编 码 一4 0 KD 33个 氨 基 酸 的跨 膜 糖 蛋 白 , 名为 多囊蛋 白 1有 1 个跨 膜 区 , 命 , 1 预期 分 子量 为 4 0k) 6 I。它 的 N一 端位 于细胞 外 , 括两个 富含 末 包
( ci)区 ,6个 免 疫 球 蛋 白样 区 , 个 L L A 样 1t e n 1 一 D2
区。它 的羧基 端位 于胞浆 侧 , 含有 一个 疏 水 的螺旋 一 螺旋 区 。多囊蛋 白整 个大 分子 与 已知所 有 的蛋 白质 家族均 无 同源性 , 测 多 囊蛋 白可 能 涉及 细 胞 与 细 推
1 AD KD 致病基 因的定 位和 克隆 P 目前 已知 有 3种基 因 的 突变 导 致 了 A K DP D,
白一 参与 E c hr 1 — d e n和 a ct i a i B一 e n复合物的形成 , an
这些 研究 表 明 多囊 蛋 白一 信 号 转 导 通 路 中起 重 1在 要 作用 , 控制 细胞 的增殖 和分 化 。 12 P 2 P D . KD K 2定 位在 42—3 大约有 1 % ~ q 12 , 0 1 % 的 AD KD病人是 由 P 2突变 所致 。该 基 因 5 P KD 跨 度 6 b 由 1 8 k, 5个 外 显 子 组 成 。 它 不 同 于 6 J P KD1在人 类基 因组 中是单 拷 贝 , 不 含多 嘧啶 区 , , 并 但 它 的第 一 个 外 显 子 富含 G 从 而 使 得 此 处 易 于 C, 突变 。P 2编 码 的蛋 白质被 命 名 为 多囊 蛋 白 一2 KD , 含 98个 氨 基 酸 , 是 一 种跨 膜 蛋 白, 跨 膜 6次 , 6 亦 共
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
常染色体显性遗传多囊肾研究进展
摘要常染色体显性遗传多囊肾(ADPKD)是一种发病率高、预后差的疾病,它在发病机制、治疗等方面有很多进展。
关键词常染色体显性遗传多囊肾;瞬时受体势;Max作用因子1;雷帕霉素靶蛋白
常染色体显性遗传多囊肾(autosomal dominant polycystic kidney disease,ADPKD)是一种常见的遗传性肾病,是导致肾衰竭的重要疾病。
现在已经发现3个基因(PKD1、PKD2、PKD3)与此病有关,其中PKD1定位于染色体16p13.3,其突变而导致ADPKD约占85%,PKD2定位于染色体4q21-23,其突变约占15%。
PKD3突变仅在几个家族中发现,目前尚未定位[1]。
ADPKD病变以双肾多发性进行性充液囊泡为主要特征。
囊泡损伤肾组织,引起肾功能改变,出现血尿、蛋白尿等临床症状,最终导致肾衰竭。
ADPKD除累及肾脏外,还可引起肝脏囊肿、胰腺囊肿、心脏瓣膜病、结肠憩室和颅内动脉瘤等肾外病变[2],给患者、家属及社会带来沉重负担。
故揭示其发病机理,研究新的治疗方法有很重要的意义。
本文对这些进展作以综述。
1 ADPKD的发病机制
1. 1 多囊蛋白PC PKD1基因的蛋白产物被称为多囊蛋白-1(polycystin-1,PC1),又叫做TRPP1,多囊蛋白-1是一种跨膜蛋白,分布广泛,可以与多种蛋白(如PC2)、糖、脂类结合并发生交互作用,从而发挥功能。
PC1还与Wnt信号途径、JAK-STAT途径、转录因子AP-1和G蛋白偶联等信号途径有关。
PKD2基因的蛋白产物被称为多囊蛋白-2(polycystin-2,PC2),又叫做TRPP2,是TRP家族的一员,为非选择性钙离子通道。
它不同于PKD1,在人类基因组中是单拷贝,并不含多嘧啶区,但它的第一个外显子富含GC,从而使得此处易于突变[3]。
瞬时受体势(transient receptor potential,TRP)通道虽然最初发现于感受器,主要参与神经传导,但随着研究的深入,近年来人们发现该通道在肾脏病的发生、发展中也发挥了作用。
瞬时受体势C,即TRPC,为传统型,被认为是最可能的钙库操纵性钙通道和受体操纵性钙通道的分子基础[4]。
PKD2蛋白当被G偶联蛋白或钙库耗竭激活时,与TRPC1蛋白连接形成蛋白复合物并引起钙离子动态平衡失调,引起胞浆及内质网内钙水平降低,发生生物学作用。
1. 2 Mxi1 Max作用因子1(MAX-interacting protein 1,Mxi1)是抑癌基因Mxi1编码的蛋白。
Mxi1可以竞争性的使Max与Myc结合,抑制Myc的负调节作用,也就起到了抑癌的作用。
Max是Myc-associated factor X,作用是调控myc信号通路。
故以往的研究多在肿瘤方面,近年来发现Mxi1与肾脏病的发生发展有很大作用。
Mxi1(又称为mad2、mxd2)为Mad 转录家族的成员之一,Mxil 基因敲除小鼠,PKD1、PKD2表达上调[5]。
肾脏出现了ADPKD特征性病理改变[6] 。
在发生多囊肾的Mxi1 基因敲除小鼠肾组中,Mxi1 失活可
导致肾小管上皮细胞纤毛缩短和功能丢失,并伴有B-Raf/MEK/ERK增殖信号通路异常活化,说明Mxi1 可通过影响肾小管上皮细胞的纤毛形成参与到ADPKD 的发病过程中[7] 。
2 治疗
2. 1 免疫抑制剂PKD基因突变会激活哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)的信号通路,促使其上皮细胞增殖和肾囊肿扩张。
雷帕霉素是一种强效免疫抑制剂,可通过抑制mTOR 的信号途径,抑制组织细胞增殖和多囊肾模型中囊肿的增长,并能延缓肾衰竭。
既往的研究显示PKD1突变上调mTOR信号途径的活性,但PKD2突变与mTOR信号途径是否相关没有证实。
相关学者研究证实,PC1,即PKD1基因的蛋白产物缺陷上调mTOR信号途径活性,而不是PC2。
2. 2 酪氨酸激酶抑制剂研究发现ADPKD患者肾脏细胞内cAMP浓度增加激活PKA,进一步激活Ras等信号通路的酪氨酸酶活性而促进囊泡细胞增殖。
同时囊泡上皮细胞的增殖也涉及多种生长因子,如表皮生长因子(EGF)、血清肌酐(Scr)、MEK等的激酶活性有关。
因此酪氨酸激酶抑制剂可以抑制ADPKD 的发展。
2. 3 金属蛋白酶抑制剂金属蛋白酶(MMPs)是一组锌依赖性酶,以往多研究其在肿瘤的增殖中及肾脏纤维化中的作用。
ADPKD囊肿生成的重要病理生理机制是细胞外基质的异常,肾脏囊泡上皮细胞异常增殖。
过度增生的小管向外扩张必须降解已形成的细胞外基质并形成新的细胞外基质。
基质金属蛋白酶和组织金属蛋白酶抑制剂可能在其中发挥重要作用,其机制与抑制金属蛋白酶参与的细胞外基质的降解有关。
随着对多囊肾病基质生成及降解确切机制的研究深入,一种或多种金属蛋白酶抑制剂可能会证实其在多囊肾病治疗中的疗效[8]。
2. 4 基因治疗基因治疗是最理想的治疗方法,这些药物主要针对细胞增殖失调、细胞分化、凋亡和囊液分泌异常以及异常细胞信号传递途径。
从基因水平治疗ADPKD的突变,从而使其编码功能正常的蛋白,发挥正常生物学作用,是人类一直研究及追求的梦想。
Harsh 等[9]用PKD/Mhm(cy/+)大鼠模型,在分析mRNA和microRNA基因芯片探索其在ADPKD功能调节方式中发现一些miRNAs与ADPKD的调节途径有关联。
可以指导研究人员用它们进行人ADPKD 基因组的治疗,或者抗纤维化的治疗。
2. 5 中医治疗雷公藤是从天然草本植物分离出来的药物,在肾脏病领域应用多年。
Leuenroth等[10]发现雷公藤内酯可通过促进PC2介导的细胞内钙的释放和促进p21的表达,从而发挥抑制细胞增殖的作用,此过程不依赖PC1的表达,提示雷公藤内酯可用于治疗ADPKD。
3 小结
ADPKD的发病机制及相关治疗研究近年来有了较大的进展,ADPKD的发病涉及多个信号传导通路,作用靶点的选择及基因水平的治疗仍然是目前研究的热点内容。
ADPKD的临床治疗目前多局限于继发病变的治疗,随着ADPKD
发病机制在细胞和分子水平的逐渐阐明,多靶点抑制囊泡生成,延缓囊泡生长,控制继发病变发生将成为治疗多囊肾病的优选方案。
参考文献
[1] 王文娟,李莉,杨建一. 常染色体显性遗传型多囊肾病的研究进展. 中国优生与遗传杂志,2011(1):127-128.
[2] 梅长林,李林. 常染色体显性遗传型多囊肾病的诊断及治疗现况. 中国中西医结合肾病杂志,2002,3(12):685-686.
[3] Abramowitz J,Birnbaumer L. Physiology and pathophysiology of canonical transient receptor potential channels. Faseb Journal,2009,23(2):297-328.
[4] Wechsler D S,Hawkins A L,Li X,et al. Localization of the human Mxi1 transcription factor gene (MXI1)to chromosome 10q24-q25. Genomics,1994,21(3):669-672.
[5] Yoo KH,Sung YH,Yang MH,et al. Inactivation of Mxil induces IL-8 secretion activation in polycystic kidney. BBRC,2007,356(1):85-90.
[6] Pirity M,Blanck J K,Schreiber A N. Lessons learned from Myc/Max/Mad knockout mice . CTMI,2006(302):205-234.
[7] Ko JY,Yoo KH,Song SA,et al. Inactivation of max-interacting protein 1 induces renal cilia disassembly through reduction in levels of intraflagellar transport 20 in polycystic kidney. Journal of Biological Chemistry,2013,288(9):6488-97.
[8] 崔心刚,安瑞华,王立明,等. 基质金属蛋白酶1/组织金属蛋白酶抑制因子1在常染色体显性遗传性多囊肾组织中的表达. 第二军医大学学报,2006,27(11):1174-1177.
[9] Harsh D,Carsten S,Asawari K,et al. Parallel Analysis of mRNA and microRNA Microarray Profiles to Explore Functional Regulatory Patterns in Polycystic Kidney Disease:Using PKD/Mhm Rat Model . Journal List,2013,8(1):e53780.
[10] Louenroth SJ,Okuhara D,Shotwell JD,et a1. Triptolide is a traditional Chinese medicine-derived inhibitor of polycystic kidney disease. PNAS,2007,104(11):4389-4394.。