discover分子动力学软件使用攻略
forcite分子动力学 流程

forcite分子动力学流程Forcite分子动力学流程。
一、基础了解。
Forcite可是分子动力学模拟里超厉害的一个工具呢。
分子动力学嘛,简单说就是看分子们在一段时间里是怎么跑来跑去、相互作用的。
那Forcite能做这个事儿,咱们就得先知道它的一些基本情况。
它在很多材料科学、化学这些领域都能用得上。
比如说研究新的材料,想知道分子之间的力怎么影响材料的性能呀,或者研究化学反应过程中分子的运动轨迹,这时候Forcite就像一个小侦探一样,可以帮我们找到答案。
二、准备工作。
在开始用Forcite做分子动力学模拟之前,得先准备好分子结构。
这就像是要做饭,得先把食材准备好一样。
我们得知道要研究的分子长啥样,原子之间是怎么连接的。
有时候这个分子结构可能是从数据库里找出来的,就像去超市买菜一样方便。
但有时候呢,得自己构建,这就有点考验咱们的本事啦。
比如说用一些建模软件把分子一点一点搭起来,就像搭积木一样。
而且呀,还得确定这个分子的初始状态,是静止的呢,还是已经有了一定的速度和方向。
这就好比是决定这个分子是在原地休息,还是已经开始奔跑了。
三、选择力场。
力场这个东西呢,对于Forcite分子动力学模拟可重要啦。
力场就像是分子们遵循的一套规则,告诉它们怎么相互作用。
有各种各样的力场可以选择,像COMPASS力场就很常用。
不同的力场适用于不同类型的分子。
就好像不同的游戏有不同的规则一样。
如果选错了力场,那模拟出来的结果可能就会很奇怪,就像在足球比赛里用篮球的规则一样,那肯定是不行的。
所以呢,得根据自己要研究的分子特性来选择合适的力场。
这就需要我们对分子和力场都有一定的了解,就像了解自己的朋友有什么喜好一样。
四、设定模拟参数。
这一步也很关键哦。
要设定好多参数呢,比如说模拟的时间步长。
这个时间步长就像是我们拍照的间隔时间。
如果时间步长太大,就像隔了很久才拍一张照片,可能会错过分子之间一些很精彩的瞬间,模拟结果就不准确了。
Discovery操作手册全.

Discovery软件操作手册GNT国际公司目录一.工区建立与管理 (1)二.数据加载 (3)三.微机地质应用 (10)四. 微机三维地震解释综合应用 (19)五.微机单井测井解释及多井评价 (39)六.储层管理与预测 (54)七.smartSECTION模块 (63)八.正演建模 (69)一.工区建立与管理1.1工区目录(home)建立我们一般习惯把Discovery工区放在一个文件夹下,这样可方便进行管理。
1) 利用Windows资源管理器,建立一个文件夹,如proj-dis,同时可以把其它机器上的Discovery工区放在次目录下。
2) 在桌面上双击GeoGraphix Discovery图标,打开工区管理模块ProjectExplorer。
3) 进入ProjectExplorer > File> New> Home。
4) 通过浏览器确定proj-dis文件夹,点击确定按钮。
5) 点击下一步按钮,直至完成。
6) 在工区管理模块ProjectExplorer左侧,出现proj-dis工区目录,这样就可以在该目录下建立Discovery工区。
1.2工区(project)创建一般来说,任何一个地学软件工作流程的第一步都是建立一个工区,Discovery 软件也不例外。
工区的建立包括为工区名、路径、坐标系统和工区范围等。
1) 在桌面上点击GeoGraphix Discovery 图标,启动ProjectExplorer2) 在工具栏点击New Project 按钮或从菜单条选File > New> Project。
出现NewProject Wizard(新工区向导)的工区对话框。
3) 在Project Name(工区名)框中,输入工区名称。
4) 在Description(描述)框中,输入对工区的描述(如:位置、远景区等)。
5) 压力和深度单位,在中国,选择米制(这对底图和数据库坐标系统都没有影响)。
Discovery Studio Visualizer简易教程

Discovery Studio Visualizer精简教程哈尔滨医科大学解鸿波PDB: 3K9F工具栏工具导航栏主窗口Ctrl+H氨基酸残基氨基酸相应性质Ctrl+D线型显示必须先棍型显示选择原子球棍显示线型显示棍型显示必须先选择原线型条带子条带改变背景颜色View > Toolbars > View View > Toolbars > Sketching 区旋平放画分子工具域转移大(不推荐使用)选缩择小PDB编号代表晶格侧链编号按字母顺序排列*不同部分蛋白(或DNA等大分子)点加号+查看侧链所含有氨基酸残基点加号+查看氨基酸所含有原子氨基酸及编号字母重新循环配体分子去除对勾表示隐藏其他分子例如水等显示配体和周围形成的氢键作用2. Structure > Monitor > HBonds1. 仅显示配体分子,并双击选中,分子显示为黄色。
3. 显示和配体可以形成氢键的氨基酸序号及相应原子将氨基酸以棍状形态显示,Stick默认氢键为绿色size调整为0.10和配体以示区别,并不显示其带状二级结构选中分子,查看其他相互作用Tools > Analyze Complexes > Show 2D DiagramStructure > Surface > AddStructure > Monitor > Distance 选中两个原子,测距离单位为(Å)选中两根化学键,测角度查看蛋白序列Chart > Ramachandran Plot选择原子> 右键> Attributes of … 查看原子坐标Build and Edit Nucleic Acid工具Build and Edit Protein工具(不是蛋白三维结构,需要做从头预测)Create Pharmacophore Manully工具(根据已知分子构建药效团,但不实用)写字板打开*.pdb文件查看信息,内附原子坐标,原子类型及氨基酸序号等信息。
Discovery Studio官方教程--拉伸动力学计算结合自由能

拉伸动力学计算结合自由能介绍拉伸分子动力学模拟可以使原来在微妙至秒时间范围内发生的生物物理过程在纳秒尺度内进行模拟,从而动态再现目前实验所无法提供的一些过程,如蛋白去折叠、配体解离和构象变化等过程。
上述这些过程可以通过在拉伸动力学模拟中让事件按照指定方向主动发生,而不像在标准分子动力学模拟中需要被动等待事件的发生。
本教程中,我们使用T4溶菌酶蛋白作为研究目标,模拟配体2-丙基苯酚从活性口袋的解离的过程。
具体见参考文献(J. Mol. Biol.2009, 394, 747-763). 本教程假定你已经熟悉建立和运行标准分子动力学模拟的过程。
对于该体系的SMD模拟比较复杂,因为配体的位置位于结合口袋的深处,而且将配体从结合口袋里拉出来的同时伴随着蛋白的散开,退出配体和蛋白的复原等过程。
1.准备蛋白配体模拟体系从菜单File | Open URL,在ID框中填入3HTB之后点击Open。
选择Macromolecules | Protein Report工具栏,点击Protein Report工具栏下方的Protein Report,通过蛋白报告可以检查出该蛋白是否有缺失残疾或其它需要校正的地方。
然后点击Prepare Protein工具栏下方的Clean Protein按钮,对蛋白质进行简单清理,该过程主要是包括删除蛋白文件中多余的构象,为蛋白加上N端和C端等过程。
接下来我们将手动删除蛋白中的晶体水分子和盐。
通过Ctrl+H打开Hierarchy View窗口,见下图,选择Water点击键盘上的Delete删除水分子。
另外第二个A链是磷酸根基团、BME残基和JZ4配体。
删除磷酸根基团和BME残基,保留JZ4配体和蛋白。
到这里,体系已经准备好了,接下来是给体系加入立场参数和水盒子,然后准备动力学模拟。
在做拉伸动力学之前需要使用标准动力学方法将复合物进行能量最小化,短的平衡过程和动力学模拟过程。
选择Simulation | Change Forcefield工具栏,在其下方的Forcefield中选择CHARMm力场,Partial Charge中选择Momany-Rone电荷,之后点击Apply Forcefield应用所选的力场和电荷。
Discovery Studio 2.5操作教程

蛋白质结构预测技术简介简介蛋白质结构的解析对其功能的理解至关重要。
然而,由于技术手段的限制,利用实验方法(主要为X-ray,NMR)解析蛋白质结构投入大、周期长、风险大。
对于某些膜蛋白,只利用现有技术条件,其结构甚至无法解析。
另一方面,随着分子生物学技术的成熟及高通量测序技术的发展,越来越多的基因序列可以轻松被找到。
这造成了现代蛋白质科学中一个奇怪的现象:蛋白质序列数据的累积量及积累速度远远超过蛋白质结构。
这种序列与结构间不平衡的现象极大地限制了我们对蛋白质功能及其相关作用机理的理解。
所以我们需要一种能够简单、快速且相对准确的技术来确定蛋白质的空间结构。
蛋白质建模技术可以很好的解决上面的问题。
该方法利用信息技术的手段,可以直接从蛋白的一级结构(氨基酸序列)预测蛋白质的高级结构(主要为三级结构)。
根据最新一届国际建模大赛(CASP)的分类,目前主要的蛋白质建模方法包括两种:基于模板的建模(Template-based Modeling)和自由建模(Free Modeling)。
前者又包括两种方法:同源建模法(Homology Modeling)和“穿线法”(Threading)。
后者主要以从头计算法(ab initio)为主。
所有的建模方法中,以同源建模法(Homology Modeling)使用最为广泛,预测结果的准确性最大。
同源建模的理论基础为蛋白质三级结构的保守性远远超过一级序列的保守性。
因此,人们可以通过使用一个或多个已知结构的蛋白(模板蛋白,template)来构建未知结构蛋白(目标蛋白,target)的空间结构。
其主要的步骤包括:1.搜索用于建模的template(s)2.将target与templates进行比较3.将步骤(2)中的比较信息用于建模Discovery Studio为用户提供了一整套利用Homology Modeling方法自动预测蛋白质空间结构的工具。
用户只需要提供蛋白质的氨基酸序列就可以轻松完成模型构建及模型可信度评估的工作。
Materials Studio操作步骤(本人原创)
第3章铁基块体非晶合金-纳米晶转变的动力学模拟过程3.1 Discover模块动力学模拟3.1.1 原子力场的分配在使用Discover模块建立基于力场的计算中,涉及几个步骤。
主要有:选择力场、指定原子类型、计算或指定电荷、选择non-bond cutoffs。
在这些步骤中,指定原子类型和计算电荷一般是自动执行的。
然而,在某些情形下需要手动指定原子类型。
原子定型使用预定义的规则对结构中的每个原子指定原子类型。
在为特定的系统确定能量和力时,定型原子使工作者能使用正确的力场参数。
通常,原子定型由Discover使用定型引擎的基本规则来自动执行,所以不需要手动原子定型。
然而,在特殊情形下,人们不得不手动的定型原子,以确保它们被正确地设置。
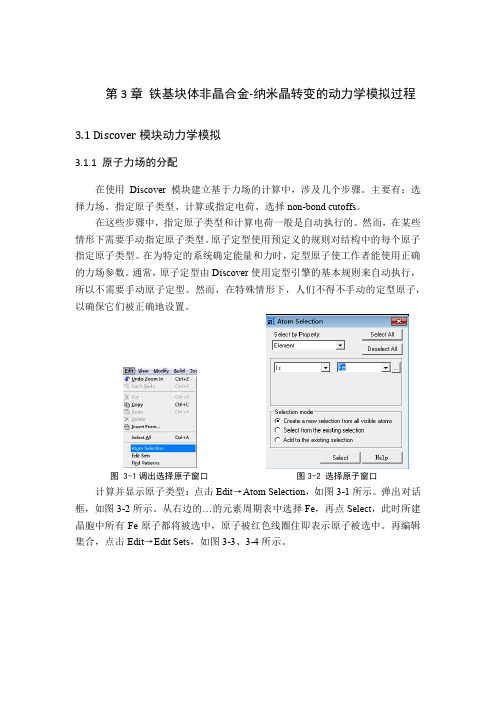
图 3-1调出选择原子窗口图3-2 选择原子窗口计算并显示原子类型:点击Edit→Atom Selection,如图3-1所示。
弹出对话框,如图3-2所示。
从右边的…的元素周期表中选择Fe,再点Select,此时所建晶胞中所有Fe原子都将被选中,原子被红色线圈住即表示原子被选中。
再编辑集合,点击Edit→Edit Sets,如图3-3、3-4所示。
图3-3 编辑集合图3-4 设定新集合弹出对话框见图3-4,点击New...,给原子集合设定一个名字。
这里设置为Fe,则3D视图中会显示“Fe”字样,再分配力场:在工具栏上点击Discover按钮,从下拉列表中选择Setup,显示Discover Setup对话框,选择Typing选项卡,见图3-5。
图3-5 给原子添加力场在Forcefield types里选择相应原子力场,再点Assign(分配)按钮进行原子力场分配。
注意原子力场中的价态要与Properties Project里的原子价态(Formalcharge)一致。
3.1.2体系力场的选择点击Energy选项卡,见图3-6。
图3-6 Energy选项卡图3-7 力场下拉菜单力场的选择:力场是经典模拟计算的核心,因为它代表着结构中每种类型的原子与围绕着它的原子是如何相互作用的。
Discovery Studio官方教程--预测蛋白质聚集、水溶性、粘度、可开发性等性质
预测蛋白聚集位点(Protein Aggregation)教程介绍抗体等具有治疗功能的蛋白,如果处于比较高的浓度下,会有发生聚集的趋势。
这会导致抗体的活性下降,并引起免疫反应。
抗体的聚集趋势计算是一种衡量蛋白表面氨基酸聚集倾向性的指标。
具有比较高的聚集趋势得分的位点表明了该区域的氨基酸倾向于发生聚集。
因此这些位点的预测,使得我们可以通过氨基酸定点突变的方法来改造蛋白,增强其稳定性。
本教程使用Calculate Aggregation Scores对一个全长IgG1抗体分子(PDB号为1h2h)进行蛋白聚集位点的预测计算,并分析预测的结果。
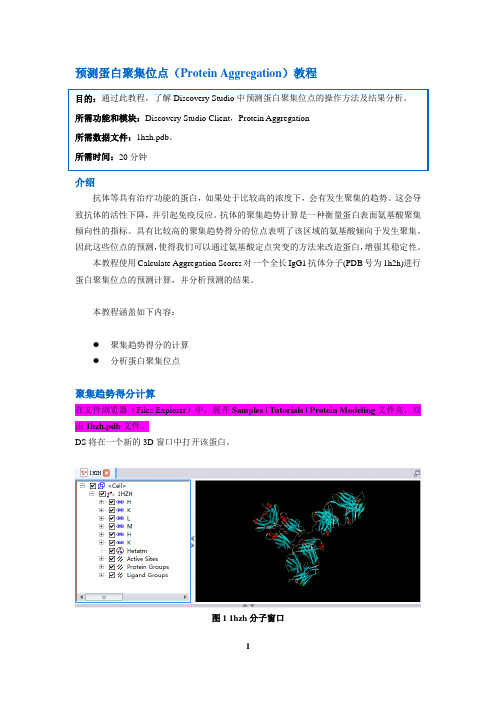
本教程涵盖如下内容:●聚集趋势得分的计算●分析蛋白聚集位点聚集趋势得分计算在文件浏览器(Files Explorer)中,展开Samples | Tutorials | Protein Modeling文件夹,双击1hzh.pdb文件。
DS将在一个新的3D窗口中打开该蛋白。
图1 1hzh分子窗口Ctrl+H打开Hierarchy窗口,然后选中Water,点击Delete删除蛋白结构中的结晶水分子。
在工具浏览器(Tools Explorer)中,展开Simulation | Change Forcefield,将Forcefield设为CHARMm Polar H,然后点击Apply Forcefield,这将为蛋白赋上CHARMm Polar H力场。
图2 Apply Forcefield设置界面在工具浏览器(Tools Explorer)中,展开Macromolecules | Predict Protein Aggregation工具面板,点击Calculate Aggregation Scores。
在弹出的参数设置界面中,将Input Typed Protein设为1hzh:1HZH,将Cutoff Radius设为5,7,10。
点击Run运行该任务。
该任务在奔腾4,2Gb内存和2.8GHz的计算机平台上运行大约需要3分钟。
discovery studio libdock打分 -回复

discovery studio libdock打分-回复Discovery Studio LibDock是一种分子对接软件,用于评估蛋白质和小分子之间的相互作用。
本文将详细介绍如何使用Discovery Studio LibDock 进行分子对接,并解释其在药物设计和筛选中的应用。
一、介绍Discovery Studio是一款常用的分子模拟和药物设计软件套件,其中包括许多强大的模块,如LibDock、CDOCKER、Glide等。
LibDock模块是其中一个用于分子对接的工具,主要用于预测蛋白质与小分子之间的结合能力。
二、准备工作在使用Discovery Studio LibDock之前,需要准备以下文件:1. 蛋白质文件(PDB或MOL2格式),包含蛋白质的三维结构信息。
2. 小分子文件(SDF或MOL2格式),包含待筛选的小分子信息。
三、创建LibDock项目1. 打开Discovery Studio软件,点击菜单栏中的“LibDock”。
2. 在弹出的LibDock窗口中,点击“New Project”按钮。
3. 在新建项目的对话框中,选择蛋白质文件和小分子文件,点击“Next”按钮。
4. 在下一个页面中,设置项目名称和保存路径,点击“Next”按钮。
5. 在后续页面中,可以选择是否输入额外的参数和限制条件,根据需要进行设置。
四、定义活性位点为了进行蛋白质与小分子之间的相互作用预测,需要明确活性位点的位置。
1. 在LibDock窗口中,点击“Define Sites”按钮。
2. 在蛋白质结构中选择活性位点的残基,并点击鼠标右键,选择“Add to Site”。
3. 重复以上步骤,直到所有活性位点都添加完毕。
4. 点击“Done”按钮以保存活性位点设置。
五、设置搜索参数在进行分子对接之前,需要设置搜索参数来指导LibDock算法进行搜索。
1. 在LibDock窗口中点击“Define Search Parameters”按钮。
Discovery简单使用手册

修改曲线的左右 值,将其调整到 界面的中部,大 部分值都以正值 显示(正反值颜 色不同,将在下 节具体叙述)
根据数据修改单位
声波值: 伽马值: 密度值:
3.2 修改曲线颜色
选择正负值的颜色,色差尽量大,便 于分辨。负值的出现是因为井头的值 域选择不当,这时就要修改井头值域, 调整到大部分值都是正值的情况,偶 然出现的几个跳值,可考虑为误差值
先单击白框,再在曲线上选择重叠好的一段, 在最好的一处单击,深度值会自动读取。
3.6 拼接曲线
选择刚截取 的那条曲线
不关闭截取的对话框, 以便和移动的对话框
进行数据对照
数据 相等
负值上移 正值下移
3.6 拼接曲线
在截好后,将两条曲线拼接起
将A(SONIC)曲 线拼接为B
(SONIC2)曲线
3.7 合成地震记录
插入时,原来的岩性会下移, 将顺序打乱,使得原来模型中 填充好的岩性发生混乱。可以 先一次性添加完所有要用的岩 性,或是在添加时用新的岩性
替换某个不用的岩性
选择使用的曲线
选择合成记录 的曲线依据
合成多少道记录
3.7 合成地震记录
选择进行合成的子波
合成时用的子波只有一个,不然 会出错,为方便查看区别,不用 的最好删除或用新的替换掉。
选择子波类型,一般为Ricker, 为更加准确可以用井旁道提取的
子波(下节详述)
3.7 合成地震记录
点空 白处 加入 合成 记录
36拼接曲线不关闭截取的对话框以便和移动的对话框进行数据对照选择刚截取的那条曲线数据相等负值上移正值下移36拼接曲线在截好后将两条曲线拼接起将asonic曲线拼接为bsonic2曲线37合成地震记录选择使用的曲线选择合成记录的曲线依据合成多少道记录37合成地震记录选择进行合成的子波选择子波类型一般为ricker为更加准确可以用井旁道提取的子波下节详述合成时用的子波只有一个不然会出错为方便查看区别不用的最好删除或用新的替换掉
discovery studio libdock打分 -回复

discovery studio libdock打分-回复Discovery Studio LibDock打分是一种分子对接技术,用于评估小分子与蛋白质之间的结合能力和亲和力。
这种技术可以用于药物发现和设计,有助于研究人员预测分子之间的相互作用。
LibDock打分是一种基于模拟计算的分子对接方法。
它能够模拟小分子与蛋白质之间的结合情况,并为每个结合形式生成一个打分。
这个打分可以反映出小分子与蛋白质之间的亲和力,从而帮助研究人员评估候选药物的结合潜力。
LibDock的打分过程可以分为以下几个步骤:第一步:准备蛋白质和小分子的结构在进行LibDock打分之前,首先需要准备蛋白质和小分子的结构。
蛋白质结构可以从蛋白质数据库或其他来源中获取,而小分子结构可以通过化学绘图软件进行设计或从化学数据库中获取。
这两者的结构需要以适当的格式保存,以便进行后续的计算。
第二步:为蛋白质和小分子设置计算参数在进行LibDock打分之前,需要为蛋白质和小分子设置一些计算参数。
这些参数包括计算类型、模型的名称、搜索空间的大小等等。
这些参数将影响后续计算的结果。
第三步:运行LibDock模拟当所有的参数设置完毕后,可以运行LibDock模拟。
LibDock模拟将通过计算来预测小分子与蛋白质之间的结合方式,并为每个结合方式生成一个打分。
这个过程将涉及到分子力场的计算、碰撞检测以及评估结合位点等操作。
第四步:分析和解释结果一旦LibDock模拟运行完毕,就可以对结果进行分析和解释。
这包括查看每个结合位点的打分情况,比较不同结合位点的亲和力等。
通过对结果的分析,可以评估候选药物与目标蛋白质之间的结合情况,并为药物发现和设计提供有用的信息。
在使用Discovery Studio LibDock进行分子对接打分时,有一些注意事项需要考虑。
首先,计算结果仅供参考,需要进一步实验验证确认。
其次,LibDock打分是基于计算模型和力场的,存在一定的误差。
DiscoveryStudio4教程

Copyright ©2014, Neotrident Technology Ltd. All rights reserved.DS CDOCKERCopyright ©2014, Neotrident Technology Ltd. All rights reservedDiscovery Studio CDOCKER 教程CDOCKER - 精准的分子对接技术所需功能和模块:Discovery Studio Client, DS CDOCKER.所需数据文件:1EQG.dsv, 1EQD-ibuprofen-conf.sd, 1EQG-ibuprofen.sd所需时间:15分钟分钟介绍CDOCKER 是基于CHARMm 力场的分子对接方法,这种方法可以产生高精度的对接结果。
在本教程中,天然布洛芬配体分子对接回COX-1受体的结合位点中,得到的对接构象和X-ray 衍射得到的晶体结构中的配体天然构象进行比较。
本教程包括:∙ 准备对接体系准备对接体系∙ 运行CDOCKER∙ CDOCKER 结果分析结果分析准备对接体系在文件浏览器(Files Explorer )中,找到并双击打开Samples | Tutorials | Receptor-Ligand Interactions| 1EQG.dsv 。
在分子窗口中将打开一个带有活性位点的蛋白质三维结构(图1)。
)。
图1蛋白质三维结构示意图在工具浏览器(Tools Explorer )中,展开Receptor-Ligand Interactions | Define and Edit Binding Site ,依次点击Show/Hide Residues Outside Sphere 和Show/Hide Sphere 。
展开菜单栏View|Transform ,点击Fit To Screen 将结合位点的氨基酸在窗口中居中显示(图2)。
Discovery Studio官方教程--ADMET预测药物代谢动力学

使用Discovery Studio进行化合物ADMET性质预测教程介绍ADMET性质是指分子在有机体内的吸收、分布、代谢、排泄和毒性等性质。
如果在药物研发的早期阶段就能依据化合物的ADMET性质对先导化合物进行有针对性的选取和优化改造,这对提高药物研发的成功率以及减少药物研发后期过程中由于ADMET性质问题所造成的资金浪费问题,是非常必要的。
ADMET描述符可以有助于及早排除ADMET性质不好的化合物从而避免后期耗资巨大的结构改造,同时也可以评价结构优化的效果,是否确实改善了ADMET属性,从而避免合成所支出的过多资源。
本教程主要介绍了在Discovery Studio中如何进行化合物ADMET性质的预测,以及预测结果的分析。
本教程包括:•运行ADMET性质计算流程•分析ADMET性质预测结果Discovery Studio中可以计算的ADMET性质包括:•25摄氏度下水溶解度(aqueous solubility)•血脑屏障通透性(Blood brain barrier penetration,BBB)•细胞色素P450 2D6抑制性(Cytochrome P4502D6 inhibition)•肝毒性(hepatotoxicity)•人类肠道吸收性(human intestinal absorption,HIA)•血浆蛋白结合率(plasma protein binding)运行ADMET性质计算流程1. 导入小分子化合物文件在文件浏览器(Files Explorer)中,展开Samples | Tutorials | QSAR,双击打开pk-test.sd文件。
在表格浏览器中可以看到一共有20个丙酮酸盐激酶抑制剂。
2. 选择计算性质,运行计算流程在工具浏览器(Tools Explorer)中,展开Small Molecules | Calculate Molecular Properties,点击ADMET Descriptors,打开ADMET Descriptors对话框。
Discovery Studio官方教程(Help-Tutorials) 分子动力学模拟

Discovery Studio Molecular Dynamics教程Molecular Dynamics –分子动力学方法介绍分子动力学(Molecular Dynamics, MD)是分子模拟中最常用的方法之一。
该方法基于分子力场,能够动态的描述分子的运动状况,继而描述生命的动态过程。
分子动力学在生命科学领域中的应用非常广泛,如蛋白质折叠的机理研究、酶催化反应的机理研究、与功能相关蛋白质的运动研究、生物大分子大范围构象变化的研究等等。
近几十年来,分子动力学方法已经成功的运用于大分子体系低能量构象的模建、X射线晶体衍射以及NMR实验结果的处理。
[1,2]现在分子动力学方法已经成了理论生物学研究中必不可少的方法之一。
[3,4]分子动力学模拟主要包括如下几个步骤:1.模拟体系升温过程(Heating Stage)2.模拟体系平衡过程(Equilibrium Stage)3.模拟体系采样过程(Production Stage)4.分析体系的目标性质(Analysis)然而,由于一般的待模拟体系的初始结构或多或少的存在缺陷(比如,初始结构不完整或存在不合理的结构区域),所以我们往往需要对初始结构进行预处理才能进行分子动力学模拟。
一个完整的分子动力学模拟过程应包括如下几个步骤:1.初始结构检查及预处理2.给模拟体系赋力场参数3.考虑溶剂效应4.初始结构能量最小化5.动力学模拟(包括升温、平衡、采样)6.结果分析目前,常用的分子动力学模拟软件都基于Unix(Linux)操作系统。
实施每一个模拟的步骤都需要特定的命令来调用相关的程序对模拟体系进行处理。
同时,模拟者还需要熟知每个参数的意义并定义相关的参数值。
最终分析过程也需要配合其他软件才能完成。
这对初级分子动力学模拟者而言是非常困难的(模拟者不仅需要掌握分子动力学软件的使用命令,还需要掌握操作系统相关的命令)。
为了解决这样的问题,Discovery Studio为用户提供了基于窗口的分子动力学模拟工具。
Discovery软件操作
新建工区 上次运行的工区
取消也可以
工区建立
在某目录下先建 一个工区文件夹
在刚刚建立的文件夹中建立工区目录(即建立一个home)
指定目录路径
右键点击刚刚建立的home,在该home中建立新工区project
新建工区
工区名字 工区描述(可不
填)
深度制式:选公制
选择坐标系统
第2个 选20带 最后一个
按此键查看下一 口井数据
按此键查看全部 井数据
井斜数据导入
井斜数据的 导入
选3 选此项目
运行后出现下框
点此按钮后出现数据.
或查看井轨迹
显示直井数据
按此按钮计算 斜井轨迹
对井斜资料进行批量计算 批量计算井斜轨迹
加载数据源
分层数据的加载
分层数据加载
加名字
加载分层数据
在此编辑
第二基准面为基础调整.
保存合成记录到 WILLBASE
点击
建立层位
建立两个追踪层位
建立一条主剖面
??????????
两个剖 面间隔
追踪主剖面层位1,上下方向键
按此 进行 追踪
层位1
追踪联络测线,左右方向键
工区内的层 位追踪完毕。
提取地震属性
做切片
以4_top层为中心, 向上偏移5毫秒
合成记录制作
合成记录的制作
输入测井曲线
数据源位置
更新井数据
做合成记录
选择基准面和高程
设置高程及替换速度
高程值 替换速度
重新做一遍
粗调 细调
粗调,先点这里
在点这里,二 者就对齐了
细调,首先以粗调 后的基准面为准
点击要微调的点
Discovery操作手册全

Discovery软件操作手册GNT国际公司目录一.工区建立与管理 (1)二.数据加载 (3)三.微机地质应用 (10)四. 微机三维地震解释综合应用 (19)五.微机单井测井解释及多井评价 (39)六.储层管理与预测 (54)七.smartSECTION模块 (63)八.正演建模 (69)一.工区建立与管理1.1工区目录(home)建立我们一般习惯把Discovery工区放在一个文件夹下,这样可方便进行管理。
1) 利用Windows资源管理器,建立一个文件夹,如proj-dis,同时可以把其它机器上的Discovery工区放在次目录下。
2) 在桌面上双击GeoGraphix Discovery图标,打开工区管理模块ProjectExplorer。
3) 进入ProjectExplorer > File> New> Home。
4) 通过浏览器确定proj-dis文件夹,点击确定按钮。
5) 点击下一步按钮,直至完成。
6) 在工区管理模块ProjectExplorer左侧,出现proj-dis工区目录,这样就可以在该目录下建立Discovery工区。
1.2工区(project)创建一般来说,任何一个地学软件工作流程的第一步都是建立一个工区,Discovery 软件也不例外。
工区的建立包括为工区名、路径、坐标系统和工区范围等。
1) 在桌面上点击GeoGraphix Discovery 图标,启动ProjectExplorer2) 在工具栏点击New Project 按钮或从菜单条选File > New> Project。
出现NewProject Wizard(新工区向导)的工区对话框。
3) 在Project Name(工区名)框中,输入工区名称。
4) 在Description(描述)框中,输入对工区的描述(如:位置、远景区等)。
5) 压力和深度单位,在中国,选择米制(这对底图和数据库坐标系统都没有影响)。
discoverystudio4.5教程

discoverystudio4.5教程
Discovery Studio 4.5是一款生物信息学和计算生物学软件套件,由Accelrys(现在是BIOVIA)开发。
它提供了一系列功能强
大的工具和功能,可用于蛋白质建模、分析、虚拟筛选、药物设计等任务。
要学习Discovery Studio 4.5的使用,可以参考以下教程:
1. 官方教程:Accelrys官方网站上提供了一些针对Discovery Studio 4.5的教程和在线帮助文档,可以登录官方网站查找相
关资源。
2. 在线教程:在互联网上可以找到一些免费的在线教程,如YouTube上的视频教程、论坛上的帖子等,这些教程可以帮助你了解Discovery Studio 4.5的基本操作和功能。
3. 书籍和教材:有一些专门针对Discovery Studio的书籍和教材,可以购买或借阅这些资料来深入学习和理解软件的使用。
4. 培训课程:有些机构或专业培训机构提供Discovery Studio
的培训课程,可以参加这些课程来系统地学习软件的使用技巧和应用。
5. 学术论文和研究文章:可以阅读与Discovery Studio相关的
学术论文和研究文章,了解其他研究者是如何使用该软件进行科学研究的。
请注意,Discovery Studio 4.5是商业软件,购买和使用需要相关许可证。
DISCOVERY操作步骤

DISCOVERY操作步骤一、安装完成。
二、井符号位置:(自定义的井符号可拷贝到此处)→C:\programdata\geographix\data\symbols\→三、定义HOME和PROJECT:1、点开DISCOVERY程序→DISCOVERY主界面→NEW→HOME→OK(完成)2、点开DISCOVERY程序→DISCOVERY主界面→NEW→PROJCET →下一步→定义名字→ASIA→RUSSIA→GAUSS-KRUGER B (SYSTEM)→11:60E-66E LONGITUDE(ZONE)(哈国)→(WGS 1984)(DATUM)→下一步→OK(完成)四、数据的测井曲线准备:1、井头数据(HEADER)坐标需要带上经纬度带(比如哈国X坐标必须加上经度11000000)2、分层数据(TOPS)3、射孔数据(COMPLETION)必须有NUM列(OBSERV ATION NUMBER)4、测井曲线5、各数据表中的WELL ID必须和测井曲线的WELL ID一致。
五、数据和测井曲线的载入:(数据和测井曲线)数据载入:1、DISCOVERY主界面→FILE→IMPORTS→SPREADSHEET(点中)→载入EXCEL文件→选中(HEADER、TOPS、COMPLETION)→定义表中各列→点选项旁边的半边小太阳→OK(完成)2、EXCEL文件导入→主菜单→工具→加载宏→点中左框中的GEOGRAPHIX SPREADSHEET TRANSACTIONS→半边小太阳(OK)→选中(HEADER、TOPS、COMPLETION)→定义表中各列→点选项旁边的半边小太阳→OK(完成)测井曲线载入:点开PRIZM→IMPORTS→LAS(ASCII)→选中所要加载的测井曲线→OK(完成)六,作井位图1、在WELLBASE主界面→TOOLS→WELL SYMBOL MANAGER→新定义一个STATUS TOLIBRARY(自己命名)→选好各井别代表的SYMBOL→SA VE→OK(完成)2、点开GEOATLAS图标→主界面→LAYERS→CREATE LAYER→WELLBASE→NORMAL→OK→左上框中的WELL HEADER→WELL ID→拖入右框井圈旁→在井圈上点右键→STRAIGHT HOLE WELL SYMBOL(第一个)→选自己定义的STATUS(LIBRARY)→自己命名图名→OK(完成)3、在上面作图的基础上→作图界面→FILTER→勾中APPLY FILTER →选中自己需要的FILTER→RERUN FILTER→确定→OK(完成)FILTER的制作:点开FILTER→左框中的WELL HEADER→WELL ID→拖入右下框→选CRITERIA 中的IN(#,#,#…)→将IN括号中的#用所选井替换→点主界面下的RUN QUERY→OK(完成)如何获得IN(#,#,#…)中的井号:EXCEL表→选中小层的所有井号→复制到新EXCEL表中(数值+转置)→井号变成横行→另存为*.csv文件→用记事本打开→选中所有井号(复制)→IN(#,#,#…)→替换IN括号中#→OK(完成)七、测井系列曲线的定义:PRIZM→主界面→PROJECT→DEFAULT SETTINGS(第一个)→北将测井系列在表中对应的曲线输好(整理好的“测井曲线类型”)→OK(完成)。
discovery studio libdock打分 -回复

discovery studio libdock打分-回复关于Discovery Studio LibDock打分的文章引言:近年来,计算机辅助药物设计(Computer-Aided Drug Design, CADD)在新药发现领域起到了重要作用。
其中,虚拟筛选是一种常用的CADD方法,通过计算机模拟来预测和优选化潜在的药物候选分子。
在虚拟筛选中,分子对接(molecular docking)方法常被用于评估分子之间的相互作用,并对候选药物进行定量分析。
Discovery Studio LibDock是一款功能强大的分子对接软件,本文将介绍Discovery Studio LibDock打分的原理和使用步骤。
第一部分:Discovery Studio LibDock打分的原理1. 蛋白质的准备:在进行分子对接之前,首先需要准备蛋白质的三维结构。
通常,可以从蛋白质数据库(如PDB)中获取已经解析的蛋白质结构,或者通过蛋白质结构预测方法得到目标蛋白的结构信息。
2. 分子的准备:除了蛋白质之外,还需要准备待筛选的小分子化合物。
这些小分子通常可以从化合物数据库中获取,或者通过化学合成的方式得到。
3. 分子对接:将小分子与蛋白质进行对接,预测它们的相互作用方式。
在Discovery Studio LibDock中,采用的是基于粒子群算法(particle swarm optimization)的LibDock算法。
该算法能够搜索最佳的小分子-蛋白质结合构象,并给出相应的打分。
4. 分子打分:根据对接得到的小分子-蛋白质结合构象,使用各种打分函数对其进行评估。
Discovery Studio LibDock提供了多种打分函数,包括LigScore、PLP等。
这些打分函数可以衡量分子在蛋白质的结合位点中的稳定性和亲和性。
第二部分:使用Discovery Studio LibDock进行分子对接和打分的步骤1. 导入蛋白质结构:首先,在Discovery Studio软件界面中导入蛋白质的结构文件。
MS软件中Discover模块建模的基本流程

MS软件中Discover模块建模的基本流程
一、建模过程
1.构建表面:输入晶体结构,并劈下所需晶面,如001面。
2.优化表面:分配力场(表面离子键需存在),定义charge group;
表层以下的原子固定,删除离子键,并用Minimize优化表面。
3.增大表面机并改变周期性:构造超胞(Supercell)来增大表
面积;使用Builder Vacuum Slab 将周期性从2D改为3D。
4.有机分子建模:生成有机物分子,分配力场,定义charge
group;利用Minimize优化有机物分子结构;将分子复制到
已建好的表面内,调整其构型(或者利用
amorphous/construction 建立层结构,然后使用分层建模工
具Builder\Layer将层结构加到表面上)。
5.利用分子动力学模拟:设置好温度(压力)、步长、步数、
保存数据类型以及输出帧数等参数,并且冻结表面原子坐标,
然后进行动力学模拟。
二、计算相互作用能:E interaction=E total-(E surface+E molecule)
1.选择所有原子取消固定,利用Set up\Energy计算总势能
(total potential energy)。
2.计算有机分子的势能E molecule。
3.计算表面总势能(取消固定)E surface。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
1、现在发现,好像最好不要进行SHAKE处理。
那么步长就为0.001.
2、并且,如果你想画从时间为0开始的图的话,你就需要不要把最后一个构象拷贝到新窗口,只需打开上一步的输出结果,然后把最后一个构象在table中的conformation点中最左边数字,使其成为active。
3、把保存的频率改为1000也许文件更小些,更好点。
4、equilibration时间尽量长点,也许500ps更合适。
你要做总能量对时间作图,看稳定否,做temperature 对time作图,看稳定否。
有时,后者变化一直很大。
并且,我还遇到一个问题,就是做production 经常出错。
不知道什么原因。
5、希望大家伙继续补充个人使用经验。
再次更新(也许是last):
先加溶剂分子,记住要加抗衡离子。
Cell shape选择加水子最少的也许好点,记住最好把Minimum Distance From Boundary设置大一点,如12。
不过计算会变慢。
然后把多肽进行Steepest Descent,即最陡下降法进行minimization,先限制多肽,用tools中的simulation 中的constraints中的create fix atom constraints处理,其中把模建最好的结果用CHARMm力场,作为input typed molecular的分子,max steps选择6000,electrostatics选择particle mesh ewald,algorithm 为Steepest Descent,number of processors选择2,使RMS Gradient为0.1,用最陡下降法优化水分子6000步。
然后把结果用Conjugate Gradient即共轭梯度法优化6000步,限制为fix,rms gradient设为0.001,electrostatics选择particle mesh ewald,number of processors选择2,其它参数默认。
然后把fix删除。
即在hierarchy中用delete删除。
然后,进行Steepest Descent,即最陡下降法进行minimization,max steps选择6000,electrostatics 选择particle mesh ewald,number of processors选择2,使RMS Gradient为0.1,用最陡下降法优化6000步。
做完之后,用tools中的protein modeling中的protein health中的check structure处理,看看结构变好没有。
有时候结果会很坏,那么就希望在以后的优化中,结果会越来越好。
主要是把时间设置长一点。
然后用共轭梯度法优化6000步,rms gradient设为0.001(看实验而定),electrostatics选择particle mesh ewald,number of processors选择2,其它参数默认。
做完之后,用protein model中的check structure 处理,看看结构变好没有。
有时候结果会很坏,那么就希望在以后的优化中,结果会越来越好。
然后进行dynamics(heating or cooling),其中input typed molecular为上步conjugate gradient法得到的outputfile(包含水分子和离子),steps设置为100000,save results frequency为1000,electrostatics为particle mesh ewald,heating save restart file为true,number of processors选择2,没有限制常数,其它
参数默认。
温度当然要看你的试验情况了。
做完之后,用protein
model中的check structure处理最后一个构象(需要把最后构象复制到新窗口,即选中最后一列数字,然后在hierarchy中点击右键选择copy,然后选择file-new-3d windows,然后paste),看看结构变好没有。
然后进行dynamics(equilibration),其中input typed molecular为上步dynamics(heating or cooling) 中的output中结果(要包括水分子和离子,不用复制最后构象到新窗口,只要打开含有全部构象的文件就行),记住把最后一个构象变为active(即选中最后一列数字),温度当然要看你的试验情况了,steps 设置为500000,save results frequency为1000,constant pressure为true(根据实验而定),electrostatics 为particle mesh ewald,equilibration save restart file为true,其中equilibration restart file为上步得到的output中的rst文件,number of processors选择2,其它参数默认。
然后进行dynamics(production),其中的input type molecular为上步dynamics(equilibration)中output 中的结果(要包括水分子和离子,不用复制最后构象到新窗口,只要打开含有全部构象的文件就行),记住要把最后一个构象选中(即选中最后一列数字),使其变为active,温度当然要看你的试验情况了,steps设置为1000000,save results frequency为1000,production type为NPT(根据你实验情况而定),electrostatics为particle mesh ewald,production restartfile为上步得到的output中的rst文件,production save restart file为true,number of processors选择2,其它参数默认。
用protein
model中的check structure处理最后一个构象(需要把最后构象复制到新窗口),看看结构变好没有。
时间可以设置更长,现在好象要求越来越长了。
不过,我一般开始先跑500ps,然后才1ns。
主要怕出错。
这是我的基本步骤,主要处理蛋白质的,并且加了水分子和离子,不知道有什么不足。
对于小分子,可能没有这么麻烦,可以用最简单的方法进行优化(即进行minimization时,algorithm选择smart minimizer处理,其中implicit solvent model选择GBSW,max steps选择5000,其它默认)。
对于蛋白质用隐形模型处理的话,Implicit Solvent Model最好选择GBSW, Electrostatics最好为Spherical Cutoff。
综上,也许有不对之处请指出。
谢谢!
说句题外话,分区格式最好为NTFS,因为FAT32最大支持文件为4G。
更新:
好像跑平衡跑500ps太少,我打算先跑2ns,再跑production。
因为我做的东西总是production出错。