中科大-Materials-Studio-培训教程4PPT课件
合集下载
中科大MaterialsStudio培训教学课件

此时按下 ALT key 单击鼠标左键,则出现一个具有 with resonant bonds 的 芳香环aromatic ring)
Sketch Atom 按钮 可以画任意元素的原子,默认画碳原子。下面要将双碳 原子侧链接到环上。
在绘图工具栏(Sketch toolbar) 上单击Sketch Atom 按钮 ,松开,然后鼠 标移动到环的3D文档中,这时鼠标看起来象一只“铅笔”。将“铅笔”移到环的一 个碳原子上,这个碳原子变蓝。左键单击此碳原子,将键连接到该原子上。移动“ 铅笔”并在3D的合适位置单击左键,设置另一个碳原子。键自动加在此碳原子与环 之间。将“铅笔”移到另一位置,双击左键,又画出一个碳原子。这样就作出了连 到碳环上的一个双碳原子链。
选择相应的对象单击
* 在TON 结构上单击选中的原子此原子颜色改变,说明被选中
* 单击一个键此键的颜色改变,说明被选中
键被
选中
原子被选中
* 按住鼠标左键沿斜线托拽,可以选择一定区域内的所有对象,包括原子和键
此区域的 原子和键 都被选中
* 在结构中的某个原子或键上双击鼠标可以选择整个结构 * 在3D Viewer 上无TON 结构的地方单击或双击鼠标则取消对象的选择。 * 需要将结构保存为project的一部分时,单击3D Viewer 的 ,再按 Yes 按钮。 * File / Save Project ,Windows / Close All
To monitor and adjust distances
下面建造苯甲酰胺结构:
1. 生成新的3D文档 在菜单上选择File / New并且选择3D Atomistic 后单击OK此时文件名称出现在左
侧的Project Explorer 中,名称为3D Atomistic.xsd,在其上单击鼠标右键,选择 Rename ,将名称改为my-benzamide。
Sketch Atom 按钮 可以画任意元素的原子,默认画碳原子。下面要将双碳 原子侧链接到环上。
在绘图工具栏(Sketch toolbar) 上单击Sketch Atom 按钮 ,松开,然后鼠 标移动到环的3D文档中,这时鼠标看起来象一只“铅笔”。将“铅笔”移到环的一 个碳原子上,这个碳原子变蓝。左键单击此碳原子,将键连接到该原子上。移动“ 铅笔”并在3D的合适位置单击左键,设置另一个碳原子。键自动加在此碳原子与环 之间。将“铅笔”移到另一位置,双击左键,又画出一个碳原子。这样就作出了连 到碳环上的一个双碳原子链。
选择相应的对象单击
* 在TON 结构上单击选中的原子此原子颜色改变,说明被选中
* 单击一个键此键的颜色改变,说明被选中
键被
选中
原子被选中
* 按住鼠标左键沿斜线托拽,可以选择一定区域内的所有对象,包括原子和键
此区域的 原子和键 都被选中
* 在结构中的某个原子或键上双击鼠标可以选择整个结构 * 在3D Viewer 上无TON 结构的地方单击或双击鼠标则取消对象的选择。 * 需要将结构保存为project的一部分时,单击3D Viewer 的 ,再按 Yes 按钮。 * File / Save Project ,Windows / Close All
To monitor and adjust distances
下面建造苯甲酰胺结构:
1. 生成新的3D文档 在菜单上选择File / New并且选择3D Atomistic 后单击OK此时文件名称出现在左
侧的Project Explorer 中,名称为3D Atomistic.xsd,在其上单击鼠标右键,选择 Rename ,将名称改为my-benzamide。
中科大MaterialsStudio培训教程4

激活。
如果动画(Animation)工具条 是不可见的,
则按右侧的操作,使用观看 (View)菜单让它显示。
设置显示方式,按播放键 。 B-O近似,体系的电子能量是核构型的函数。
在继续工作之前,需要关闭Materials Visualizer 中的所有文件。 关闭DMol3 计算对话框。选择File\Save Project,然后Window\Close All。 双击几何优化子文件夹中的reactant.xsd 和product.xsd。 现在工作区域中只有两个优化了的结构。
运用DMol3的 LST/QST功能来搜索过渡态,需要 在反应物和产物之间创建一条通道,这也是DMol3 计 算时所要求的输入文件。
在反应预览(Reaction Preview)对话框中,把桢数 提高到100;勾选上Superimpose structures;单击 Preview按钮,关闭反应预览(Reaction Preview)对话 框。
电子Hamiltonian 的设置与几何优化计 算的设置一样。
这次需要计算频率(Frequency)相关的性质。 点击Properties 标签栏,勾选上Frequency。
最后,需要对工作描述(Job Description)加以设 置。
点击Job Control 标签,确认Automatic 没有被勾 选上;在Job Description 一栏里打上TS。
点击Set Match,还剩下5H、7H未匹配。重复这个过程,继续点击Set Match来匹配 剩下的没有配对的原子。
反应物、产物的原子已配对。现在可以预览一下反应物和产物之间原子的匹配 情况。
点击反应物或者产物栏中列表里的任意一个原子,可以看到匹配的另一原子。 考察匹配情况,直到满意为止。关闭Find Equivalent Atoms 对话框。
如果动画(Animation)工具条 是不可见的,
则按右侧的操作,使用观看 (View)菜单让它显示。
设置显示方式,按播放键 。 B-O近似,体系的电子能量是核构型的函数。
在继续工作之前,需要关闭Materials Visualizer 中的所有文件。 关闭DMol3 计算对话框。选择File\Save Project,然后Window\Close All。 双击几何优化子文件夹中的reactant.xsd 和product.xsd。 现在工作区域中只有两个优化了的结构。
运用DMol3的 LST/QST功能来搜索过渡态,需要 在反应物和产物之间创建一条通道,这也是DMol3 计 算时所要求的输入文件。
在反应预览(Reaction Preview)对话框中,把桢数 提高到100;勾选上Superimpose structures;单击 Preview按钮,关闭反应预览(Reaction Preview)对话 框。
电子Hamiltonian 的设置与几何优化计 算的设置一样。
这次需要计算频率(Frequency)相关的性质。 点击Properties 标签栏,勾选上Frequency。
最后,需要对工作描述(Job Description)加以设 置。
点击Job Control 标签,确认Automatic 没有被勾 选上;在Job Description 一栏里打上TS。
点击Set Match,还剩下5H、7H未匹配。重复这个过程,继续点击Set Match来匹配 剩下的没有配对的原子。
反应物、产物的原子已配对。现在可以预览一下反应物和产物之间原子的匹配 情况。
点击反应物或者产物栏中列表里的任意一个原子,可以看到匹配的另一原子。 考察匹配情况,直到满意为止。关闭Find Equivalent Atoms 对话框。
Materials Studio软件简介及基本操作PPT课件

计算机模拟方法可以根据相应基本理论,在计算机虚拟 环境下从纳观、微观、介观、宏观尺度对材料进行多层次 研究,进而实现材料服役性能的改善和材料设计。
目前,已成为与实验研究、理论研究具有同样重要地位 的研究手段。
哈勃望远镜观察到的宇宙
三体-该片根据刘慈欣同名小说改编,讲述了在红岸基地人类文明初次向宇宙 发出啼鸣后,开启了与计划殖民地球的三体文明间生存之战
诺贝尔化学奖得主(自左向右):亚利耶·瓦谢尔、马丁·卡普拉斯和迈克尔·莱维特。
随着科学技术的不断发展,科学研究的体系越来越复杂, 理论研究往往不能给出复杂体系解析表达,或者即使能够给 出解析表达也常常不能求解,传统的解析推导方法已不敷应 用,也就失去了对实验研究的指导意义。反之,失去了理论 指导的实验研究,也只能在原有的工作基础上,根据科研人 员的经验理解、分析与判断,在各种工艺条件下反复摸索, 反复实验,最终造成理论研究和实验研究相互脱节。
分子力学中用力场来描述分子中各原子间的相互作用。 所谓力场是指描述各种形式的相互作用对分子能量影响的函 数,其有关参数、常数和表达式通常称为力场。
一般力场的表达式为:
E E str e E b tc e h n E td or E svio d E n w el e .c ......
Estretch. 为键的伸缩能; E bend . 为键的弯曲能,二者均采用谐振子模型;
最后可以对轨迹进行各种结构、能量、热力学、动力学、 力学等的分析。
④ 耗散动力学方法 1992年,Hoogerbrugge和Koelman提出了一种新型分子模拟
方法,他们把分子动力学与格子气体自动控制方法有机地结合起 来,提出了针对复杂流体介观层次上的模拟方法,被称为耗散粒 子动力学(DPD)方法。通过保留体系运动方程积分的主要部分而 首先积分出最小的空间自由度,找到了一个能够在介观的时间与 空间尺度上模拟复杂流体的方法。在DPD体系中,珠子通过软势 与其它珠子之间发生相互作用,其中每一个珠子表示体系中的一 个小区域。并假设其运动遵从牛顿定律,即珠子上的合力为其直 接相互作用及它与其它珠子之间的耗散力和随机力之和。通过对 其运动方程积分,得到体系的动力学行为沿着一个通过相空间的 抛物线运动,利用柔性(soft)势能函数进行能量计算,平衡性质可 由沿该轨迹作适当平均计算得出。
目前,已成为与实验研究、理论研究具有同样重要地位 的研究手段。
哈勃望远镜观察到的宇宙
三体-该片根据刘慈欣同名小说改编,讲述了在红岸基地人类文明初次向宇宙 发出啼鸣后,开启了与计划殖民地球的三体文明间生存之战
诺贝尔化学奖得主(自左向右):亚利耶·瓦谢尔、马丁·卡普拉斯和迈克尔·莱维特。
随着科学技术的不断发展,科学研究的体系越来越复杂, 理论研究往往不能给出复杂体系解析表达,或者即使能够给 出解析表达也常常不能求解,传统的解析推导方法已不敷应 用,也就失去了对实验研究的指导意义。反之,失去了理论 指导的实验研究,也只能在原有的工作基础上,根据科研人 员的经验理解、分析与判断,在各种工艺条件下反复摸索, 反复实验,最终造成理论研究和实验研究相互脱节。
分子力学中用力场来描述分子中各原子间的相互作用。 所谓力场是指描述各种形式的相互作用对分子能量影响的函 数,其有关参数、常数和表达式通常称为力场。
一般力场的表达式为:
E E str e E b tc e h n E td or E svio d E n w el e .c ......
Estretch. 为键的伸缩能; E bend . 为键的弯曲能,二者均采用谐振子模型;
最后可以对轨迹进行各种结构、能量、热力学、动力学、 力学等的分析。
④ 耗散动力学方法 1992年,Hoogerbrugge和Koelman提出了一种新型分子模拟
方法,他们把分子动力学与格子气体自动控制方法有机地结合起 来,提出了针对复杂流体介观层次上的模拟方法,被称为耗散粒 子动力学(DPD)方法。通过保留体系运动方程积分的主要部分而 首先积分出最小的空间自由度,找到了一个能够在介观的时间与 空间尺度上模拟复杂流体的方法。在DPD体系中,珠子通过软势 与其它珠子之间发生相互作用,其中每一个珠子表示体系中的一 个小区域。并假设其运动遵从牛顿定律,即珠子上的合力为其直 接相互作用及它与其它珠子之间的耗散力和随机力之和。通过对 其运动方程积分,得到体系的动力学行为沿着一个通过相空间的 抛物线运动,利用柔性(soft)势能函数进行能量计算,平衡性质可 由沿该轨迹作适当平均计算得出。
Materials Studio 培训教程4(包你学会!)请将这一系列全看完,一定有收获。
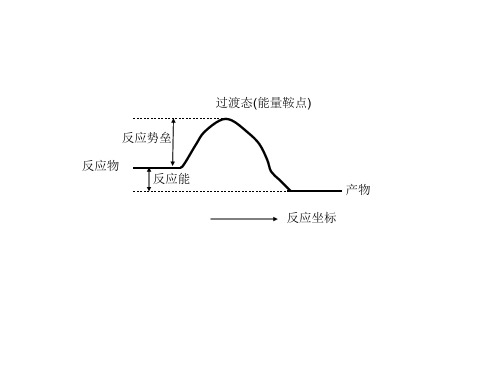
过渡态(能量鞍点)
反应势垒 反应物 反应能 反应坐标
产物
用LST/QST 搜索过渡态
目的: 介绍如何使用 DMol3 和 Reaction Preview 工具进行过渡态搜索的计算。 对简 单反应,这种方法是有效的。 模块: Materials Visualizer, DMol3 前提: 用局域内坐标对固体进行结构优化。 背景 对任何反应的势能面的探索都要求知道反应进程中每一步的结构和能量, 或者动力学和热动力学的快照(snapshots)。特别重要的是决定反应速率的那一步, 这通常需要找到那些难以捕获的过渡态结构。有一些方法对找到过渡态的结构是 很有效果的,其中比较知名的就是线性同步度越(linear synchronous transit, LST)和二次同步度越(quadratic synchronous transit,QST)。 本例中,我们将介绍DMol 中的LST /QST 工具的使用,将会看到如何使用 LST/QST 搜索乙烯醇转变为乙醛的H转移反应 的过渡态结构。 CH2CHOH → CH3CHO 本例包括以下内容: ������ 1. 建立一个计算模型 ������ 2. 优化分子结构 ������ 3.定义原子对 ������ 4.用LST/QST 的方法计算过渡态 ������ 5.优化过渡态结构
1.建立一个计算模型 选择 creating a new project,建立名为vinylOH 的project 。
在本单元中,你要在两个不同的3D Atomistic 界面中建立反应物和产物模型。第 一步就是打开一个新的3D Atomistic界面,构建反应物乙烯醇(vinyl alcohol)。 点击工具栏里的New button,选择3D Atomistic。
点击碳-碳键一次,选中。点击Sketch工 具条上的Modify Bond Type 键 ,选 择双键,从而把单键变成双键。点击别处, 取消选择碳-碳键。
反应势垒 反应物 反应能 反应坐标
产物
用LST/QST 搜索过渡态
目的: 介绍如何使用 DMol3 和 Reaction Preview 工具进行过渡态搜索的计算。 对简 单反应,这种方法是有效的。 模块: Materials Visualizer, DMol3 前提: 用局域内坐标对固体进行结构优化。 背景 对任何反应的势能面的探索都要求知道反应进程中每一步的结构和能量, 或者动力学和热动力学的快照(snapshots)。特别重要的是决定反应速率的那一步, 这通常需要找到那些难以捕获的过渡态结构。有一些方法对找到过渡态的结构是 很有效果的,其中比较知名的就是线性同步度越(linear synchronous transit, LST)和二次同步度越(quadratic synchronous transit,QST)。 本例中,我们将介绍DMol 中的LST /QST 工具的使用,将会看到如何使用 LST/QST 搜索乙烯醇转变为乙醛的H转移反应 的过渡态结构。 CH2CHOH → CH3CHO 本例包括以下内容: ������ 1. 建立一个计算模型 ������ 2. 优化分子结构 ������ 3.定义原子对 ������ 4.用LST/QST 的方法计算过渡态 ������ 5.优化过渡态结构
1.建立一个计算模型 选择 creating a new project,建立名为vinylOH 的project 。
在本单元中,你要在两个不同的3D Atomistic 界面中建立反应物和产物模型。第 一步就是打开一个新的3D Atomistic界面,构建反应物乙烯醇(vinyl alcohol)。 点击工具栏里的New button,选择3D Atomistic。
点击碳-碳键一次,选中。点击Sketch工 具条上的Modify Bond Type 键 ,选 择双键,从而把单键变成双键。点击别处, 取消选择碳-碳键。
materialstudio教程PPT课件

.
2
1. 生成Projects
.
3
生成Projects
(1).运行Material studio (2).生成Project
.
4
2.打开并且观察3D 文档
.
5
打开并且观察3D 文档
(1)调整显示风格 在 3D 结构上单击右键 并选择Display Style
对话框中的各选项的意义如下 Atom栏 Line:线状模型 Stick:棍状模型 Ball and stick:球棍模型 CPK:球堆砌模型 Polyhedron:多面体堆积模型(晶体) Lattice 栏: Display:显示单个晶胞或者元胞 Range:显示在X、Y、Z 方向上晶胞的数量 Lattice:显示晶胞边界的风格
.
18
Байду номын сангаас
4.观察并且处理研究表格文档
.
19
观察并且处理研究表格文档
.
8
3.绘制苯甲酰胺分子
.
9
绘制苯甲酰胺分子
下面是要建造的苯甲酰胺结构:
(1).生成3D 文档
➢ 在菜单上选择 New,并且选择3D Atomistic Document 后单 击OK此时文件名称出现。
➢ 在左侧的Project Explorer 中,名称为3D Atomistic Document.xsd,在其上单击鼠标右键,选择ReName 进行改 名并进行保存。
单击原子将键连接到该原子上,然后移动鼠标并在合适 位置单击设置另一个原子。
要结束绘制,请在最后一个原子上双击鼠标左健或者按
下键盘上的 Esc 键。
.
11
绘制苯甲酰胺分子
(3).将分子改变为球棍模型
Materials-Studio-快速入门教程PPT课件

* 在3D Viewer 上无TON 结构的地方单击或双击鼠标,则取消对象的选择。
* 需要将结构保存为project的一部分时,单击3D Viewer 的 ,再按 Yes 按钮。
* File / Save Project ,Windows / Close 2A0l2l 1
12
三. 绘制苯甲酰胺分子
话框。在Display Style中选择Ball and stick 。单击 Close button 关闭 Default Atom Style 对话框。
这样在本project 中,默认显示方式被设置为 ball and stick。
2021
15
3. 绘制分子环和原子链
Sketch toolbar 在绘图工具栏(Sketch toolbar) 上单击Sketch Ring 按钮 ,松开,然后鼠标 移动到3D文档中。这时鼠标看起来象一只铅笔,其右侧的数字表示将要绘制的环上 的原子数(可以在键盘上按下数字键3-8 来改变环的大小)。这里键入6,在3D Viewer上单击左键,则出现一个六边形的碳环。
目的: 介绍Materials Visualizer 中的绘图工具sketching tools 模块: Materials Visualizer 前提: 已生成一个Project 引言 化学家不得不每天处理大量的小分子和化学中间体。快速生成该类分子对于 每一个分子建模环境都是非常重要的。苯甲酰胺分子就是这样一种小分子, 在下边我们将以该分子作为例子,进行研究工作。
To monitor and adjust distances
2021
13
下面建造苯甲酰胺结构:
1. 生成新的3D文档 在菜单上选择File / New,并且选择3D Atomistic 后单击OK。此时文件名称出现