普通口服固体制剂参比制剂选择和确定的指导原则
普通口服固体制剂溶出度试验技术指导原则

普通口服固体制剂溶出度试验技术指导原则普通口服固体制剂溶出度试验是通过测定药物在一定时间内从固体制剂中溶出的含量来评价制剂的质量和性能。
这个试验在药物研发和生产中具有重要的意义,可以用来比较不同制剂的溶出速率和溶出度,从而指导制剂的优化和改进。
以下是普通口服固体制剂溶出度试验的技术指导原则:1.药物选取:选择溶出介质和试验条件时,应考虑药物的溶解性和性质。
药物的溶解度可以通过文献资料或初步试验来确定。
2.溶出介质:根据药物的特性选择合适的溶出介质,一般选用仿生体液模拟消化道环境,如pH1.2酸性液体、pH4.5醋酸缓冲液或pH6.8磷酸盐缓冲液。
如果药物在上述介质中溶解度较低,可以添加适量的表面活性剂。
3.试验仪器:常见的溶出度试验仪器有旋转篮法、双槽法、胶囊法等。
根据药物的特性选择合适的试验仪器。
4.试验条件:试验温度和搅拌速度是影响溶出度试验结果的重要因素。
一般可选择37℃恒温水浴,并设置适当的搅拌速度,通常为50-100转/分钟。
5.取样与检测:根据试验要求和所测药物的特性,确定取样时间和方法。
离线法和在线法都可以用于取样与测定。
离线法通常是取制剂各个时间点的溶液样品,通过适当的手段对样品进行分析。
在线法则是实时测定溶解度。
可通过利用紫外光谱仪、高效液相色谱仪等仪器对取得的样品进行检测。
6.数据处理:溶出度试验通常得到的是药物释放曲线,对于不同制剂的溶出度试验结果进行统计学处理,如计算平均溶出度和标准差等。
7.结果评价:根据药物的特性和要求,对于结果可以进行合理的评价。
可以比较不同制剂的溶出速率和溶出度;也可以将实验数据与相关标准进行比较,如中国药典中对于溶出度的规定。
总之,普通口服固体制剂溶出度试验技术指导原则包括药物选取、溶出介质选择、试验仪器选择、试验条件确定、取样与检测方法选择、数据处理和结果评价等。
根据这些原则,可以进行准确、可靠地评价固体制剂的溶出度,为制剂的优化和改进提供基础依据。
生物等效性试验实施中的注意事项

生物等效性试验生物等效性定义如下:在相似的试验条件下单次或多次给予相同剂量的试验药物后,受试制剂中药物的吸收速度和吸收程度与参比制剂的差异在可接受范围内。
以药动学参数为终点评价指标的生物等效性研究通常采用Cmax 和AUC进行评价。
生物等效的接受标准:一般情况下,Cmax和AUC 几何均值比值的90%置信区间数值应落在80.00%-125.00%范围内,对于窄治疗窗药物,应根据药物的特性适当缩小90%置信区间范围。
一个完整的生物等效性研究包括研究设计、生物样本分析、统计分析、结果评价四个方面内容。
生物等效性研究方法按照研究方法评价效力,其优先顺序为药代动力学研究、药效动力学研究、临床研究(以临床疗效为终点)和体外研究。
研究设计1、两制剂、单次给药、交叉试验设计;2、两制剂、单次给药、平行试验设计:适用于半衰期较长的药物;3、重复试验设计:适用于部分高变异药物(个体内变异≥30%)。
受试者选择1、年龄在18周岁或以上,同一批受试者年龄相差不宜超过10岁;2、如果研究药物拟用于两种性别的人群,一般情况下,研究入选的受试者应有适当的性别比例;3、为避免其他药物干扰,试验前两周内及试验期间禁服任何其他药物,试验期间禁烟、酒及含咖啡因的饮料;4、如果研究药物主要拟用于老年人群,应尽可能多地入选60岁以上的受试者;5、当入选健康受试者参与试验可能面临安全性方面的风险时,则建议入选试验药物拟适用的患者人群,并且在试验期间应保证患者病情稳定。
参比制剂的选择:1、尽可能选择原研产品;2、国际公认的同种药品,指在欧盟、美国上市并获得参比制剂地位。
给药方案:1、推荐采用单次给药药代动力学研究方法评价生物等效性;2、若出于安全性考虑,需入选正在进行药物治疗,且治疗不可间断的患者时,可选择多次给药;3、给药剂量一般应与临床单次用药剂量一致,不得超过临床推荐的单次最大剂量或已经证明的安全剂量。
食物影响1、通常需进行空腹和餐后生物等效性研究;2、如果参比制剂说明书中明确说明该药物仅可空腹服用(饭前1小时或饭后2小时服用)时,则可不进行餐后生物等效性研究;3、仅能与食物同服的口服常释制剂,除了空腹服用可能有严重安全性方面风险的情况外,均建议进行空腹和餐后两种条件下的生物等效性研究;4、如有资料充分说明空腹服药可能有严重安全性风险,则仅需进行餐后生物等效性研究;5、对于口服调释制剂,建议进行空腹和餐后生物等效性研究。
人体生物等效性研究--常见问题及其相关考虑

科
High Permeability
High Permeability
Low Permeability
迈
医
药
三、其他类型的BE豁免
BE豁免--例外
对于副作用太大,无法用健康受试者做高规格制剂 BE试验的药物,若 Low Solubility High Solubility
制剂为含量属于小浓度(当浓度小于 5%),高效能的药品,可以低规 High Permeability High Permeability 格通过BE,则高规格获得BE豁免(需满足上述BE豁免三要素)。 【例】 盐酸普拉克索片BE(治疗特发性帕金森氏症)
4. BCS Ⅲ类药物受试制剂与参比制剂的辅料性质相同、含量相似
泰
科
High Permeability
迈
医
Low Solubility
药
二、基于BCS分类的BE豁免
溶出度:具有快速溶出能力High Permeability
溶解性:在250 mL或更少的介质中溶解 渗透性:体内吸收程度≥85%
基于BCS分类的 BE 豁免不适用于口服缓释(extended release,ER) Low Permeability
Low Permeability
或控释(control release,CR)制剂, ER和CR制剂不得BE豁免; 只有制剂中的API属于BCS I类或BCS III类药物才可以考虑BE豁免。
Low Solubility 糖醇在胃肠道的吸收较差,可增加肠道渗透压,影响胃肠 Low Permeability
道中水分的输送和肠道运药时间,从而影响药物吸收。表 面活性剂、脂肪酸、壳聚糖等辅料可能改变肠道通透性。
固体制剂研发技术及法规要点汇总

口服固体仿制药及一致性评价技术要点1.固体仿制药背景仿制药一致性评价工作就是要将我国仿制药品质由满足最低层次(主成分无误、含量符合规定)提升至广大患者用药有效安全的高品质层次。
通俗地讲,就是要达到“吃药不是吃含量,而是吃生物利用度”的目的。
2.法规2.1.CTD撰写指导原则《化学药品仿制药口服固体制剂质量和疗效一致性评价申报资料要求(试行)》《化学药品新注册分类申报资料要求(试行)》《M4:人用药物注册申请通用技术文档(CTD)》2.2.制剂工艺指导原则《化学仿制药口服片剂功能性刻痕设计和研究技术指导原则(试行)》《化学仿制药晶型研究技术指导原则(试行)》《化学药物口服缓释制剂药学研究技术指导原则》《化药口服固体制剂混合均匀度和中控剂量单位均匀度研究技术指导原则(试行)》《化学药物(原料药和制剂)稳定性研究技术指导原则》《化学药物制剂研究基本技术指导原则》2.3.参比制剂确定《普通口服固体制剂参比制剂选择和确定指导原则》《仿制药参比制剂目录》2.4.制剂质量指导原则《普通口服固体制剂溶出度试验技术指导原则》《普通口服固体制剂溶出曲线测定与比较指导原则》《化学药物杂质研究的技术指导原则》《中国药典》2020版2.5.BE指导原则《人体生物等效性试验豁免指导原则》《化学药物制剂人体生物利用度和生物等效性研究技术指导原则》《注册分类4、5.2类化学仿制药(口服固体制剂)生物等效性研究批次样品批量的一般要求》《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》3.固体制剂研究思路根据ICH Q8(R2)中“QbD质量源于设计”理念,处方前研究显得尤为重要,甚至可达事半功倍的效果。
通过对原料药、辅料、参比制剂的充分研究,筛选出会影响制剂处方和工艺、制备过程和质量的有关的各项因素,然后以预先设定的目标产品质量概况(QTPP)为研发起点,在确定产品关键质量属性(CQA)基础上,基于风险评估和实验研究,确定关键物料属性(CMA)和关键工艺参数(CPP),进而建立能满足产品性能且工艺稳健的控制策略,并实施产品和工艺的生命周期管理和持续改进。
2022年江西省执业药师继续教育试题与答案2

2022年江西省执业药师继续教育试题与答案2选择题(每题有一个或一个以上最佳选项,请选出最符合题意的答案)1、O/在WHHAI提出的非政府雇员最低工资法(LPGW),是计算药品费用相当于国家政府工作人员日(A)的天数。
:(1分)某A最低工资B平均工资C最高工资D绩效工资2、以下关于中位价格比值(MPR)的说法不正确的是(D):(1分)某A某药品的MPR是调查机构中该药品最高价格与该药品国际参考价格的比值B可用于衡量某地区或某国药品价格水平与国际药品价格水平的差异CMPR<1,表示调查地区某药品的价格低于国际参考价格DMPR<1,表示调查地区某药品的价格高于国际参考价格3、按照WHO药品价格调查方法,每一个调查地区要选择公立医院()家,零售药店/药房()家,民营医院()家。
:(1分)某AA555B777C666D4444、按照WHO药品价格调查指南,调查地区需在确定的行政区中选择()个符合一定条件的地区。
:(1分)某AA6B7C5D45、根据历史经验,判断总体标准差为15,总体单位数总为1000个医疗机构,希望的平均误差在3元之间,调查结果要求在95%的置信范围内,则抽样调查的样本含量为()。
:(1分)某AA88B85C92D976、通常,不考虑医疗保障的情况下,消费者(患者)面对的药品价格是(A):(1分)某A零售价格B出厂价格C批发价格D国家最高零售限价7、以下方法中,(A)不是概率抽样方法。
:(1分)某A定额抽样B系统抽样C分层抽样D整群抽样8、下列研究方法中,(A)是一种前瞻性研究。
:(1分)某A队列研究B现况研究C病例对照研究D病例研究9、药品价格是指药品作为一种商品的(A)表现形式。
:(1分)某A价值B使用价值C劳动价值D生产成本10、生物转化(ABC):(1分)某A.主要在肝脏进行B.第一步为氧化、还原或水解,第二步为结合C.与排泄统称为消除D.使多数药物药理活性增强,并转化为极性高的水溶性代谢物E.主要在肾脏进行11、影响药物作用因素包括(ABCDE):(1分)某A.年龄,遗传异常B.性别,病理情况C.心理因素,药物耐受D.机体生理功能时间节律性,药物的时间节律性E.药物剂型,配伍禁忌12、临床用硝苯地平治疗心绞痛发作根据昼夜节律特点为(ad):(1分)某A.心肌梗死、短暂心肌缺血的发病频数,在昼夜间的波动,呈现早晨(6:00时)到中午(12:00时)之间的高峰,在午夜后4:00时左右低谷。
口服固体制剂溶出曲线测定与比较指导原则

口服固体制剂溶出曲线测定与比较指导原则1. 引言口服固体制剂溶出曲线测定与比较是药物研发和生产过程中的重要环节。
通过测定药物在给定时间内从固体制剂中溶出的速度和程度,可以评估药物的释放特性和溶出动力学,为药物的质量控制、生物等效性评价和临床应用提供依据。
本文将介绍口服固体制剂溶出曲线测定与比较的原则、方法和注意事项。
2. 原则口服固体制剂溶出曲线测定与比较需要遵循以下原则:2.1 确定测试条件在进行溶出曲线测定前,需要确定一系列测试条件,包括: - 溶媒选择:根据药物的特性选择适当的溶媒,常用的有酸性介质、中性介质和碱性介质。
- 温度选择:根据药物的特性和临床应用条件选择合适的温度。
- 搅拌速度:搅拌速度对于药物释放速率有较大影响,需要根据实际情况进行调整。
- 采样时间间隔:根据药物的释放速度和溶出动力学选择合适的采样时间间隔。
2.2 使用合适的仪器设备口服固体制剂溶出曲线测定与比较需要使用专用的仪器设备,包括: - 溶出仪:常见的有旋转式溶出仪、流动式溶出仪等,根据药物特性和测试要求选择合适的仪器。
- 采样器:用于定时自动采集样品。
2.3 合理选择样品数量为了获得可靠和准确的结果,需要合理选择样品数量。
一般情况下,每个制剂至少应测定3个批次,并进行统计分析。
2.4 数据处理与分析口服固体制剂溶出曲线测定得到的数据需要进行处理与分析。
常见的方法包括计算药物在不同时间点的累积释放量、计算释放度等指标,并绘制溶出曲线图。
同时,还可以通过统计学方法对多个批次之间进行比较,评估其相似性或差异性。
3. 方法口服固体制剂溶出曲线测定的一般方法如下:3.1 样品准备按照药物制剂的规定方法准备样品,包括粉碎、筛选和称量等步骤。
3.2 溶媒选择根据药物的特性选择合适的溶媒,常见的有酸性介质、中性介质和碱性介质。
在选择时需要考虑药物的溶解度、稳定性和生物相容性等因素。
3.3 测试条件确定根据实际情况确定测试条件,包括温度、搅拌速度和采样时间间隔等参数。
仿制药质量和疗效一致性评价参比制剂与BE备案

BE试验备案和程序
2.属于下列情形的化学药,如需开展BE试验,可按照《药品注册管理办法》的有关规定申报受理和审评审批1)放射性药品、麻醉药品、第一类精神药品、第二类精神药品和药品类易制毒化学品;2)细胞毒类药品;3)不适用BE试验方法验证与参比制剂质量和疗效一致的药品;
备案范围
BE试验备案和程序
征求意见时间
2015.10.30-2015.11.20
发布时间
2016.03.18
参比制剂遴选
企业找不到且无法确定参比制剂,需开展临床有效性试验。
总局关于落实《国务院办公厅关于开展仿制药质量和疗效一致性评价的意见》有关事项的公告(食品药品监管总局公告2016年第106号)总局药品审评中心正在拟定一致性评价临床技术指南
参比制剂遴选
公 开 公 布
公布目录
参比制剂遴选
国际公认的同种药物定义: 在欧盟、美国、日本获准上市并获得参比制剂地位的仿制药。 ---《普通口服固体制剂参比制剂选择和确定指导原则》参比制剂办理一次性进口的要求?原研地产化如何申报参比制剂?国产药品可否作为参比制剂?国内上市改剂型、改酸根、碱基的制剂,如何选择参比制剂?
企业用户备案
企业用户备案
化学药BE试验备案信息平台简介
1.承诺书 首先需要签署承诺书:查看相应的承诺书条款,清楚了解要求后,在同意签署以上承诺的地方选择“是”,点击下一步进入上传伦理委员会批件页面。
企业用户备案
企业用户备案
化学药BE试验备案信息平台简介
2.上传伦理委员会批件 填写伦理委员会批件号,并上传批件的PDF文件。上传成功后点击“下一步”进入备案申请表页面。此时备案记录已经生成,备案状态为“待提交申请表” 。
选 择 原 则
仿制药质量和疗效一致性评价工作中改剂型药品(普通

附件仿制药质量与疗效一致性评价工作中改剂型药品(普通口服固体制剂)评价一般考虑(征求意见稿)为贯彻《国务院关于改革药品医疗器械审评审批制度的意见》(国发〔2015〕44号)以及《国务院办公厅关于开展仿制药质量和疗效一致性评价的意见》(国办发〔2016〕8号)的精神,国家食品药品监督管理总局发布了《关于落实〈国务院办公厅关于开展仿制药质量和疗效一致性评价的意见〉有关事项的公告》(2016年第106号)和《普通口服固体制剂参比制剂选择和确定指导原则》等多个技术指导原则,本文是上述指导原则的补充文件。
改剂型药品是指该剂型在美国、欧盟或日本均未获准上市,或无法确定同剂型参比制剂的药品。
本文件适用于仿制药质量和疗效一致性评价工作中普通口服固体制剂改剂型且不改变给药途径药品的评价,包括但不限于本文件中描述的内容。
一、概述普通口服固体制剂改剂型药品,从药代动力学角度可分为不显著改变药代动力学行为和改变药代动力学行为两类。
本文所指的不显著改变药代动力学行为的改变剂型,包括片剂(普通片、吞服给药分散片、咀嚼片等)、胶囊剂(硬胶囊和软胶囊)、干混悬剂和颗粒剂(混悬型)等剂型间的改变,肠溶片剂与肠溶胶囊间改变。
改变药代动力学行为的改变剂型,包括:普通口服固体制剂改为缓控释制剂、普通口服固体制剂与速释制剂间的改变。
二、评价内容(一)改剂型的科学性和合理性从药物的理化性质、生物学性质、临床需要、患者的依从性、药物经济学、与原研剂型参比制剂的优劣比较等方面分析论证改剂型药品的科学性、合理性和必要性。
(二)药学评价与原研剂型参比制剂系统进行对比试验,评价两者的异同与优劣。
改剂型药品质量应与原研剂型参比制剂质量相当。
评价内容主要包括以下几个方面:1.处方筛选与工艺优化:对照原研剂型参比制剂与所改剂型的要求,进行处方筛选、生产工艺优化,包装材料选择与验证。
2.原辅料的控制:2.1分析检测原料药的关键理化特性(如:晶型、不同pH 条件下溶解度、粒度与粒度分布、pKa、logP等);原料药和辅料的相容性试验及结果。
6.仿制药口服固体制剂质量和疗效一致性评价药学研究关键技术要求和常见问题

(二)药学研究-8.3 生产信息
工艺验证:
基本要求 适用于工艺有改变的品种(实际无变更的产品也会提交)。 验证报告\或验证方案+空白批生产记录+验证承诺。 工艺验证方案和报告\空白批生产记录是整个工艺过程信息的重要载 体,应充分的重视。 常见问题 • 工艺验证方案和报告\批生产记录不完善-应有编号及版本号,且应由 合适人员签署。 • 多个亚批次颗粒混合压片的情况,应充分验证各亚批中间体的质量一 致。中间体放置时间的验证容易被忽略。
药的体外研究。
13
(二)药学研究-8.2. 产品再评价研究
处方工艺的再研究
基本要求:
论证本品剂型、处方组成、生产工艺、包装材料选择和确定的合理性。
处方工艺的尽可能接近,对于保证仿制品与参比品的各项质量属性一致 是非常有利的。
差异是审评关注的重点。(FDAQ&A:是否存在潜在影响与原研药治疗等 效性的差异?)
目标
• 全面提 高仿制 药的质 量
• 与国际 先进水 平接轨
5
一、概述
一致性评价品种的主要特点
• 优势 • 参比制剂比较明确,可获得信息相对较多。 • 待评价产品已获得批准,有较长时间的生产经验,对 产品的关键质量属性了解比较充分。
• 问题 • 变更?不变更?--需要评估,难度不同。 • 补课有可能更困难。
9
主要内容
一、概述 二、药学研究的技术支持性文件 三、关键技术要求和常见问题
三、关键技术要求和常见问题
化学药品仿制药口服固体制剂质量和疗效一致性评价申报资料要求
药学研究 申报资料
概要
药学研究 体外评价
(一)概要
基本要求: 应提供原研产品上市背景信息,申报产品批准上市及上市后变更 情况,变更前后的处方和工艺对比。 常见问题: • 关键信息收集不够充分(如处方成分,原研品上市历史)。 • 上市后变更资料提供不全,处方工艺、生产场地变更的相关批准 信息缺失。
普通口服固体制剂参比制剂选择和确定的指导原则

附件1普通口服固体制剂参比制剂选择和确定指导原则一、概述为推进仿制药与原研药品质量和疗效一致性评价工作的开展,根据《国务院关于改革药品医疗器械审评审批制度的意见》(国发〔2015〕44号)要求,制定本指导原则。
仿制药是指与被仿制药具有相同的活性成分、剂型、给药途径和治疗作用的药品。
参比制剂是指用于仿制药质量一致性评价的对照药品,可为原研药品或国际公认的同种药物。
原研药品是指在全球市场率先上市的,拥有或曾经拥有相关专利、或获得了专利授权的原创性药品。
国际公认的同种药物是指在欧盟、美国获准上市并获得参比制剂地位的仿制药。
原研药品和国际公认的同种药物通常具有完善的临床研究数据或生物等效性研究数据。
本指导原则适用于普通口服固体制剂仿制药质量一致性评价研究用参比制剂的选择与备案。
二、选择原则参比制剂首选原研药品,若确实无法获得原研药品或有证据证明原研药品不适合评价方法要求时,也可以选用国际公认的同种药物作为参比制剂。
(一)首选国内上市的原研药品作为参比制剂。
如原研企业同时有进口和地产化药品的上市许可,优先选择进口原研药品作为参比制剂。
若原研药品未在国内上市,可选择在国外上市的原研药品。
优先选择在欧盟、美国上市并被列为参比制剂的原研药品。
(二)国际公认的同种药物首选国内上市药品。
如企业同时有进口和地产化药品的上市许可,优先选择进口药品作为参比制剂。
若国际公认的同种药物未在国内上市,则选择在欧盟、美国上市并被列为参比制剂的同种药物。
(三)参比制剂的质量及均一性应满足药品评价要求。
三、产生方式(一)药品生产企业应按照上述要求,明确所产仿制药的参比制剂,报食品药品监管总局备案。
(二)行业协会可以组织同品种企业提出参比制剂的意见,报食品药品监管总局审核确定。
(三)食品药品监管总局可以推荐参比制剂,供药品生产企业参考。
四、备案和审核(一)药品生产企业应根据国家仿制药质量一致性评价的任务要求和拟评价品种的情况,开展先期研究,拟定参比制剂,填写参比制剂备案申请表,报食品药品监督管理总局备案。
中国药品管理和准入政策介绍

()药品研制(注册审批)方面
《药品注册管理办法》 现行办法于年月日实施。明确了在中华 人民共和国境内申请药物临床试验、药品 生产和药品进口,以及进行药品审批、注 册检验和监督管理等方面的要求。 相关法规:《药物非临床研究质量管理规 范》、《药物临床试验质量管理规范》、 《直接接触药品的包装材料和容器管理办 法》和《药品说明书和标签管理规定》等
.生物等效性()试验审批改备案制
年月日, 发布《关于化学药生物等效 性试验实行备案管理的公告》。至此, 我国化学药生物等效性试验由审批制改 为备案制。 试验备案制也有助于解决 仿制药注册申请积压的问题。
. 保健品食品注册与备案管理办法
年月日,发布《保健食品注册与备案管理办法》,年月 日实施,重点内容: )将保健食品产品上市的管理模式由原来的单一注册制, 调整为注册与备案相结合的管理模式。 )规定生产使用保健食品原料目录以外原料的保健食品, 以及首次进口的保健食品(属于补充维生素、矿物质等营 养物质的保健食品除外)必须通过产品注册。 )重申优化保健食品注册程序,强化保健食品注册证书的 管理,该办法规定保健食品注册证书有效期为年。
场核查和抽样检验的时限。
谢谢!
中国药品管理 和准入政策介绍
副会长单位 天津医药集团总工程师
张平
主要内容
一、中国药品管理体系 二、中国药品准入政策 三、最新的药品管理制度变化
一、中国药品管理体系
. 中国药品监督管理机构 . 中国药事法律制度体系
省政府 市政府 县政府
.中国药品监督管理机构
国家食品药品监督管理总局() 中国食品药品检定研究院
)国内药品生产企业已在欧盟、美国和日本获 准上市的仿制药,可以国外注册申报的相关资 料为基础,按照化学药品新注册分类申报药品 上市,批准上市后视同通过一致性评价;在中 国境内用同一生产线生产上市并在欧盟、美国 和日本获准上市的药品,视同通过一致性评价 。
刚刚!!!NMPA和FDA近三年来法规文件及指导原则

刚刚NMPA和FDA近三年来法规文件及指导原则以下是2016年和2017年NMPA内容:2017年10月8日,中共中央办公厅、国务院办公厅印发了《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》,并发出通知。
这是继2015年8月《国务院关于改革药品医疗器械审评审批制度的意见》之后,又一个深化药品医疗器械审评审批制度改革的纲领性文件,对我国医药产业创新发展具有里程碑意义。
本文以《创新意见》为线索,对CFDA及CDE自2015年7月22日以来发布的重要文件进行梳理,给各位同仁研究相关政策提供一份参考。
1. 改革临床试验管理•临床试验机构资格认定实行备案管理•支持临床试验机构和人员开展临床试验•完善伦理委员会机制•提高伦理审查效率•优化临床试验审批程序•接受境外临床试验数据•支持拓展性临床试验•严肃查处数据造假行为1.1 法规政策•2016年3月23日,CFDA、卫计委委员会发布《医疗器械临床试验质量管理规范》(国家食品药品监督管理总局中华人民共和国国家卫生和计划生育委员会令第25号)•2016年4月7日,CFDA发布关于贯彻实施《医疗器械临床试验质量管理规范》的通知(食药监办械管〔2016〕41号)•2016年12月2日,总局办公厅公开征求《药物临床试验质量管理规范(修订稿)》的意见•2017年3月17日,总局公开征求《国家食品药品监督管理总局关于调整进口药品注册管理有关事项的决定(征求意见稿)》意见的通知•2017年5月11日,总局关于征求《关于鼓励药品医疗器械创新改革临床试验管理的相关政策》(征求意见稿)意见的公告(2017年第53号)•2017年8月4日,关于征求《医疗器械临床试验机构条件和备案管理办法(征求意见稿)》意见的函(食药监械管便函〔2017〕42号)•2017年10月20日,CDE关于《接受境外临床试验数据的技术要求》征求意见通知1.2 指导原则•2015年8月4日,CDE关于征求《儿科人群药物临床试验技术指导原则》意见的通知•2016年3月7日,总局关于发布儿科人群药物临床试验技术指导原则的通告(2016年第48号)•2015年8月21日,CDE关于征求《药物临床试验的生物统计学指导原则》意见的通知o2016年6月3日,总局关于发布药物临床试验的生物统计学指导原则的通告(2016年第93号)•2015年11月3日,国家食品药品监督管理总局关于发布中药新药临床研究一般原则等4个技术指导原则的通告(2015年第83号)•2015年12月24日,CDE关于《药物临床试验的一般考虑》指导原则征求意见的通知•2017年1月20日,总局关于发布药物临床试验的一般考虑指导原则的通告(2017年第11号)•2016年1月29日,关于《临床试验数据管理工作技术指南》、《临床试验的电子数据采集(EDC)技术指导原则》和《药物临床试验数据管理和统计分析的计划和报告指导原则》征求意见的通知o2016年7月29日,总局关于发布临床试验的电子数据采集技术指导原则的通告(2016年第114号)o2016年7月29日,总局关于发布药物临床试验数据管理与统计分析的计划和报告指导原则的通告(2016年第113号)o2016年7月29日,总局关于发布临床试验数据管理工作技术指南的通告(2016年第112号)•2016年9月30日,CDE关于征求《新药I期临床试验申请技术指南(草案)》意见的通知•2016年10月11日,总局关于发布中药新药治疗流行性感冒临床研究技术指导原则的通告(2016年第136号)•2016年10月29日,CDE关于征求《成人用药数据外推在儿科人群药物临床试验及相关信息使用的技术指导原则》意见的通知o2017年5月18日,总局关于发布成人用药数据外推至儿科人群的技术指导原则的通告(2017年第79号)•2017年7月3日,关于《创新药(化学药)Ⅲ期临床试验药学研究信息指南(草案)》征求意见的通知2. 加快上市审评审批•加快临床急需药品医疗器械审评审批•支持罕见病治疗药品医疗器械研发•严格药品注射剂审评审批•实行药品与药用原辅料和包装材料关联审批•支持中药传承和创新•建立专利强制许可药品优先审评审批制度2.1 法规政策•2015年11月13日,国家食品药品监督管理总局关于征求《关于解决药品注册申请积压实行优先审评审批的意见(征求意见稿)》意见的公告(2015年第227号)•2015年12月24日,总局关于征求《中药配方颗粒管理办法(征求意见稿)》意见的公告(2015年第283号)•2015年12月31日,国家食品药品监督管理总局关于落实中药提取和提取物监督管理有关规定的公告(2015年第286号) •2016年2月26日,CFDA发布关于解决药品注册申请积压实行优先审评审批的意见(食药监药化管〔2016〕19号)•2016年3月18日,总局关于取消中药材生产质量管理规范认证有关事宜的公告(2016年第72号)•2016年1月12日,总局关于征求药包材和药用辅料关联审评审批申报资料要求(征求意见稿)意见的公告(2016年第3号)o2016年11月28日,总局关于发布药包材药用辅料申报资料要求(试行)的通告(2016年第155号)•2016年5月12日,总局办公厅公开征求关于药包材药用辅料与药品关联审评审批有关事项的公告(征求意见稿)意见o2016年8月10日,总局关于药包材药用辅料与药品关联审评审批有关事项的公告(2016年第134号)•2016年6月21日,关于征求医疗器械优先审批程序意见的函(食药监械管便函〔2016〕40号)o2016年10月26日,总局关于发布医疗器械优先审批程序的公告(2016年第168号)•2016年7月21日,CDE关于征求《“首仿”品种实行优先审评评定的基本原则》的意见•2017年7月21日,总局办公厅公开征求《关于对医疗机构应用传统工艺配制中药制剂实施备案管理的公告(征求意见稿)》意见•2017年10月9日,总局办公厅公开征求《中药经典名方复方制剂简化注册审批管理规定(征求意见稿)》及申报资料要求(征求意见稿)意见2.2 指导原则•2017年1月11日,总局办公厅公开征求中成药通用名称命名技术指导原则(征求意见稿)的意见•2017年2月16日,总局关于发布医疗器械优先审批申报资料编写指南(试行)的通告(2017年第28号)•2017年3月3日,CDE关于征求《儿科用药非临床安全性研究技术指导原则》意见的通知•2017年3月28日,CDE关于再次征求《新药I期临床试验申请技术指南》意见的通知•2017年4月13日,CDE关于6个中药新药临床研究技术指导原则上网征求意见的通知•2017年10月11日,总局办公厅公开征求《中药资源评估技术指导原则》意见•2017年10月11日,总局办公厅公开征求《中成药规格表述技术指导原则(征求意见稿)》意见3. 促进药品创新和仿制药发展•建立上市药品目录集•探索药品专利链接制度•开展药品专利期限补偿制度试点•完善和落实药品试验数据保护制度•促进药品仿制生产•发挥企业的创新主体作用•支持新药临床应用3.1 创新药3.1.1 法规政策•2015年11月6日,总局关于征求药品上市许可持有人制度试点方案和化学药品注册分类改革工作方案两个征求意见稿意见的公告(2015年第220号)o2016年3月9日,总局关于发布化学药品注册分类改革工作方案的公告(2016年第51号)o2016年5月4日,总局关于发布化学药品新注册分类申报资料要求(试行)的通告(2016年第80号)•2017年5月11日,总局关于征求《关于鼓励药品医疗器械创新加快新药医疗器械上市审评审批的相关政策》(征求意见稿)意见的公告(2017年第52号)•2017年5月12日,总局关于征求《关于鼓励药品医疗器械创新保护创新者权益的相关政策(征求意见稿)》意见的公告(2017年第55号)3.1.2 指导原则•2017年5月30日,药品审评中心关于征求《药品电子通用技术文档结构(征求意见稿)》意见的通知o2017年10月17日,药品审评中心关于再次征求《药品电子通用技术文档结构》意见的通知3.2 仿制药3.2.1 法规政策•2015年11月6日,国家食品药品监督管理总局关于征求化学仿制药生物等效性试验备案管理规定(征求意见稿)意见的公告(2015年第221号)•2015年11月18日,国家食品药品监督管理总局关于征求《关于开展仿制药质量和疗效一致性评价的意见(征求意见稿)》意见的公告(2015年第231号)•2016年3月5日,国务院办公厅印发关于开展仿制药质量和疗效一致性评价的意见(国办发〔2016〕8号)•2016年3月10日,总局办公厅公开征求研制过程中所需研究用对照药品一次性进口有关事宜的意见o2016年7月1日,总局关于研制过程中所需研究用对照药品一次性进口有关事宜的公告(2016年第120号)•2016年3月28日,总局办公厅公开征求仿制药质量和疗效一致性评价工作程序及化学药品仿制药口服固体制剂一致性评价申报资料要求意见o2016年8月17日,总局关于发布化学药品仿制药口服固体制剂质量和疗效一致性评价申报资料要求(试行)的通告(2016年第120号)•2016年4月1日,总局办公厅公开征求关于落实《国务院办公厅关于开展仿制药质量和疗效一致性评价的意见》的有关事项的意见o2016年5月26日,总局关于落实《国务院办公厅关于开展仿制药质量和疗效一致性评价的意见》有关事项的公告(2016年第106号)•2016年5月26日,总局关于发布仿制药质量和疗效一致性评价工作程序的公告(2016年第105号)•2016年4月12日,食品药品监管总局办公厅公开征求仿制药质量和疗效一致性评价参比制剂备案与推荐程序的意见o2016年5月19日,总局关于发布仿制药质量和疗效一致性评价参比制剂备案与推荐程序的公告(2016年第99号)•2017年6月9日,总局办公厅公开征求《关于仿制药质量和疗效一致性评价工作有关事项的公告(征求意见稿)》意见•2017年8月25日,总局关于仿制药质量和疗效一致性评价工作有关事项的公告(2017年第100号)•2017年9月4日,CDE关于公开征求《中国上市药品目录集》框架意见的通知•2017年9月22日,仿制药质量与疗效一致性评价办公室关于发布《仿制药质量和疗效一致性评价申报资料立卷审查技术标准(暂行)》的通知•2017年10月13日,国家食品药品监督管理总局、国家卫生和计划生育委员会关于药物临床试验机构开展人体生物等效性试验的公告(2017年第119号)3.2.1 指导原则•2015年10月30日,1.普通口服固体制剂参比制剂选择和确定指导原则(征求意见稿);2.普通口服固体制剂溶出曲线测定与比较指导原则(征求意见稿);3.仿制药质量一致性评价人体生物等效性研究技术指导原则(征求意见稿)o2016年3月18日,总局发布了《普通口服固体制剂参比制剂选择和确定指导原则》《普通口服固体制剂溶出曲线测定与比较指导原则》《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》•2015年11月27日,食品药品监管总局办公厅关于征求化学仿制药CTD格式申报资料撰写要求意见的通知(食药监办药化管函〔2015〕737号)•2015年11月27日,CDE《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》上网征求意见的通知•2016年4月8日,总局办公厅公开征求人体生物等效性试验豁免指导原则的意见o2016年5月19日,总局关于发布人体生物等效性试验豁免指导原则的通告(2016年第87号)•2016年9月13日,总局办公厅公开征求仿制药质量和疗效一致性评价改规格药品评价一般考虑的意见•2016年9月14日,总局办公厅公开征求仿制药质量和疗效一致性评价临床有效性试验一般考虑的意见o2017年2月7日,总局关于发布仿制药质量和疗效一致性评价临床有效性试验一般考虑的通告(2017年第18号)•2016年11月7日,总局办公厅公开征求仿制药质量和疗效一致性评价工作中改剂型药品(普通口服固体制剂)评价一般考虑的意见•2016年11月7日,总局办公厅公开征求仿制药质量和疗效一致性评价工作中改盐基药品评价一般考虑的意见•2016年11月29日,总局办公厅公开征求进一步规范仿制药质量和疗效一致性评价参比制剂选择等相关事宜的指导意见的意见•2016年11月29日,总局办公厅公开征求仿制药质量和疗效一致性评价品种分类的指导意见的意见o2017年4月5日,总局关于发布仿制药质量和疗效一致性评价品种分类指导意见的通告(2017年第49号)•2016年12月21日,总局办公厅公开征求仿制药质量和疗效一致性评价研究现场核查等指导原则的意见o2017年5月18日,总局关于发布仿制药质量和疗效一致性评价研制现场核查指导原则等4个指导原则的通告(2017年第77号)•2017年4月28日,总局办公厅公开征求化学仿制药口服固体制剂一致性评价复核检验技术指南(征求意见稿)的意见•2017年5月18日,总局办公厅公开征求胃肠道局部作用药物、电解质平衡用药仿制药质量和疗效一致性评价及特殊药品生物等效性试验申请有关事宜意见(征求意见稿)的意见o2017年5月25日,总局办公厅公开征求胃肠道局部作用药物、电解质平衡用药仿制药质量和疗效一致性评价及特殊药品生物等效性试验申请有关事宜意见(征求意见稿)的意见•2017年5月30日,药品审评中心关于征求《化学仿制药电子通用技术文档申报指导原则(征求意见稿)》意见的通知o2017年10月17日,药品审评中心关于再次征求《化学仿制药电子通用技术文档申报指导原则》意见的通知•2017年6月9日,总局办公厅公开征求《仿制药质量和疗效一致性评价受理审查指南(需一致性评价品种)(征求意见稿)》《仿制药质量和疗效一致性评价受理审查指南(境内共线生产并在欧美日上市品种)(征求意见稿)》及相关单据意见o2017年9月5日,总局关于发布《仿制药质量和疗效一致性评价受理审查指南(需一致性评价品种)》《仿制药质量和疗效一致性评价受理审查指南(境内共线生产并在欧美日上市品种)》的通告(2017年第148号)4. 加强药品医疗器械全生命周期管理•推动上市许可持有人制度全面实施•落实上市许可持有人法律责任•建立上市许可持有人直接报告不良反应和不良事件制度•开展药品注射剂再评价•完善医疗器械再评价制度•规范药品学术推广行为4.1 法规政策•2015年11月6日,总局关于征求药品上市许可持有人制度试点方案和化学药品注册分类改革工作方案两个征求意见稿意见的公告(2015年第220号)•2015年11月12日,食品药品监管总局办公厅、国家卫生计生委办公厅关于公开征求医疗器械不良事件监测和再评价管理办法(征求意见稿)意见的函(食药监办械监函〔2015〕723号)•2016年2月20日,总局公开征求对药品经营质量管理规范修订的意见•2016年5月6日,关于征求《医疗器械冷链(运输、贮存)管理指南》意见的函(食药监械监便函〔2016〕61号)o2016年9月22日,总局关于发布医疗器械冷链(运输、贮存)管理指南的公告(2016年第154号)•2016年6月6日,国务院办公厅发布关于印发药品上市许可持有人制度试点方案的通知(国办发〔2016〕41号)•2016年7月7日,CFDA发布关于做好药品上市许可持有人制度试点有关工作的通知(食药监药化管〔2016〕86号)•2016年9月31日,关于征求《医疗器械生产企业质量控制与成品放行指南(征求意见稿)》意见的函(食药监械监便函〔2016〕127号)•2016年10月31日,总局关于公开征求医疗器械不良事件监测和再评价管理办法(征求意见稿)意见的通知•2017年2月8日,CFDA发布《医疗器械召回管理办法》(国家食品药品监督管理总局令第29号)•2017年5月11日,总局关于征求《关于鼓励药品医疗器械创新实施药品医疗器械全生命周期管理的相关政策》(征求意见稿)意见的公告(2017年第54号)•2017年6月21日,总局关于公开征求《网络医疗器械经营监督管理办法(征求意见稿)》意见的通知•2017年10月17日,总局办公厅公开征求《药品生产场地变更简化注册审批管理规定(征求意见稿)》及《药品生产场地变更研究技术指导原则(征求意见稿)》意见•2017年10月27日, 总局办公厅公开征求《中药材生产质量管理规范(修订稿)》意见4.2 指导原则•2017年1月10日,CDE关于《已上市化学药品生产工艺变更研究技术指导原则》征求意见的通知•2017年8月29日,总局关于发布已上市化学药品生产工艺变更研究技术指导原则的通告(2017年第140号)•2017年3月6日,CDE关于《已上市中药生产工艺变更研究技术指导原则》征求意见的通知•2017年8月24日,总局关于发布已上市中药生产工艺变更研究技术指导原则的通告(2017年第141号)5. 提升技术支撑能力•完善技术审评制度•落实相关工作人员保密责任•加强审评检查能力建设•落实全过程检查责任•加强国际合作5.1 配套支持•2015年12月10日,国家食品药品监督管理总局关于征求《生物制品批签发管理办法》(修订稿)意见的公告(2015年第263号)•2016年12月13日,总局公开征求生物制品批签发管理办法修订草案意见的通知•2016年5月31日,总局关于药物非临床研究质量管理规范认证和药物临床试验机构资格认定施行电子申请受理的公告(2016年第110号)•2016年6月6日,总局关于发布药物研发与技术审评沟通交流管理办法(试行)的通告(2016年第94号)•2016年7月25日,总局办公厅公开征求《药品注册管理办法(修订稿)》意见o2017年10月23日,总局办公厅公开征求《药品注册管理办法(修订稿)》意见•2016年8月19日,总局办公厅公开征求《药物非临床研究质量管理规范(修订稿)》意见o2017年8月2日,CFDA发布《药物非临床研究质量管理规范》(国家食品药品监督管理总局令第34号)•2016年9月20日,总局办公厅公开征求《进口药材管理办法(修订稿)》意见•2016年9月21日,关于征求《体外诊断试剂注册管理办法》修正案意见的函(食药监械管便函〔2016〕62号)•2016年9月30日,总局办公厅关于征求医疗器械分类目录(修订稿)意见的函•2017年9月4日,总局关于发布医疗器械分类目录的公告(2017年第104号)o2017年9月4日,总局关于实施《医疗器械分类目录》有关事项的通告(2017年第143号)•2016年10月10日,食品药品审核查验中心公开征求《药品数据管理规范》的意见o2017年8月25日,总局办公厅公开征求《药品数据管理规范(征求意见稿)》意见o2016年11月22日,总局办公厅公开征求药品标准管理办法(征求意见稿)意见•2016年10月31日,总局关于公开征求《医疗器械标准管理办法》(征求意见稿)意见的通知•2016年12月6日,CDE关于《药品审评项目管理办法》征求意见的通知•2016年12月29日,总局关于调整部分行政审批事项审批程序决定公开征求意见的通知o2017年4月5日,CFDA发布《国家食品药品监督管理总局关于调整部分药品行政审批事项审批程序的决定》(国家食品药品监督管理总局令第31号)o2017年4月6日,CFDA发布《国家食品药品监督管理总局关于调整部分医疗器械行政审批事项审批程序的决定》(国家食品药品监督管理总局令第32号)•2016年12月29日,总局关于《国家食品药品监督管理总局药品医疗器械审评审批保密管理办法﹙征求意见稿﹚》公开征求意见的通知•2017年2月23日,总局办公厅公开征求《关于药品再注册有关事项的公告(征求意见稿)》意见•2017年3月9日,总局关于发布药品注册审评专家咨询委员会管理办法(试行)的公告(2017年第27号)•2017年8月31日,总局办公厅公开征求《药品境外检查规定(征求意见稿)》意见•2017年9月13日,总局办公厅公开征求《关于调整药品注册受理工作的公告(征求意见稿)》意见•2017年10月23日,总局办公厅公开征求《〈中华人民共和国药品管理法〉修正案(草案征求意见稿)》意见Clindata目前提供数据管理、生物统计、随机、医学核查、医学编码、独立数据委员会6大服务:Data Management数据管理Fast delivery of high quality and meaningful data means that you can make well informed decisions earlier in the trial. This also ensures patient protection and reliable trial results.迅速发送高质量和有意义的数据意味着您能在试验早期做出正确的决策,也可以确保保护受试者和可信的试验结果。
仿制药一致性评价生物等效性试验临床有效性解读

普通口服固体制剂参比制剂选择和确定指导原则 以药动学参数为终点评价指标的化学药物仿制药人体生物等效性
研究技术指导原则 …
3
背景
推进仿制药质量一致性评价。对已经批准上市的仿制药, 按与原研药品质量和疗效一致的原则,分期分批进行质量 一致性评价。(国发[2015]44号)
当入选健康受试者参与试验可能面临安全性方面的风险 时,则建议入选试验药物拟适用的患者人群,并且在试 验期间应保证患者病情稳定。
10
参比制剂选择
用于仿制药一致性评价的对照药品,可为原研药 品或国际公认的同种药品。
国际公认的同种药物是指在欧盟、美国获准上市 并获得参比制剂地位的仿制药。
原研药品和国际公认的同种药物通常具有完善的 临床研究数据或生物等效性研究数据。
15
特殊药物的生物等效性
高变异药物(个体内变异≥30%):可根据参比制剂的 个体内变异,将等效性评价标准作适当比例的调整,但 调整应有充分的依据。
窄治疗窗药物:应根据药物的特性适当缩小90%置信区 间范围。
16
特殊情况
检测物质(原形药/代谢产物、对映体/消旋体) 长半衰期药物 内源性化合物 口服给药发挥局部作用的药物
20
其他:试验制剂留样
强调试验机构应对试验制剂及参比制剂按相关要 求留样(而不是退回申请人)。
试验制剂应留样保存至药品获准上市后2年。
21
仿制药临床有效性试验一般考虑
实施对象:企业找不到且无法确定参比制剂的, 由药品生产企业开展临床有效性试验。
遵从《药物临床试验的一般考虑》和《药物临床 试验的生物统计指导原则》。
一致性评价改剂型

附件2仿制药质量与疗效一致性评价工作中改剂型药品(口服固体制剂)评价一般考虑为贯彻《国务院关于改革药品医疗器械审评审批制度的意见》(国发〔2015〕44号)和《国务院办公厅关于开展仿制药质量和疗效一致性评价的意见》(国办发〔2016〕8号)的精神,国家食品药品监督管理总局发布了《关于落实〈国务院办公厅关于开展仿制药质量和疗效一致性评价的意见〉有关事项的公告》(2016年第106号)和《普通口服固体制剂参比制剂选择和确定指导原则》(2016年第61号通告)等多个技术指导原则,本文是上述指导原则的补充文件。
改剂型药品是指该剂型在美国、欧盟或日本均未获准上市,或无法确定同剂型参比制剂的药品。
本文件适用于仿制药质量和疗效一致性评价工作中口服固体制剂改剂型且不显著改变给药途径药品的评价。
评价工作包括但不限于本文件中描述的内容。
一、概述口服固体制剂改剂型药品,从药代动力学角度可分为不显著—1—改变药代动力学行为和显著改变药代动力学行为两类。
本文件所指的不显著改变药代动力学行为的改变剂型,如:片剂、胶囊剂、干混悬剂和颗粒剂等剂型间的改变;显著改变药代动力学行为的改变剂型,如:普通口服固体制剂改为缓控释制剂等剂型间的改变。
二、评价内容(一)改剂型的科学性和合理性从药物的理化性质、生物学性质、临床需要、患者的依从性、药物经济学、与原研剂型参比制剂的优劣比较等方面分析论证改剂型药品的科学性、合理性和必要性。
(二)药学评价与原研剂型参比制剂系统进行对比试验,评价两者的异同与优劣。
改剂型药品质量应与原研剂型参比制剂质量相当。
评价内容主要包括以下几个方面:1.处方筛选与工艺优化:对照原研剂型参比制剂与所改剂型的要求,进行处方筛选、生产工艺优化,包装材料选择与验证。
2.原辅料的控制:(1)分析检测原料药的关键理化特性(如:晶型、不同pH 条件下溶解度、粒度与粒度分布、pKa、logP等);原料药和辅料的相容性试验及结果。
—2—(2)分析检测辅料与制剂性能相关的关键特性。
《普通口服固体制剂溶出曲线测定与比较指导原则(草案)》

采用相似因子(f2)法比较溶出曲线相似性时,除另有规定外, 两条溶出曲线相似因子(f2)数值不小于50,可认为具有相似性。 5 其他
下列条件:
4
4.1.1 应在完全相同的条件下对仿制制剂和参比制剂的溶出曲线进 行测定。 4.1.2 两条溶出曲线的取样点应相同。时间点的选取应尽可能以溶出 量等分为原则,并兼顾整数时间点,但溶出量在 85%以上的时间点仅 能选取 1 个。 4.1.3 选取的第一个时间点溶出结果的相对标准偏差不得过 20%,自 第二个时间点至最后时间点溶出结果的相对标准偏差不得过 10%。 4.2 溶出曲线相似性判定标准
以下任何一个条件均可作为考察截止时间点选择的依据。 a. 连续两点溶出量均达 85%以上,且差值在 5%以内。 b. 一般在酸性溶出介质(pH 值 1.0~3.0)中考察时间不超过 2 小
时。肠溶制剂也可选择 pH 值 4.5 的溶出介质替代酸性溶出介 质进行考察。 c. 在其它各 pH 值溶出介质中考察时间不超过 6 小时。 3.4 溶出条件的优化
醋酸盐缓冲溶液的配制
3.8
4.0
4.5
0.67 1.22 2.99
22.6 20.5 14.0
5.5 5.98
3.0
5.8 6.23
2.1
3 磷酸盐缓冲液 0.2mol/L 磷酸二氢钾溶液:取 27.22g 磷酸二氢钾,用水溶解并 稀释至 1000ml。 0.2mol/L 氢氧化钠溶液:取 8.00g 氢氧化钠,用水溶解并稀释至 1000ml。 取 250ml 0.2mol/L 磷酸二氢钾溶液与下表中规定量的 0.2mol/L 氢氧化钠溶液混合后,再加水稀释至 1000ml,摇匀,即得,见表 3。
仿制药质量一致性评价发布政策法规汇总
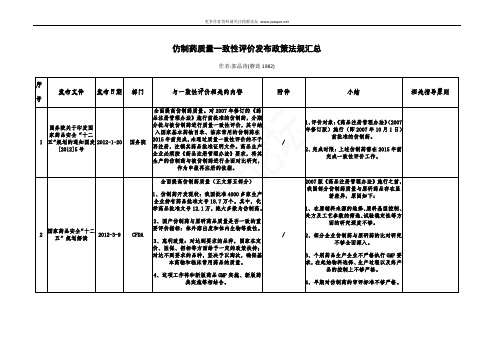
导原则(征求意见稿)
荐使用不少于 3 种 pH 值的溶出介质)及
通口服固体制剂参
2015-10-3
10 比制剂选择和确定
CFDA
0
3.仿制药质量一致性评价人体生物等效性研
体积选择(500ml、900ml 或 1000ml)3、 1-4
溶出曲线时间点选择 4、条件优化(在截
1-3
指导原则等意见的
究技术指导原则(征求意见稿)
局。
7、拟定了由中检院组织专家委员会在审 查一致性评价资料时一并审查处方、工艺 变更的工作流程。药审中心将参加专家委
员会,一并负责相关技术审查工作。
8、省局进行生产现场核查,连续抽取三 批样品送指定的药检所复核。
9、省局收到药检所复核结果后,将研究 资料及检验报告一并报专项办公室。
更多作者资料请关注药群论坛
国家食品药品监督 管理总局公开征求 6 仿制药质量一致性 2014-1-29 评价有关指导原则
等意见
CFDA CFDA
根据《国家食品药品监督管理局关于开展仿制 2013 年度仿制药
药质量一致性评价工作的通知》(国食药监注 质量一致性评价 涉及 75 个化学仿制药,制定评价方法的
〔2013〕34 号)要求,对 2013 年度仿制药质量 品种名单和方法 任务分发给中国食品药品检定研究院、其
2、对注册分类调整前受理的药品注册申请, 继续按照原规定进行审评审批,
国务院关于改革药
3、开展药品上市许可持有人制度试点。
品医疗器械审评审
ห้องสมุดไป่ตู้
4、改进药品临床试验审批。
批制度的意见 国发
9
2015-8-18 国务院
〔2015〕44 号
5、落实申请人主体责任。
化学药品仿制药口服固体制剂质量和疗效一致性 评价申报资料要求(试行)

化学药品仿制药口服固体制剂质量和疗效一致性评价申报资料要求(试行)一、申报资料项目(一)概要1.历史沿革2.批准及上市情况3.自评估报告4.临床信息及不良反应5.最终确定的处方组成及生产工艺情况6.生物药剂学分类(二)药学研究资料7.(2.3.P,注:括号内为CTD格式的编号,以下同)制剂药学研究信息汇总表8.(3.2.P)制剂药学申报资料8.1.(3.2.P.1)剂型与产品组成8.2.(3.2.P.2)产品再评价研究8.2.1.(3.2.P.2.1)处方组成8.2.2.(3.2.P.2.2)制剂再研发(适用于处方、工艺有改变的品种)8.2.2.1.(3.2.P.2.2.1)处方再研发8.2.2.2.(3.2.P.2.2.2)工艺再研发8.3.(3.2.P.3)生产信息8.3.1.(3.2.P.3.1)生产商8.3.2.(3.2.P.3.2)批处方8.3.3.(3.2.P.3.3)生产工艺和工艺控制8.3.4.(3.2.P.3.4)关键工艺步骤和中间体的控制8.3.5.(3.2.P.3.5)工艺验证和评价(适用于工艺有改变的品种)8.3.6.(3.2.P.3.6)临床试验/生物等效性(BE)样品的生产情况8.4.(3.2.P.4)原辅料的控制8.5.(3.2.P.5)制剂的质量控制8.5.1.(3.2.P.5.1)质量标准8.5.2.(3.2.P.5.2)分析方法8.5.3.(3.2.P.5.3)分析方法的验证8.5.4.(3.2.P.5.4)批检验报告8.5.5.(3.2.P.5.5)杂质谱分析8.5.6.(3.2.P.5.6)质量标准制定依据8.6.(3.2.P.6)对照品8.7.(3.2.P.7)包装材料8.8.(3.2.P.8)稳定性8.8.1.(3.2.P.8.1)稳定性总结8.8.2.(3.2.P.8.2)后续稳定性承诺和稳定性方案(适用于处方、工艺有改变的品种)8.8.3.(3.2.P.8.3)稳定性数据(三)体外评价9.参比制剂9.1.参比制剂的选择9.2.基本信息9.3.质量考察9.4.溶出曲线考察9.5.溶出曲线稳定性考察(适用于理化性质不稳定品种)10.质量一致性评价10.1.国内外质量标准收载情况比较10.2.关键质量属性研究10.3.参比制剂与被评价制剂的检验结果11.溶出曲线相似性评价11.1.建立体外溶出试验方法11.2.批内与批间差异考察11.3.溶出曲线相似性比较结果(四)体内评价12.(2.5. P)制剂临床试验信息汇总表13.制剂临床试验申报资料13.1.(5.2)临床试验项目汇总表13.2.(5.3)生物等效性试验报告13.2.1.(5.3.1.2.1)空腹生物等效性试验报告13.2.2.(5.3.1.2.2)餐后生物等效性试验报告13.2.3.(5.3.1.4)方法学验证及生物样品分析报告13.3.(5.3.5.4)其他临床试验报告14.参考文献及相关实验数据研究资料二、申报资料项目说明工艺如有变化,应以文字或列表方式说明变更前、后生产工艺主要变化(包括对于仅涉及工艺变化、未涉及处方变化的品种,也应列出制剂处方,并核实与已批准的处方的一致性。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
附件1
普通口服固体制剂参比制剂
选择和确定指导原则
一、概述
为推进仿制药与原研药品质量和疗效一致性评价工作的开展,根据《国务院关于改革药品医疗器械审评审批制度的意见》(国发〔2015〕44号)要求,制定本指导原则。
仿制药是指与被仿制药具有相同的活性成分、剂型、给药途径和治疗作用的药品。
参比制剂是指用于仿制药质量一致性评价的对照药品,可为原研药品或国际公认的同种药物。
原研药品是指在全球市场率先上市的,拥有或曾经拥有相关专利、或获得了专利授权的原创性药品。
国际公认的同种药物是指在欧盟、美国获准上市并获得参比制剂地位的仿制药。
原研药品和国际公认的同种药物通常具有完善的临床研究数据或生物等效性研究数据。
本指导原则适用于普通口服固体制剂仿制药质量一致性评价研究用参比制剂的选择与备案。
二、选择原则
参比制剂首选原研药品,若确实无法获得原研药品或有证据证明原研药品不适合评价方法要求时,也可以选用国际公认的同种药物作为参比制剂。
(一)首选国内上市的原研药品作为参比制剂。
如原研企业同时有进口和地产化药品的上市许可,优先选择进口原研药品作为参比制剂。
若原研药品未在国内上市,可选择在国外上市的原研药品。
优先选择在欧盟、美国上市并被列为参比制剂的原研药品。
(二)国际公认的同种药物首选国内上市药品。
如企业同时有进口和地产化药品的上市许可,优先选择进口药品作为参比制剂。
若国际公认的同种药物未在国内上市,则选择在欧盟、美国上市并被列为参比制剂的同种药物。
(三)参比制剂的质量及均一性应满足药品评价要求。
三、产生方式
(一)药品生产企业应按照上述要求,明确所产仿制药的参比制剂,报食品药品监管总局备案。
(二)行业协会可以组织同品种企业提出参比制剂的意见,报食品药品监管总局审核确定。
(三)食品药品监管总局可以推荐参比制剂,供药品生产企业参考。
四、备案和审核
(一)药品生产企业应根据国家仿制药质量一致性评价的任务要求和拟评价品种的情况,开展先期研究,拟定参比制剂,填写参比制剂备案申请表,报食品药品监督管理总局备案。
食品药品监督管理总局如有异议,应当在接到备案文件20个工作日内作出。
(二)当参比制剂难以确定时,企业应将相关情况和建议报食品药品监督管理总局,经征询专家意见后审核确定。
(三)由行业协会提出和总局推荐的参比制剂,经征询专家意见后审核确定。
五、参比制剂的研究
(一)通过备案或审核确定的参比制剂,由企业自行购买,并对参比制剂开展研究。
(二)参比制剂应有合法或明确来源,其批次和数量应满足企业仿制药质量一致性评价研究及药品检验机构检验复核的需求。
(三)企业应对参比制剂和仿制药开展全面对比研究。
附:仿制药质量一致性评价参比制剂备案表
附
仿制药质量一致性评价参比制剂备案表编号[1]:。