MEGA6使用教程——进化树的构建
进化树构建方法-MEGA

利用MEGA 来构建进化树(molecular evolutionary genetics analysis 分子进化遗传分析)打开mega5,选择Align----edit/built alignment----create a new alignment—OK选择DNA/protein出现新的对话框Open------选择已经保存好的用clustalx 经过比对保存的以.aln格式的文件打开之后,出现下面的页面双击文件名可以进行修改的。
我的就是从这里开始修改把A,B,C 都去掉,只留号码就好右键菜单点击delete 删除带※的那一行。
得到下面的图示,点击保存,重新起名字。
之后点击此图内的Alignment 选择Align by clustalW即可。
默认设置即可,点击OK就进行比对了,此后会出现一个过渡对话框,显示的是两两比对和多序列比对的过程之后回到初始页面,就是这个页面之后点File---点开,把刚才保留的文件点开然后出现下面的页面多了几个内容,点击TA的那个框框。
之后出现这样的框框图片然后在主程序中选择phylogeny---construct/test neighbor-joining tree,然后出现下面的页面黄色框框处的的参数是可以改变的,该图为我已经改变好的,把Bootstrap 的值改为1000 Methods根据文献上的参考改为了Kimura2-parameter model.之后点击compute,就出现了,而且还带有必需的支持率即自展值,是用来检验你所计算的进化树分支可信度的。
简单地讲就是把序列的位点都重排,重排后的序列再用相同的办法构树,如果原来树的分枝在重排后构的树中也出现了,就给这个分枝打上一分,如果没出现就给0分,这样经过你给定的repetitions 次(至少1000次)重排构树打分后,每个分枝就都得出分值,计算机会给你换算成bootstrap值。
重排的序列有很多组合,值越小说明分枝的可信度越低,最好根据数据的情况选用不同的构树方法和模型。
系统进化树绘制

MEGA:系统进化树绘制1.从测序公司获取可以用TXT格式打开的菌株16SrDNA序列文本信息;2.打开NCBI网站(https:///),依次点击BlAST—>Microbes,进入最下图所示界面。
3.将序列信息粘贴到黄色文本框内,点击BLAST按钮,进入比对结果页面,根据所需选择20条(参考)相似序列信息,点击Download,下载FASTA(aligned sequences)格式序列信息到电脑;4.将测序所得序列信息与Blast所得序列信息合并到同一个Text文件中;5.打开MEGA软件(以MEGA6.06为例),点击Align—>Edit/Build Alignment,选择Creat a new alignment并点击OK,点击DNA,进入最下图界面,最大化子界面;6.点击下图红线圈出的图标或通过Edit—>Insert Sequence From File Ctrl+I,进入第二个图所示界面,将文件格式由ABI改为Text,选择所选序列信息文件,点击打开;7.按住Shift,鼠标点击首条和最后一条多余的序列信息,即可选择某一需要删除的序列信息区域,点击Delete删除多余序列信息,并编辑各序列名称,点击保存编辑好的序列信息;8.点击Data—>Phylogenetic Analysis(系统进化分析),点击“Yes”完成系统进化分析;9.回到MEGA主页面,依次点击Analysis—>Phylogeny—>Construct/Test Neighbor-Joining Tree...,在跳出的界面中点击“Yes”,接着将跳出页面中的Test of Phylogeny项的None改为Bootstrap method,点击Compute,系统完成运算,生成系统进化树;10.依次点击Image—>Save as PDF file,保存成PDF格式系统发育树图谱。
Mega的使用以及进化树的绘制

1.MEGA构建系统进化树的步骤2.CLUSTALX进行序列比对1.MEGA构建系统进化树的步骤1. 将要用于构建系统进化树的所有序列合并到同一个fasta格式文件,注意:所有序列的方向都要保持一致( 5’-3’)。
如图:2. 打开MEGA软件,选择"Alignment" - "Alignment Explorer/CLUSTAL",在对话框中选择Retrieve sequences from a file, 然后点OK,找到准备好的序列文件并打开,如图:。
3. 在打开的窗口中选择”Alignment”-“Align by ClustalX” 进行对齐,对齐过程需要一段时间,对齐完成后,最好将序列两端切齐,选择两端不齐的部分,单击右键,选择delete即可,如图:。
4. 关闭当前窗口,关闭的时候会提示两次否保存,第一次无所谓,保存不保存都可以,第二次一定要保存,保存的文件格式是.meg。
根据提示输入Title,然后会出现一个对话框询问是否是Protein-coding nucleotide sequence data, 根据情况选择Yes或No。
最后出现一个对话框询问是否打开,选择Yes,如图:。
5. 回到MEGA主窗口,在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” -“Neighbor-joining”,打开一个窗口,里面有很多参数可以设置,如何设置这些参数请参考详细的MEGA说明书,不会设置就暂且使用默认值,不要修改,点击下面的Compute按钮,系统进化树就画出来了,如图:在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” –“Minimun-evolution”,如图:在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” –“Maximun-parsimony”,如图:在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” –“UPGMA”,如图:6. 最后,使用TreeExplorer窗口中提供的一些功能可以对生成的系统进化树进行调整和美化。
用MEGA构建进化树

如何用MEGA构建进化树MEGA3.1是一个关于序列分析以及比较统计的工具包,其中包括有距离建树法和MP建树法;可自动或手动进行序列比对,推断进化树,估算分子进化率,进行进化假设测验,还能联机的Web数据库检索。
下载后可直接使用,主要包括几个方面的功能软件:i)DNA和蛋白质序列数据的分析软件。
ii)序列数据转变成距离数据后,对距离数据分析的软件。
iii)对基因频率和连续的元素分析的软件。
iv)把序列的每个碱基/氨基酸独立看待(碱基/氨基酸只有0和1的状态)时,对序列进行分析的软件。
v)绘制和修改进化树的软件,进行网上blast搜索。
用MEGA构建进化树有以下步骤:1. 16S rDNA测序和参考序列选取从环境中分离到单克隆,去重复后扩增16S rDNA 序列并测序,然后与数据库/blast/Blast.cgi比对,找到相似度最高的几个序列,确定一下你分离的细菌大约属于哪个科哪个属,如果相似度达到百分之百那基本可以确定你分离得到的就是Blast到的那个,然后找一到两个同科的,再找一到两个同目的,再找一到两个同纲的细菌,把序列全部下下来,以FSATA形式整合在TXT文档中,如>TS1 GCAGTCGAACGATGAAGCCCAGCTTGCTGGGT GGATTAGTGGCGAACGGGTGAGTAACACGTGG GTGATCTGCCCTGCACTTCGGGATAAGCCTGGG AAACTGGGTCTAATACCGGATAGGACCTCGGGA TGCATGTTCCGGGGTGGAAAGGTTTTCCGGTG CAGGATGGGCC>gi|117572706|gb|EF028124.1| Rhodococcus sp. Atl25 16S ribosomal RNA gene, partial sequence CGATTAGAGTTTGATCCTGGCTCAGGACGAACG CTGGCGGCGTGCTTAACACATGCAAGTCGAACG ATGAAGCCCAGCTTGCTGGGTGGATTAGTGGC GAACGGGTGAGTAACACGTGGGTGATCTGCCC TGCACTTCGGGATAAGCCTGGGAAACTGGGTCTAATACCGGAT>TS2 TGCAAGTCGAGCGAATGGATTAAGAGCTTGCTC TTATGAAGTTAGCGGCGGACGGGTGAGTAACA CGTGGGTAACCTGCCCATAAGACTGGGATAACT CCGGGAAACCGGGGCTAATACCGGATAACATTT TGAACTGCATGGTTCGAAATTGAAAGGCGGCTT CGGCTGTCACT>gi|56383044|emb|AJ809498.1| Bacillus cereus partial 16S rRNA gene, strain TMW 2.383 GATGAACGCTGGCGGCGTGCCTAATACATGCAA GTCGAGCGAATGGATTAAGAGCTTGCTCTTATG AAGTTAGCGGCGGACGGGTGAGTAACACGTGG GTAACCTGCCCATAAGACTGGGATAACTCCGGG AAACCGGGGCTAATACCGGATAACATTTTGAAC YGCATGGTTC………………………….………………………….参考序列选择有几个原则:a,不选非培养(unclutured)微生物为参比;b,所选参考序列要正确,里面无错误碱基;c,在保证同属的前提下,优先选择16S rDNA全长测序或全基因组测序的种;d,每个种属选择一个参考序列,如果自己的序列中同一属的较多,可适当选择两个参考序列。
进化树软件MEGA最新6.06说明书

第一步:打开软件下面介绍菜单的使用:Data菜单:Creat a new :创建一个新的数据比对文件,也就是说当我们比对完一组后,想接着比对另一组,那么使用它就可以不用退出直接把数据文件导入;Open :打开先前已经比对并保存好的文件,它包含两个子菜单:retive sequence from file 和saved aligment session ;Close: 关闭当前的比对数据文件;Save session :保存当前比对结果,可以给比对的结果一个文件名;Export alignment :将当前的序列比对结果输出到指定文件,有两种输入格式可供选择:MGTA 和FASTA.DNA sequence :使用它来选择输入的数据DNA 序列,这里需要说明的是如果你输入的数据是氨基酸序列的话,比对窗口只显示一个标签,若是DNA 序列的话则显示两个标签,一个是DNA 序列的,另一个是氨基酸序列的。
Protein sequences :选择输入的氨基酸序列,选择后,所以的位点就被当作氨基酸残基位点来对待。
Translate/untranslate :只有比对的序列是编码蛋白的DNA序列的时候才可用。
它可以根据指定的遗传密码表将DNA 序列翻译成特定的氨基酸序列。
Select genetic code table :使用它将编码蛋白的DNA 翻译成特定的蛋白序列。
R everse complement :将选择的一整行的DNA 序列变为与之互补配对碱基序列。
Exit alignment explorer :退出序列比对的资源管理窗口Edit 菜单:使用这个菜单可以对我们的比对序列进行想要的一些编辑工作具体为Undo:撤销上一步操作;Copy:复制;Cut:剪切;Paste:粘贴;这三个操作都可以只针对一个碱基或氨基酸残基也可以是一段甚至是整个序列;Delete:从比对表格中删除一段序列;Delete gaps:去掉序列中的空缺;Insert blank sequence:重新插入一空行;标签和序列都是空的;Insert sequence from file :从已保存的文件中插入新的序列;Select sites :选择一列序列,与点击比对表上方的灰白空格作用类似;Select sequence:选择一行序列,与点击比对表格左侧的标签名作用类似;Select all:全选;Allow base editing :只读保护,只有选择后才能对序列进行编辑操作,否则所以的序列为只读格式,不能进行任何编辑操作。
MEGA 软件——系统发育树构建方法

• 双击图标
,
• 下载下来的序列片段保存文件为FASTA格式,打开方式为TXT格式。 • 将Blast对比后所Download的序列筛选后,构建系统发育进化树。 • 构建系统发育树需要测序所得序列1个,Blast对比得出序列5-10个, 构建出的为单枝系统发育进化树。通过这个对比可以确定出所测 定的序列最相似物种,当相似度为99%甚至100%时,基本可以确 定所测定的基因序列所属物种。
ME待测PCR产物送测序后,一个星期左右会得到生物 公司发的邮件,里面包含测序结果的附件,下载后得到文件压缩 包 ,将文件包解压,可以看到文件夹里有文件
• 测序公司会提供一款解读软件Chromas,免安装类型,能够直接 打开测序结果并通过软件直接进入NCBI数据库进行Blast搜索。
• 可培养真菌可以选定对比物种种属后构建大型系统发育进化树, 不可培养真菌则可直接构建系统发育进化树。
• 双击
后打开MEGA软件
手把手教你构建系统进化树

9、要学生做的事,教职员躬亲共做; 要学生 学的知 识,教 职员躬 亲共学 ;要学 生守的 规则, 教职员 躬亲共 守。2021/6/292021/6/29Tuesday, June 29, 2021
10、阅读一切好书如同和过去最杰出 的人谈 话。2021/6/292021/6/292021/6/296/29/2021 8:10:36 AM
以外米缀蛾的cds为例,点击cdsTA格式,如何保 存见下图
一般情况下点
击该页的右上 角有send 图标, 选择后点击 create file 即 可下载。Txt可 以打开。
该图显示的是
序列全长的 FASTA格式下 载。
因为我采取基于氨
17、儿童是中心,教育的措施便围绕 他们而 组织起 来。2021/6/292021/6/292021/6/292021/6/29
2、Our destiny offers not only the cup of despair, but the chalice of opportunity. (Richard Nixon, American President )命运给予我们的不是失望之酒,而是机会之杯。二〇二一年六月十七日2021年6月17日星期四 3、Patience is bitter, but its fruit is sweet. (Jean Jacques Rousseau , French thinker)忍耐是痛苦的,但它的果实是甜蜜的。10:516.17.202110:516.17.202110:5110:51:196.17.202110:516.17.2021 4、All that you do, do with your might; things done by halves are never done right. ----R.H. Stoddard, American poet做一切事都应尽力而为,半途而废永远不行6.17.20216.17.202110:5110:5110:51:1910:51:19 5、You have to believe in yourself. That's the secret of success. ----Charles Chaplin人必须相信自己,这是成功的秘诀。-Thursday, June 17, 2021June 21Thursday, June 17, 20216/17/2021
MEGA软件——系统发育树构建方法
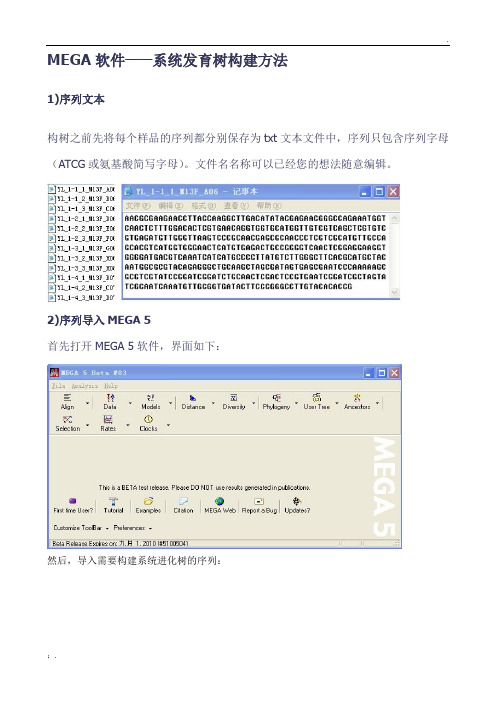
MEGA软件——系统发育树构建方法1)序列文本构树之前先将每个样品的序列都分别保存为txt文本文件中,序列只包含序列字母(ATCG或氨基酸简写字母)。
文件名名称可以已经您的想法随意编辑。
2)序列导入MEGA 5首先打开MEGA 5软件,界面如下:然后,导入需要构建系统进化树的序列:点击OK出现新的对话框,创建新的数据文件导入成功3)序列比对分析点击W,开始比对。
比对完成后删除序列两端不能完全对其的碱基。
系统分析然后,关闭该窗口,在弹出的对话框中选择保存文件,文件名随便去,比如保存为1。
4)系统发育树构建以NJ为例Bootstrap选择1000,点Computer,开始计算计算完毕后,生成系统发育树。
以下“系统发育树树的修饰”方法沿用斑竹brightfuture01的方法5)树的修饰建好树之后,往往需要对树做一些美化。
这个工作完全可以在word中完成,达到发表文章的要求。
点击image,copy to clipboard。
新建一个word文档,选择粘贴。
见下图:在图上点击右键-编辑图片,就可以对文字的字体大小,倾斜等做出修饰。
见下图:这个时候可以通过Adobe professional 对其进行图像导出:先将此word文档打印成PDF,见下图:将打印出来的PDF保存在桌面上,打开,如下图:此时,点击工具,高级编辑工具,裁剪工具,如下图所示:选择需要的区域以删除周围的空白区,双击发育树,会出现下图:点击确定,出现下图(把空边切掉了):点击文件,另存为,在保存类型一栏中选择TIFF格式,点击确定后会生成下面这个图片,所生成图片绝对可以满足文章的发表:OK,结束了,自己玩一把吧。
用MEGA构建进化树

如何用MEGA构建进化树是一个关于序列分析以及比较统计的工具包,其中包括有距离建树法和MP建树法;可自动或手动进行序列比对,推断进化树,估算分子进化率,进行进化假设测验,还能联机的Web 数据库检索;下载后可直接使用,主要包括几个方面的功能软件:iDNA和蛋白质序列数据的分析软件;ii序列数据转变成距离数据后,对距离数据分析的软件; iii对基因频率和连续的元素分析的软件;iv把序列的每个碱基/氨基酸独立看待碱基/氨基酸只有0和1的状态时,对序列进行分析的软件;v绘制和修改进化树的软件,进行网上blast搜索;用MEGA构建进化树有以下步骤:1. 16S rDNA测序和参考序列选取从环境中分离到单克隆,去重复后扩增16S rDNA序列并测序,然后与数据库比对,找到相似度最高的几个序列,确定一下你分离的细菌大约属于哪个科哪个属,如果相似度达到百分之百那基本可以确定你分离得到的就是Blast到的那个,然后找一到两个同科的,再找一到两个同目的,再找一到两个同纲的细菌,把序列全部下下来,以FSATA形式整合在TXT文档中,如>TS1GCAGTCGAACGATGAAGCCCAGCTTGCTGGGTGGA TTAGTGGCGAACGGGTGAGTAA CACGTGGGTGATCTGCCCTGCACTTCGGGATAAGCCTGGGAAACTGGGTCTAATACCG GA TAGGACCTCGGGA TGCA TGTTCCGGGGTGGAAAGGTTTTCCGGTGCAGGATGGGCC>gi|6|gb|| Rhodococcus sp. Atl25 16S ribosomal RNA gene, partial sequence CGATTAGAGTTTGA TCCTGGCTCAGGACGAACGCTGGCGGCGTGCTTAACACATGCAA GTCGAACGATGAAGCCCAGCTTGCTGGGTGGA TTAGTGGCGAACGGGTGAGTAACAC GTGGGTGATCTGCCCTGCACTTCGGGATAAGCCTGGGAAACTGGGTCTAA TACCGGA T>TS2TGCAAGTCGAGCGAATGGA TTAAGAGCTTGCTCTTA TGAAGTTAGCGGCGGACGGGTG AGTAACACGTGGGTAACCTGCCCA TAAGACTGGGATAACTCCGGGAAACCGGGGCTAA TACCGGATAACA TTTTGAACTGCATGGTTCGAAA TTGAAAGGCGGCTTCGGCTGTCACT>gi||emb|| Bacillus cereus partial 16S rRNA gene, strain TMWGA TGAACGCTGGCGGCGTGCCTAA TACATGCAAGTCGAGCGAA TGGATTAAGAGCTTG CTCTTA TGAAGTTAGCGGCGGACGGGTGAGTAACACGTGGGTAACCTGCCCATAAGAC TGGGATAACTCCGGGAAACCGGGGCTAATACCGGATAACATTTTGAACYGCATGGTTC ………………………….………………………….参考序列选择有几个原则:a,不选非培养unclutured微生物为参比;b,所选参考序列要正确,里面无错误碱基;c,在保证同属的前提下,优先选择16S rDNA全长测序或全基因组测序的种;d,每个种属选择一个参考序列,如果自己的序列中同一属的较多,可适当选择两个参考序列;2. 序列比对将整理好的序列导入,如图接着程序自动运行,得出结果,自动输出.aln和.dnd 为后缀的两个文件; 序列比对也可以直接用MEGA来做;3. 打开程序MEGA,如下图所示:4. 只能打开meg格式的文件,但是它可以把其他格式的多序列比对文件转换过来,用.aln格式Clustal的输出文件转换.meg文件;点File:Convert to MEGA Format,打开转换文件对话框,从目的文件夹中选中Clustal 对比分析后所产生的.aln文件,点击打开;5. 转换好的meg文件,会弹出一个提示信息,点击ok;查看meg序列文件最后是否正常,若存在clustal. 行,即可删除;点存盘保存meg文件,meg文件会和aln文件保存在同一个目录;6. 关闭转换窗口,回到主窗口,现在点面板上的“Click me to activate a data file”打开刚才的meg文件;如果为蛋白质序列,选择“protein sequence”,电击“OK”,得到以下图示,数据输入之后的样子,窗口下面有序列文件名和类型;而在另外一个窗口内,出现以下数据文件点击选择和编辑数据分类图标, 可对所选择的序列进行编辑,完成后点击close即可;序列编辑完成后,可进行保存,点击保存后出现以下界面,点击ok即可;7. 构建进化树的算法主要分为两类:独立元素法discrete character methods和距离依靠法distance methods;所谓独立元素法是指进化树的拓扑形状是由序列上的每个碱基/氨基酸的状态决定的例如:一个序列上可能包含很多的酶切位点,而每个酶切位点的存在与否是由几个碱基的状态决定的,也就是说一个序列碱基的状态决定着它的酶切位点状态,当多个序列进行进化树分析时,进化树的拓扑形状也就由这些碱基的状态决定了;而距离依靠法是指进化树的拓扑形状由两两序列的进化距离决定的;进化树枝条的长度代表着进化距离;独立元素法包括最大简约性法Maximum Parsimony methods和最大可能性法Maximum Likelihood methods;距离依靠法包括除权配对法UPGMAM和邻位相连法Neighbor-joining;1 phylogeny→UPGMA2用Bootstrap构建进化树,MEGA的主要功能就是做Bootstrap验证的进化树分析,Bootstrap 验证是对进化树进行统计验证的一种方法,可以作为进化树可靠性的一个度量;各种算法虽然不同,但是操作方法基本一致;进化树的构建是一个统计学问题;我们所构建出来的进化树只是对真实的进化关系的评估或者模拟;如果我们采用了一个适当的方法,那么所构建的进化树就会接近真实的“进化树”;模拟的进化树需要一种数学方法来对其进行评估;不同的算法有不同的适用目标;一般来说,最大简约性法适用于符合以下条件的多序列:i 所要比较的序列的碱基差别小,ii 对于序列上的每一个碱基有近似相等的变异率,iii 没有过多的颠换/转换的倾向,iv 所检验的序列的碱基数目较多大于几千个碱基;用最大可能性法分析序列则不需以上的诸多条件,但是此种方法计算极其耗时;如果分析的序列较多,有可能要花上几天的时间才能计算完毕;过程如下①参数的设置:phylogeny→bootstrap test of phylogeny→NJ②系统进化树的测试方法,可以选择用Bootstrap,也可以选择不进行测试;重复次数Replications通常设定至少要大于100比较好,随机数种子可以自己随意设定,不会影响计算结果;一般选择500或1000;有许多Model供选择,默认为Kimura 2-paramete r,不同的Model有不同的算法,具体请参考专业的生物信息学书籍;设定完成,点compute,开始计算;②结果输出:这个过程所耗时间和序列的数量和长短成正比,程序就会产生这么一个树,该窗口中有两个属性页,一个是原始树,一个是bootstrap验证过的一致树;树枝上的数字表示bootstrap验证中该树枝可信度的百分比; 结果如下:8. 进化树的优化:1利用该软件可得到不同树型,如下图所示:除此之外,还可以有多种树型,根据需要来选择; 2显示建树的相关信息:点击图标i;3点击优化图标,可进行各项优化:Tree栏中,可以进行树型选择:rectangular tree/circle tree/radiation tree;每种树都可以进行长度,宽度或角度等的设定Branch:可对树枝上的信息进行修改;Lable:可对树枝的名字进行修改;Scale:标尺设置Cutoff:cut off for consensus tree;一般为50%;9、进化树的分类优化Place root on branch:可以来回转换;Flip subtree:180度翻转分枝,名字翻转180度;Swab subtree:交换分枝,名字不翻转;Compress/expand subtree与Set divergent time:可以把同一分枝的基因压缩或扩展;点击Compress/expand subtree后,在要压缩的分枝处点击,出现以下界面,在name/caption 中输入文件名例如w,其他还有很多的选项,设置好了,点击OK;所得到的结果,可以在压缩和扩展之间转换;10. 调整进化树根据所的进化树的效果,要进行调整,包括多余序列删除、不足序列添加、种属名称标注等等,还要根据投稿杂志要求在PHOTOSHOP 中修改等;完成后的进化树应包含充足的信息;本人所做进化树完成图如下:。
Mega构建进化树的方法

Open a saved alignment session:使用它可以打开一 个已经比对好的序列文件;
Retieve a sequence from a file :这种情况同第一种情
况相似。
3
选择选项1,弹出下面对话框
4
• 根据情况选择“yes”或“no”
5
从”Data”菜单中读取序列文件,如6 选择保存的fasta格式的序列文件,如7
MEGA构建进化树的方法
MEGA简介
Molecular Evolutionary Genetics Analysis Functions
• conducting automatic and manual sequence alignment • inferring phylogenetic trees • mining web-based databases • estimating rates of molecular evolution • testing evolutionary hypotheses.
9
10
在窗口10中点击“Phylogeny”下拉菜单
11
然后选择Bootstrap Test of Phylogeny。最大简 约法 Maximum Parsimony;邻接法 Neighbor Joining;最小进化法 Minimum Evolution。
三种方法中选择Neighbor Joining,出现窗口12
12
在12窗口设置好各种参数,然后,点击“Compute”进 行运算,构建进化树
结果如窗口13所示:在实际运行得到拓扑图之后, 上面有两个选项,点击 Original tree,可以选择查 看计算所得到的所有结构树。
mega操作过程-多序列比对、进化树PPT幻灯片课件

信
(neighbour joining)、phylip、dist
息
学
CORRECT DIST:决定是否做距离修正。对于小的序列歧异(<
及
10%),选择与否不会产生差异;对于大的序列歧异,需做出
应
修正。因为观察到的距离要比真实的进化距离低。
用
IGNORE GAPS:选择on,序列中的任何空位将被忽视。
Extremely slow computation.
20
Progressive Alignment Method
DbClustal:
http://igbmc.u-strasbg.fr:8080/DbClustal/dbclustal.html
基
础 Poa (Partial order alignments):
第二节 多序列比对程序及应用
基 础
Progressive Alignment Method
生
物
Iterative Alignment
信
息 学
Block-Based Alignment
及 应
DNASTAR
用
DNAMAN
12
1、Progressive Alignment Method
学
A distance matrix is built to derive a guide tree, which is
及
then used to direct a full multiple alignment using the
应
progressive approach.
用
Outperforms Clustal when aligning moderately divergent
怎样使用MEGA建立进化树

怎样使用MEGAt 立进化树如何使用MEGA4.0#立进化树 1、首先是双击软件打开如下图所示|M| ijaKMr3 valj 141 Mrhr ArgrwricQt iVvta“qplii :护 忏冲 i 二客H - I 号筍需.廿星"LIF M ■ H 、-| II ■ DKi -Mjrsrze: H r« r-r r ^c>az^ LCS2、现在是处于DNA序列,而我们要做蛋白质的进化树的话,就如下操作M4. Aligmr>&nl Explof頁H L lQnmt*Ftji Editm e祁3、接下来我们要进行序列的输入,点击左边那个红箭头,贝U出现下面的窗口刚M4: Alfgnment Explorer匚;日屯EJrt S«ar di Aflgmnenl Wfrb $e<)□ d| D ◎日「蹇輻酋1 41象Protein S^quer匚弊1|主曲色"匕色丄4、然后右击sequenee 1,修改名字,如改成DPVFrotejn Sequence?5、然后从Word里复制蛋白质序列,然后在下面的位置粘贴G 辱CopfPTCtfiT X CU,書 f sterna6则可出现如下图的序列了□ QCW1C3 iRWfl Wq^ri[ V^i>n irequ^Ki 幷册枷・1話皿讥曲佰i"—喇・ct Mgeirc 惟■ sy7、然后点击窗口上的保存图标保存 8、重复从3开始,直到你的序列输入完9、序列输入元后进行最后的保存,方法如下垂邑trit 5|讨之斗和"1 of op«r * dow亠 P TOUMT 1 <io-jrr<n接下来打开册b M 罗哥 H*lpi t X t tt b要输入ul7两次保存名字一然后关闭这个窗口出现下面这个窗口■■■MM Jfc接下来就可以建立各种样式的进化树〜乜 MdngHie-r jein^ IMJL* &? Wrigym 佔抽杓也山-« UW3ML ■> ■小h,鼻 陆01申*貝Trfl 和 Hi^Tgrn^ ,HnNkk T ivn HnM "d i-Oi^4*cflArs R 协 FriWrt '^l^diCNHE軒I 匚 fkrti tiiitanr-ri : hy A 护产就 匸沁”-嗯,只是把过程写出来,方便大家建立进化树,不足的地方,大家补充好〜。
用MEGA构建进化树

如何用MEGA构建进化树MEGA3.1是一个关于序列分析以及比较统计的工具包,其中包括有距离建树法和MP 建树法;可自动或手动进行序列比对,推断进化树,估算分子进化率,进行进化假设测验,还能联机的Web数据库检索。
下载后可直接使用,主要包括几个方面的功能软件:i)DNA 和蛋白质序列数据的分析软件。
ii)序列数据转变成距离数据后,对距离数据分析的软件。
iii)对基因频率和连续的元素分析的软件。
iv)把序列的每个碱基/氨基酸独立看待(碱基/氨基酸只有0和1的状态)时,对序列进行分析的软件。
v)绘制和修改进化树的软件,进行网上blast搜索。
用MEGA构建进化树有以下步骤:1. 16S rDNA测序和参考序列选取从环境中分离到单克隆,去重复后扩增16S rDNA序列并测序,然后与数据库比对,找到相似度最高的几个序列,确定一下你分离的细菌大约属于哪个科哪个属,如果相似度达到百分之百那基本可以确定你分离得到的就是Blast到的那个,然后找一到两个同科的,再找一到两个同目的,再找一到两个同纲的细菌,把序列全部下下来,以FSATA形式整合在TXT 文档中,如>TS1GCAGTCGAACGATGAAGCCCAGCTTGCTGGGTGGA TTAGTGGCGAACGGGTGAGTAA CACGTGGGTGATCTGCCCTGCACTTCGGGATAAGCCTGGGAAACTGGGTCTAATACCG GA TAGGACCTCGGGA TGCA TGTTCCGGGGTGGAAAGGTTTTCCGGTGCAGGATGGGCC>gi|117572706|gb|EF028124.1| Rhodococcus sp. Atl25 16S ribosomal RNA gene, partial sequence CGATTAGAGTTTGA TCCTGGCTCAGGACGAACGCTGGCGGCGTGCTTAACACATGCAA GTCGAACGATGAAGCCCAGCTTGCTGGGTGGA TTAGTGGCGAACGGGTGAGTAACAC GTGGGTGATCTGCCCTGCACTTCGGGATAAGCCTGGGAAACTGGGTCTAA TACCGGA T>TS2TGCAAGTCGAGCGAATGGA TTAAGAGCTTGCTCTTA TGAAGTTAGCGGCGGACGGGTG AGTAACACGTGGGTAACCTGCCCA TAAGACTGGGATAACTCCGGGAAACCGGGGCTAA TACCGGATAACA TTTTGAACTGCATGGTTCGAAA TTGAAAGGCGGCTTCGGCTGTCACT>gi|56383044|emb|AJ809498.1| Bacillus cereus partial 16S rRNA gene, strain TMW 2.383GA TGAACGCTGGCGGCGTGCCTAA TACATGCAAGTCGAGCGAA TGGATTAAGAGCTTG CTCTTATGAAGTTAGCGGCGGACGGGTGAGTAACACGTGGGTAACCTGCCCA TAAGAC TGGGATAACTCCGGGAAACCGGGGCTAATACCGGATAACATTTTGAACYGCATGGTTC ………………………….………………………….参考序列选择有几个原则:a,不选非培养(unclutured)微生物为参比;b,所选参考序列要正确,里面无错误碱基;c,在保证同属的前提下,优先选择16S rDNA全长测序或全基因组测序的种;d,每个种属选择一个参考序列,如果自己的序列中同一属的较多,可适当选择两个参考序列。
MEGA6使用教程——进化树的构建

MEGA6使用教程——进化树的构建首先,打开MEGA6软件。
在主界面上方的菜单栏中,选择“File”→“Open Data Directory”来选择数据目录。
在数据目录中,应该存在一个以.meg为文件格式后缀的文件,用于存储算法运行结果。
如果不存在这样的文件,可以通过“File”→“New”来创建一个新的.meg文件。
在.meg文件中,可以导入多种类型的数据,如DNA序列、蛋白质序列、线粒体DNA序列等。
点击菜单栏中的“Data”→“Import Alignment from File”来导入序列文件。
导入序列文件后,可以从菜单栏中的“Phylogeny”→“Construct/Test Maximum-Likelihood Tree”来构建最大似然进化树。
在弹出的对话框中,可以选择不同的进化模型来评估树的质量。
MEGA6提供了多种模型,如Jukes-Cantor模型、Kimura 2-parameter模型、Tamura 3-parameter模型等。
可以在下拉菜单中选择不同的模型。
计算完成后,可以在弹出的窗口上看到生成的进化树。
可以通过缩放、拖动、旋转等操作来查看树的不同部分。
此外,还可以使用MEGA6中的其他工具来分析和优化进化树。
比如,“Phylogeny”→“Switch Trees/Branches”工具可以帮助我们比较和切换不同的进化树。
此外,还可以使用“Phylogeny”→“Bootstrapping”工具来计算进化树的支持率。
Bootstrapping是一种统计方法,通过对原始数据集进行重抽样来评估进化树的支持强度。
在使用MEGA6构建进化树时,还应该注意一些问题。
首先,选择合适的进化模型对结果的准确性至关重要。
根据输入数据的特点,选择适当的模型来评估进化树会得到更可靠的结果。
其次,应该进行足够的计算重复次数,以确保所得到的进化树是可靠的。
足够的重复次数可以提高进化树的准确度和稳定性。
使用mega6做进化树
假如你要对比你所测序列E的序列与其他物质的亲缘关系,步骤如下:一,首先要先把你获得E的序列去NCBI网站进行比对,步骤如下:1.登录NCBI网站https:///2.找到右侧的BLAST,点进去;3.找到页面下方的这个图标,点Nucleotide BLAST4.将测得的序列全部粘贴到页面上的这个框里:5.找到页面最下方的Algorithm parameters,在最下面的BLAST旁边勾选“Show XXXX”后点击BLAST6.然后就会弹出另一个页面,你就得耐心等待了,因为它在比对,比对好后就会出现这样一个界面:7.然后往下拉,就看到好多序列的结果,可以选择所有的序列下载,也可以选择你想要的序列来下载(All/None可全选或都不选),选好后点击“GenBank”。
8.把所有的序列都勾选后,点右上角的“send”9.出现这个框格,File-FASTA按框格里选择好点Create File就可以批量下载内含你所选的序列的“fasta”格式的文件;11改好后打开,把自己的序列按“>名称+序列”的格式紧接在已下好的序列后面,添加好后再把后缀改回“fasta”,便可进行下一步8. 312.双击fasta文件,由MEGA6.0打开,如图13.单击W图标中的“Align DNA”,会提醒你选择序列,单击确定即可,如下图14.比对后的序列如下图。
15.然后我们需要把“*”号之外的序列全部删除,只留下"*"标注的序列,保存,保存后得到的是“mas”格式文件16.回到MEGA6.0主页面,在”DATA”中选“Open A file /Session”,然后会弹出一个选择文件的窗口,去选择刚刚保存的“mas”后缀名的文件,选择后弹出下方窗口,点“Analyze”。
17.回到MEGA5.0的主界面,在菜单栏中选择“Phylogeny”-“Construct/ Test Neighbor-Joining Tree…”-“yes”,会弹出一个窗口,里面有很多参数可以设置,以下是Bootstrap consensus tree基本的设置,可以参考:18.可能由于版本的不同,在形成的树上无相似度的数字标明,可以参照以下步骤进行标注:。
mega6构建系统发育树

燃油量控制单元 直接控制喷油量
N109
断油电磁阀
关掉点火钥匙时切断油路
N108
正时电磁阀
调整喷油正时
K29
故障灯
指示故障
N18
废气再循环电磁 控制废气再循环的量
阀
N75
增压控制电磁阀 控制增压压力
J52
预热塞继电器
控制预热塞的工作
Compresso r switch off
空调开关
给出空调工作信号判断发动机负荷是否增加
喷油量控制电磁执行器
喷油量控制电磁执行器
? 这种旋转式比例电磁阀,主要由线圈、定子、转子、 回位弹簧、转子转角传感器以及与转子一体的驱动 轴等组成。在驱动轴的下端偏心设置传动销,并与 油量控制滑套相连接。旋转式比例电磁阀是通过两 个线圈的反向信号占空比来控制流经线圈的电流, 并结合回位机构控制转子的转向和旋转位移。当 ECU控制流经线圈的电流时,转子旋转某一角度, 此时与转子一体的驱功轴下端的偏心销同步旋转, 带动油量控制活套移动、滑套的位置与转子的旋转 角度有关。
? 3 发动机转速信号 ? 4 处理后的发动机转速信
号 ? 5 计算后的开始喷射信号
BOSCH
柴油电控VE泵结构(位置型)
? 1控制套位置 传感器
? 2喷油量控制 电磁执行器
? 3断油电磁阀 ? 4分配柱塞 ? 5喷油始点电
磁阀 ? 6控制套
BOSCH
泵内传感器(位置型)
Wj
控制套位置传感器〔位移传感器〕
柴油机燃油供给系统的发展
机械是VE泵燃油系统的组成
? 1油箱 ? 2燃油滤清器 ? 3VE泵 ? 4喷油嘴 ? 5回油管 ? 6预热塞 ? 7蓄电池 ? 8预热塞和启
mega建树方法 进化树的建立过程
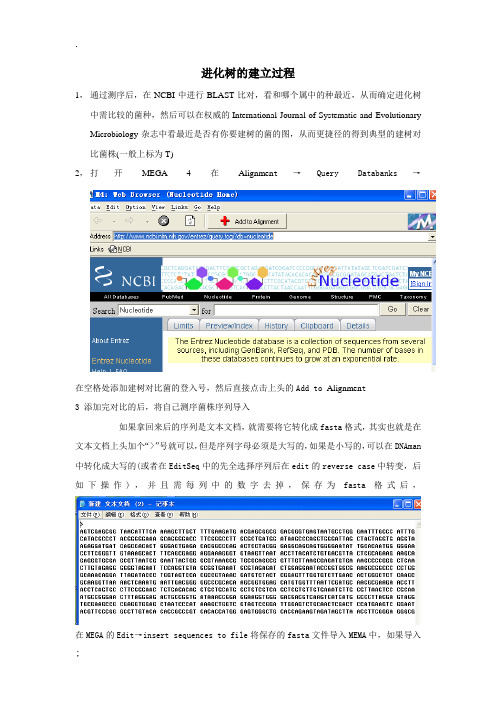
进化树的建立过程
1,通过测序后,在NCBI中进行BLAST比对,看和哪个属中的种最近,从而确定进化树中需比较的菌种,然后可以在权威的International Journal of Systematic and Evolutionary Microbiology杂志中看最近是否有你要建树的菌的图,从而更捷径的得到典型的建树对比菌株(一般上标为T)
2,打开MEGA 4在Alignmen t→Query Databanks→
在空格处添加建树对比菌的登入号,然后直接点击上头的Add to Alignmen t
3 添加完对比的后,将自己测序菌株序列导入
如果拿回来后的序列是文本文档,就需要将它转化成fasta格式,其实也就是在文本文档上头加个“>”号就可以,但是序列字母必须是大写的,如果是小写的,可以在DNAman 中转化成大写的(或者在EditSeq中的先全选择序列后在edit的reverse case中转变,后如下操作),并且需每列中的数字去掉,保存为fasta格式后,
在MEGA的Edit→insert sequences to file将保存的fasta文件导入MEMA中,如果导入
的序列是互补链的话,直接在添加的里面,点击导入链,右击后点击互补就行,选中所有的序列后,在Alignmen t选项中选中Align by clustalw让其自动分析后,在Date选项中输出格式选择为MEGA格式保存
4 再一次启动软件将上一步保存的文件打开,然后在
如上图操作就可以得出进化树,然后在上面直接修改。
手把手教你构建系统进化树

3、比对序列,比对结果转化为*.meg格式
用 Mega 6.0 的 ClustalW 做多序列联配,比对结果用 *.meg格式保存。或者用Clustal X软件进行比对,比对结果 保存为*.aln,再用Mega 6.0转化为*.meg格式。
4、构建系统进化树
打开保存的*.meg格式文件,选择邻接法构建系统发育 进化树。
以外米缀蛾的cds为例,点击cds,出现下图。
点击FASTA,出现下图。
该图为外米缀蛾的 FASTA格式,如何保 存见下图
一般情况下点 击该页的右上 角有send 图标, 选择后点击 create file 即 可下载。Txt可 以打开。 该图显示的是 序列全长的 FASTA格式下 载。
因为我采取基于氨 基酸序列比对,所 以选择coding sequences和fasta protein,下载编码 区氨基酸序列。
文件名未下载时不要更改,下下来之后再更改
MEGA6可以识别fasta格式文件。如图,将全 部-基因.txt重命名为全部-基因.fasta
•选择打开方式为MEGA6,打开全部-基因.fasta,自动跳出序列窗口 •用ClustalW做多序列联配
如何构建系统进化树
YZU.TRY
系统发生树(英文: Phylogenetic tree ) 又称为演化树( evolutionary tree ),是 表明被认为具有共同祖先的各物种间演化关 系的树。是一种亲缘分支分类方法 ( cladogram )。在树中,每个节点代表其 各分支的最近共同祖先,而节点间的线段长 度对应演化距离(如估计的演名称要么全部 斜体,要么全部不斜体,无法只让拉丁文斜体
用MEGA构建进化树

怎么样用MEGA建坐进化树之阳早格格创做是一个关于序列分解以及比较统计的工具包,其中包罗有距离建立法战MP建立法;可自动大概脚动举止序列比对付,估计进化树,估算分子进化率,举前进化假设考验,还能联机的Web数据库检索.下载后可间接使用,主要包罗几个圆里的功能硬件:i)DNA战蛋黑量序列数据的分解硬件.ii)序列数据转形成距离数据后,对付距离数据分解的硬件. iii)对付基果频次战连绝的元素分解的硬件.iv)把序列的每个碱基/氨基酸独力瞅待(碱基/氨基酸惟有0战1的状态)时,对付序列举止分解的硬件.v)画造战建矫正化树的硬件,举止网上blast搜索.用MEGA建坐进化树有以下步调:1. 16S rDNA测序战参照序列采用从环境中分散到单克隆,去沉复后扩删16S rDNA序列并测序,而后与数据库比对付,找到相似度最下的几个序列,决定一下您分散的细菌约莫属于哪个科哪个属,如果相似度达到百分之百那基础不妨决定您分散得到的便是Blast到的那个,而后找一到二个共科的,再找一到二个共脚段,再找一到二个共目的细菌,把序列齐脚下下去,以FSATA形式调整正在TXT文档中,如>TS1GCAGTCGAACGATGAAGCCCAGCTTGCTGGGTGGATT AGTGGCGAACGGGTGAGTAACACGTGGGTGATCTGCC CTGCACTTCGGGATAAGCCTGGGAAACTGGGTCTAAT ACCGGATAGGACCTCGGGATGCATGTTCCGGGGTGGA AAGGTTTTCCGGTGCAGGATGGGCC>gi|117572706|gb|EF028124.1| Rhodococcus sp. Atl25 16S ribosomal RNA gene, partial sequence CGATTAGAGTTTGATCCTGGCTCAGGACGAACGCTGGC GGCGTGCTTAACACATGCAAGTCGAACGATGAAGCCC AGCTTGCTGGGTGGATTAGTGGCGAACGGGTGAGTAA CACGTGGGTGATCTGCCCTGCACTTCGGGATAAGCCTG GGAAACTGGGTCTAATACCGGAT>TS2 TGCAAGTCGAGCGAATGGATTAAGAGCTTGCTCTTATG AAGTTAGCGGCGGACGGGTGAGTAACACGTGGGTAAC CTGCCCATAAGACTGGGATAACTCCGGGAAACCGGGG CTAATACCGGATAACATTTTGAACTGCATGGTTCGAAA TTGAAAGGCGGCTTCGGCTGTCACT GATGAACGCTGGCGGCGTGCCTAATACATGCAAGTCG AGCGAATGGATTAAGAGCTTGCTCTTATGAAGTTAGCG GCGGACGGGTGAGTAACACGTGGGTAACCTGCCCATA AGACTGGGATAACTCCGGGAAACCGGGGCTAATACCGGATAACATTTTGAA CYGCATGGTTC………………………….………………………….参照序列采用有几个准则:a,不选非培植(unclutured)微死物为参比;b,所选参照序列要精确,内里无过失碱基;c,正在包管共属的前提下,劣先采用16S rDNA齐少测序大概齐基果组测序的种;d,每个种属采用一个参照序列,如果自己的序列中共一属的较多,可适合采用二个参照序列. 2. 序列比对付将整治佳的序列导进,如图接着步调自动运止,得出截止,自动输出 .aln战.dnd 为后缀的二个文献.序列比对付也不妨间接用MEGA去搞.MEGA,如下图所示:4.只可挨启meg要领的文献,然而是它不妨把其余要领的多序列比对付文献变换过去,用.aln要领(Clustal的输出文献)变换.meg文献.面File:Convert to MEGA Format,挨启变换文献对付话框,从脚段文献夹中选中Clustal 对付比分解后所爆收的.aln文献,面打挨启.5. 变换佳的meg文献,会弹出一个提示疑息,面打ok.查看meg序列文献末尾是可平常,若存留clustal. *止,即可简略.面存盘保存meg文献,meg文献会战aln文献保存正在共一个目录.6. 关关变换窗心,回到主窗心,当前面里板上的“Click me to activate a data file”挨启刚刚才的meg文献.如果为蛋黑量序列,采用“protein sequence”,电打“OK”,得到以下图示,数据输进之后的格式,窗心底下有序列文献名战典型.而正在其余一个窗心内,出现以下数据文献面打采用战编写数据分类图标,可对付所采用的序列举止编写,完成后面打close即可.序列编写完成后,可举止保存,面打保存后出现以下界里,面打ok即可.7. 建坐进化树的算法主要分为二类:独力元素法(discrete character methods)战距离依赖法(distance methods).所谓独力元素法是指进化树的拓扑形状是由序列上的每个碱基/氨基酸的状态决断的(比圆:一个序列上大概包罗很多的酶切位面,而每个酶切位面的存留与可是由几个碱基的状态决断的,也便是道一个序列碱基的状态决断着它的酶切位面状态,当多个序列举前进化树分解时,进化树的拓扑形状也便由那些碱基的状态决断了).而距离依赖法是指进化树的拓扑形状由二二序列的进化距离决断的.进化树枝条的少度代表着进化距离.独力元素法包罗最大简约性法(Maximum Parsimony methods)战最大大概性法(Maximum Likelihood methods);距离依赖法包罗除权配对付法(UPGMAM)战邻位贯串法(Neighbor-joining).(1)phylogeny→UPGMA(2)用Bootstrap建坐进化树,MEGA的主要功能便是搞Bootstrap考证的进化树分解,Bootstrap考证是对付进化树举止统计考证的一种要领,不妨动做进化树稳当性的一个度量.百般算法虽然分歧,然而是支配要领基础普遍.进化树的建坐是一个统计教问题.咱们所建坐出去的进化树不过对付真正在的进化关系的评估大概者模拟.如果咱们采与了一个适合的要领,那么所建坐的进化树便会靠近真正在的“进化树”.模拟的进化树需要一种数教要领去对付其举止评估.分歧的算法有分歧的适用目标.普遍去道,最大简约性法适用于切合以下条件的多序列:i 所要比较的序列的碱基不共小,ii 对付于序列上的每一个碱基有近似相等的变同率,iii 不过多的颠换/变换的倾背,iv 所考验的序列的碱基数目较多(大于几千个碱基);用最大大概性法分解序列则不需以上的诸多条件,然而是此种要领估计极其耗时.如果分解的序列较多,有大概要花上几天的时间才搞估计完成.历程如下①参数的树坐:phylogeny→bootstrap test of phylogeny→NJ②系统进化树的尝试要领,不妨采用用Bootstrap,也不妨采用不举止尝试.沉复次数(Replications)常常设定起码要大于100比较佳,随机数种子不妨自己随意设定,不会做用估计截止.普遍采用500大概1000.有许多Model供采用,默认为Kimura 2-paramete r,分歧的Model 有分歧的算法,简曲请参照博业的死物疑息教书籍籍.设定完成,面compute,启初估计.②截止输出:那个历程所耗时间战序列的数量战少短成正比,步调便会爆收那样一个树,该窗心中有二个属性页,一个是本初树,一个是bootstrap考证过的普遍树.树枝上的数字表示bootstrap考证中该树枝可疑度的百分比.截止如下:8. 进化树的劣化:1)利用该硬件可得到分歧树型,如下图所示:除此除中,还不妨有多种树型,根据需要去采用.2)隐现建立的相关疑息:面打图标i.3)面打劣化图标,可举止各项劣化:Tree栏中,不妨举止树型采用:rectangular tree/circle tree/radiation tree.每种树皆不妨举止少度,宽度大概角度等的设定Branch:可对付树枝上的疑息举止建改.Lable:可对付树枝的名字举止建改.Scale:标尺树坐Cutoff:cutoff for consensus tree.普遍为50%. 9、进化树的分类劣化Place root on branch:不妨去回变换.Flip subtree:180度翻转分枝,名字翻转180度.Swab subtree:接换分枝,名字不翻转.Compress/expand subtree与Set divergent time:不妨把共一分枝的基果压缩大概扩展.面打Compress/expand subtree后,正在要压缩的分枝处面打,出现以下界里,正在name/caption 中输进文献名(比圆wwww),其余另有很多的选项,树坐佳了,面打OK.所得到的截止,不妨正在压缩战扩展之间变换. 10. 安排进化树根据所的进化树的效验,要举止安排,包罗多余序列简略、缺累序列增加、种属称呼标注等等,还要根据投稿纯志央供正在PHOTOSHOP中建改等.完成后的进化树应包罗充脚的疑息.自己所搞进化树完成图如下:。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
利用MEGA6建立进化树一.NCBI中选择Protein数据库,搜索PD-1相关蛋白
二.选择兴趣蛋白,在Send to选项中选择File-FASTA格式
三.下载安装MEGA6,刚刚下载的FASTA文件变为可读,双击用MEGA6打开
四.展开上图得到下图,至此蛋白序列成功输入MEGA
五.选择菜单栏上Alignment-Align by Clustalw进行蛋白序列比对
六.得到比对后结果,菜单栏-Data-Export Alignment输出比对结果, 得到MEGA6源文件,接下来就可以进行进化树绘制
八.返回主程序界面,菜单栏选择Phylogeny-第二个选项Neighbor,选择刚刚输出的数据,在个性化界面按需要自己的做调整,最后得到所需进化树.。