已基因组测序物种
植物物种全基因组的测序与分析

植物物种全基因组的测序与分析随着现代生物技术的不断发展和完善,越来越多的研究者开始将目光放在了植物的基因组测序和分析上。
植物物种的全基因组测序和分析可以帮助我们更好地了解植物的生长和发育规律,发现新的基因和蛋白质,促进植物育种和改良等方面的应用。
本文将从植物基因组测序和分析的意义、方法和应用等方面进行探讨。
一、植物基因组测序的意义植物基因组测序是现代遗传学和分子生物学领域的一项重要研究内容。
通过对植物基因组的测序和分析,可以为植物学、农业和生态学等方向的研究提供重要的基础数据。
首先,全基因组测序能够为我们提供大量的基因序列信息。
通过基因组测序,可以获得植物基因组的完整序列信息,为后续的基因鉴定、新基因发现、基因功能研究等提供基础,为植物学的研究提供了更全面的基础知识。
其次,基因组测序有助于发现新基因。
通过基因组测序,我们可以获取所有基因序列的信息,并进行比对分析,以发现新的、以前未知的基因,这对于数据驱动型的生物学研究具有重要的意义。
此外,基因组测序还可以促进生物信息学领域的发展。
基因组测序技术和生物信息学处理技术的结合,可以更好地研究基因与生态之间的关系,为生态学和植物保护提供更多的数据支撑。
二、植物基因组测序的方法目前,植物基因组测序主要采用Illumina高通量测序技术、 PacBio和Nanopore第三代测序技术、等温测序技术以及荧光原位杂交技术等方法。
其中,Illumina高通量测序技术是全球最为普遍的测序平台之一,其分辨率高、准确率高、数据量大,可以快速、高通量地测序,成为植物基因组测序的主流技术之一。
而PacBio和Nanopore第三代测序技术主要具有长读长和高准确性的特点,能够获得更全面的基因组序列信息,用于高质量的基因组组装。
等温测序和荧光原位杂交技术等方法也可以用于获得植物基因组信息。
在选择测序平台时,需要根据样品的特性、分辨率、数据量、费用等多个方面进行综合评估。
三、植物基因组测序的应用植物基因组测序的应用范围十分广泛,涉及到植物学、种质资源保护、农业种植和育种等多个领域。
群体进化-基于全基因组重测序

DNA样品总量: ≥3 μg 适用范围样品要求文库类型测序策略与深度分析内容项目周期 群体进化(基于全基因组重测序)标准分析时间为120天,个性化分析需根据项目实际情况进行评估HiSeq PE150推荐测序深度≥5X/个体350 bp小片段DNA文库1. 已有参考基因组序列的物种中不同亚群(自然群体)2. 各亚群间划分明显,同一亚群内的个体有一定代表性3. 每个亚群选取10个样本左右(推荐动物≥10个,植物≥15个)4. 总体不少于30个样本与参考基因组比对群体SNP检测、注释及统计系统进化树构建群体遗传结构分析群体主成分分析连锁不平衡分析选择消除分析候选基因GO和KEGG富集构建单体型图谱种群历史和有效群体大小技术参数针对已有参考基因组的物种,对其各亚种进行全基因组重测序获得基因组信息,通过与参考基因组比对,得到大量高准确性的SNP、InDel、SV等变异信息,讨论群体的遗传结构、遗传平衡和影响遗传平衡的因素,从而从分子层面揭示该物种的进化机制、环境适应性等系列问题。
该技术能精准地得到全基因组内所有遗传信息,最大程度地挖掘出群体内遗传变异。
诺禾具有丰富的群体遗传学项目经验,研究成果发表于Nature Genetics(Li, M, et al. 2013& Zhou, XM,et al. 2014)等。
参考文献[1] Li M, Tian S, Jin L, et al . Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars [J]. Nature genetics, 2013, 45(12): 1431-1438.[2] Zhan S, Zhang W, Niitepo ~ld K, et al . The genetics of monarch butterfly migration and warning colouration [J]. Nature, 2014.案例解析[案例一] 家猪和藏猪的群体进化分析[1]2013年,诺禾致源科技服务团队与四川农业大学研究者合作发表该成果。
第五章基因组测序技术(共118张PPT)

断裂产物分 别在4个泳 道电泳
G G+A T+C C
化学法测序实例
哌啶
改进的特异化学切割反应
1.基本原理
与链终止法测序原理相同,只是用不同 的荧光色彩标记ddNTP,如ddATP标记红 色荧光,ddCTP标记蓝色荧光, ddGTP标 记黄色荧光, ddTTP标记绿色荧光.由于 每种ddNTP带有各自特定的荧光颜色,而 简化为由1个泳道同时判读4种碱基.
②该酶能够用2‘,3’--双脱氧核苷三磷酸作底物并将 其聚合到新生寡核苷酸链的3‘-末端,从而终止其延 伸反应。
在DNA测序反应中,加入模板DNA,引物(特异 性引物),DNA聚合酶,dA,dT,dG,dC和一 种ddNTP。常用Klenow大片段,无5'→3'外切酶 活性。
制备单链模板
A 克隆于质粒中DNA→用碱或热变性 B M13克隆单链DNA C 噬粒克隆DNA D PCR产生单链DNA
C 参考人类基因组图,特别是大量的STS位标作为基点,进行
序列组装,排成重叠克隆群.
基于克隆群(contig-based)
鸟枪法策略
指导测序策略
遗传、物理图谱
人们对感兴趣的基因或与疾病相关的 基因优先测序.
如:人类主要组织相容性复合区位于第6号 染色体,与人类免疫系统有关,因而优先 测序.
EST是一种重要的基因组图分子标记,以EST为探针很 容易从 cDNA中筛选全基因,又可从BAC克隆中找到其
2. 人类基因组草图的完成
2000年6月26日是人类 上值得纪念的一天。人 类基因组的工作草图已 经绘制完毕并于这天向 全世界公布。最终完成 图要求测序所用的克隆 能忠实地代表常染色体 的基因组结构,序列错 误率低于万分之一。
基因测序技术在鉴定物种中的应用

基因测序技术在鉴定物种中的应用基因测序技术是指通过分析生物体的DNA序列,确定各种基因的组成和位置,进而了解基因的功能、调控和表达等生命过程。
随着科技的不断进步,基因测序技术已经广泛应用于各个领域,其中之一便是在鉴定物种方面的应用。
鉴定物种是指通过识别某个生物体属于哪个物种,从而确定它的生态地位、保护状况、进化历史等信息。
传统的鉴定物种方法依赖于形态学、生理学和生态学特征,但这种方法有其局限性,玫瑰花和葡萄藤就很容易混淆。
随着基因测序技术的出现,特别是第三代测序技术的应用,鉴定物种的精度和速度得到了大幅提升。
下面我们来看一些基因测序技术在鉴定物种中的应用案例。
DNA条形码鉴定物种DNA条形码是指将某个物种或者样本的一个特定的DNA序列作为鉴定该物种的特征码。
利用DNA条形码能够使鉴定物种更加准确,并且能够提高鉴定物种的速度,大大加快样本的处理时间。
某些低等级物种相似度较高,难以区分,采用传统鉴定物种方法极为困难,但利用DNA条形码技术则容易比对出分别。
研究人员利用DNA条形码成功鉴定了400多个鱼类物种,开发了一个名为FISH-BOL的数据库,该数据库成为全球最大的鱼类DNA条形码数据库。
另外有研究表明,利用DNA条形码可对土壤中的生物进行鉴定,并对不同土壤的生物种类进行区分,这种方法可以为土壤生态学研究提供一种高效的工具。
基因组测序鉴定物种基因组测序是指对一个物种的全部基因组进行分析,获得相应的DNA序列信息。
通过基因组测序,可以获得物种的整个DNA序列信息,包括基因的组成和位置、编码蛋白质的基因、非编码区域和整个基因组的结构等。
利用基因组测序,科研人员可以举例鉴定分辨率更高的物种,并扩展鉴定物种的范围。
海星和海参在形态上非常相似,很难进行鉴定。
但是,通过基因组测序,科研人员发现,虽然两者的外部表现相似,但它们的遗传基础是不同的,基因组序列也有明显的不同。
因此,利用基因组测序技术可以明确区分海星和海参,并防止他们被错误鉴定。
生命科学中的全基因组测序技术

生命科学中的全基因组测序技术全基因组测序技术(Whole Genome Sequencing)是指测定一个生物体所有基因组DNA的序列。
在过去的几十年里,随着高通量测序技术的不断进步和成本的降低,全基因组测序技术已经成为了研究生命科学领域的重要工具之一。
本文将从技术原理、应用领域和未来发展等方面对全基因组测序技术进行探讨。
一、技术原理全基因组测序技术的核心原理是将整个基因组DNA按照一定的长度断裂成许多小片段,使用高通量测序技术将这些小片段逐一测序,然后通过计算机算法将这些片段拼接成完整的基因组序列。
具体来说,全基因组测序技术的步骤如下:1.样品准备:首先需要从生物体的样品中提取出全部的基因组DNA,这个步骤非常关键,若提取的DNA含有杂质,后续的测序结果将会受到很大影响。
2.测序:将提取的基因组DNA分成若干小片段,通常是将DNA随机打断成200-500bp的小片段,然后使用测序仪将每个小片段的序列测出。
目前高通量测序仪种类繁多,包括Illumina、Ion Torrent、PacBio和Oxford Nanopore等,其中Illumina使用最为广泛。
3.数据分析:将测得的序列利用计算机算法进行拼接,这个步骤是全基因组测序中最困难的部分。
由于某些原因,比如测序精度不够高、片段之间存在交叉等,导致拼接出来的基因组序列并不是完整的,因此需要使用一些软件进行多次验证和修正,以确保拼接出来的序列尽量准确完整。
二、应用领域全基因组测序技术在生命科学领域的应用非常广泛,主要分为以下几个方面:1.研究基因组结构和功能:全基因组测序可以帮助研究人员了解生物体的基因组结构和功能,比如基因组大小、基因数量、基因型变异、基因表达水平等信息,从而更深入地理解生物界的进化和发展。
全基因组测序可以直接测定基因组中所有基因的序列,从而持续地帮助人们了解基因之间的相互作用和调控。
2.疾病诊断和预防:全基因组测序可以帮助诊断罕见遗传病或个体化疾病风险,同时可以预测一些患病风险,有助于促进个体化医学的发展。
如何快速查找物种间对应的同源基因

如何快速查找物种间对应的同源基因Homologous Gene ID Conversion.png有时候,大家做实验以小鼠为模型,但希望查看与之对应的人同源基因。
像这种情况,我们可以不需要进行序列比对来查找,因为比较麻烦。
使用公共数据可能更高效。
1.基于NCBI HomoloGene数据库查找物种间对应的同源基因NCBI HomoloGene数据库收集了部分已经完成基因组测序物种的同源基因数据。
数据库现包含21个物种,共44233组同源基因;HomoloGene的数据是开放的:FTPhomologene.data存放着同源基因的对应关系HID(HomoloGene group id)Taxonomy ID Gene ID Gene Symbol Protein gi Protein accession3960634ACADM160961497NP_001104286.139598469356ACADM109008502XP_001101274.131009011364Acadm6680618NP_031408.1每个物种都有一个对应的Taxonomy ID:10090 Mus musculus10116 Rattus norvegicus28985 Kluyveromyces lactis318829 Magnaporthe oryzae33169 Eremothecium gossypii3702 Arabidopsis thaliana4530 Oryza sativa4896 Schizosaccharomyces pombe4932 Saccharomyces cerevisiae5141 Neurospora crassa6239 Caenorhabditis elegans7165 Anopheles gambiae7227 Drosophila melanogaster7955 Danio rerio8364 Xenopus (Silurana) tropicalis9031 Gallus gallus9544 Macaca mulatta9598 Pan troglodytes9606 Homo sapiens9615 Canis lupus familiaris9913 Bos taurus单个基因直接检索,如Acadm:批量注释某个物种的基因对应另一个物种的同源基因,可以使用R包homologene,它调用的是c中build68的数据;homologene(genes, inTax, outTax)genes:需要查找同源基因的基因列表inTax:输入基因所属物种outTax:查找的同源基因属于那个物种例子:genelist<-c("Acadm","Eno2","Acadvl")homologene(genelist, inTax = 10090, outTax = 9606)10090 9606 10090_ID 9606_ID1 Eno2 ENO2 13807 20262 Mog MOG 17441 4340查看homologene使用的数据版本homologeneVersion[1] 682. 基于InParanoid 8数据库查找物种间对应的同源基因InParanoid 8提供的下载数据是Protein ID;构建g InParanoid 8 用到的InParanoid 4.1可以获取的,InParanoid4.1 standalone download这儿我们利用InParanoid 8提供的同源基因信息进行一个快速检索。
利用DNA测序技术鉴定物种来源的实验步骤与技巧

利用DNA测序技术鉴定物种来源的实验步骤与技巧引言:DNA测序技术在现代生物学研究中扮演着重要的角色,它不仅可以揭示生物的基因组结构和功能,还可以用于物种鉴定。
利用DNA测序技术鉴定物种来源,可以帮助我们解决生物多样性保护、犯罪侦查、食品安全等方面的问题。
本文将介绍利用DNA测序技术鉴定物种来源的实验步骤与技巧。
一、样品采集与DNA提取鉴定物种来源的第一步是采集样品,并从中提取DNA。
样品可以是动物的血液、组织或粪便,植物的叶片或根部,甚至是环境中的土壤或水样。
采集样品时,需要注意避免污染和交叉污染。
DNA提取可以使用商业化的DNA提取试剂盒,也可以根据具体样品的特点选择适当的DNA提取方法。
二、PCR扩增目标基因片段在鉴定物种来源时,通常会选择一个或多个特定的基因片段进行PCR扩增。
这些基因片段可以是线粒体DNA(mtDNA)的控制区域或核DNA的特定基因。
选择合适的引物对进行PCR扩增,确保引物的特异性和敏感性。
PCR反应条件需要根据引物的特性进行优化,包括温度、时间和酶的浓度等。
三、凝胶电泳分离与可视化PCR扩增后的产物可以通过凝胶电泳进行分离与可视化。
将PCR产物与DNA 分子量标记物一同加载到琼脂糖凝胶中,然后进行电泳分离。
根据PCR产物的大小,可以判断样品中是否存在目标基因片段,并估计其大小。
为了提高凝胶电泳的分辨率和可视化效果,可以使用荧光染料或核酸染色剂。
四、测序与序列分析如果需要进一步确定物种来源,可以将PCR产物进行测序。
测序可以使用传统的Sanger测序方法,也可以使用高通量测序技术,如Illumina测序平台。
测序后,可以通过比对测序结果与数据库中已知物种的DNA序列进行比对,从而确定物种来源。
此外,还可以利用生物信息学工具进行序列比对、系统发育分析和物种鉴定。
五、质量控制与结果解读在进行DNA测序鉴定时,需要进行质量控制,确保测序结果的准确性和可靠性。
质量控制包括检查测序片段的长度、测序深度和碱基质量等。
植物基因组的测序和解析

植物基因组的测序和解析一、引言随着基因组学技术的飞速发展,对植物基因组的测序和解析也越来越深入。
通过对植物基因组的研究,不仅能够深入了解植物生长发育和适应环境的机理,也为植物育种和农业生产提供了重要的理论和技术支持。
本文将着重介绍植物基因组的测序和解析技术及其应用。
二、植物基因组测序对于植物基因组的测序,一般采用两种主要的方法:全基因组测序(WGS)和转录组测序。
目前已经完成了大量植物的全基因组测序工作,包括拟南芥、水稻、小麦、玉米、大豆、苹果等,这些测序数据为植物基因组研究提供了基础。
而转录组测序则可以在不同生物学阶段或不同环境条件下,对植物基因表达情况做出深入分析。
1. 全基因组测序WGS是指对物种整个基因组DNA序列的测序,包括基因区域和非基因区域。
全基因组测序技术通常会采用高通量测序平台,如Illumina、PacBio等。
基因组大小和复杂性是影响测序花费和时间的主要因素。
在植物基因组测序中,由于植物基因组的大小和复杂性较高,因此一般需要使用多平台组合测序的方式。
例如,可以先使用Illumina短读长度(150bp左右)测序高覆盖度,然后用PacBio长读长度(10kb以上)来填补基因组中的重复区域、插入元件和复杂重读区域等。
2. 转录组测序转录组测序是指对某个生物在特定环境或生物阶段的mRNA进行测序,一般分为总RNA测序和mRNA测序两种。
总RNA测序可以同时得到注释基因和非编码RNA等的全面信息,而mRNA 测序则会选择性地测序已经被转录核糖体识别和选择的信息。
此外,转录组测序也包括甲基化RNA的测序,可以获得DNA甲基化的空间分布和转录水平的相关性等信息。
三、植物基因组解析植物基因组测序仅仅是一个开始,如何处理和分析这些海量的基因组数据,才能更好地理解植物基因组结构与功能呢?这就需要应用各种生物信息学分析方法来进行解析,包括基因注释、结构预测、基因家族分析、进化分析、基因功能预测等。
基因组测序与生物分类

基因组测序与生物分类随着科技的进步,基因组测序已成为现代生物学的重要工具之一。
通过对生物体中的基因序列进行测定和分析,科学家们可以揭示生物之间的关系,并为生物的分类提供新的启示。
本文将探讨基因组测序对生物分类的意义和应用。
一、基因组测序的基本原理基因组测序是指对生物体中的基因组进行测量和分析,以获得基因序列的信息。
这项技术的发展使得我们能够更深入地了解生物的遗传信息,以及不同物种之间的差异和联系。
在基因组测序过程中,科学家们首先需要提取生物体中的DNA,并将其纯化。
然后,DNA会被切割成小片段,并通过PCR扩增等技术制备成文库。
接下来,这些片段会被测序仪逐个读取,得到一系列的DNA序列片段。
最后,通过对这些序列片段进行拼接和比对,就可以获得完整的基因组序列信息。
二、基因组测序在生物分类中的应用1. 生物进化关系的研究通过基因组测序,科学家们可以获取相关物种的基因组序列信息,并进行比较分析。
通过比较物种之间的基因组序列差异,可以推测它们之间的进化关系。
这有助于揭示生物多样性的起源和发展过程,为生物的分类提供了重要线索。
2. 物种鉴定和系统分类传统的物种鉴定和分类方法主要依赖于形态特征的比较。
然而,有些生物在形态上差异较小,很难通过外部特征来准确鉴定和分类。
基因组测序技术可以提供更客观、准确的鉴定依据。
通过对物种的DNA进行测序和比对,可以准确地判断其所属物种,并揭示物种之间的亲缘关系。
3. 新物种的发现基因组测序技术不仅可以帮助科学家们对已知物种进行分类,还可以用于发现新物种。
通过对环境中的DNA进行测序,科学家们可以发现一些微生物、真菌和其他微小生物,这些生物以前由于其微小体积而难以察觉。
基因组测序揭示了一种全新的物种鉴定和分类方法。
4. 药物研发和医学应用基因组测序可以帮助科学家们揭示基因与疾病之间的关系,并发现与疾病相关的基因突变。
基于这些突变,可以开发针对性的药物,并为疾病的诊断和治疗提供新的方法。
已完成基因组测序的生物(植物部分)

水稻、玉米、大豆、甘蓝、白菜、高粱、黄瓜、西瓜、马铃薯、番茄、拟南芥、杨树、麻风树、苹果、桃、葡萄、花生拟南芥籼稻粳稻葡萄番木瓜高粱黄瓜玉米栽培大豆苹果蓖麻野草莓马铃薯白菜野生番茄番茄梨甜瓜香蕉亚麻大麦普通小麦西瓜甜橙陆地棉梅毛竹桃芝麻杨树麻风树卷柏狗尾草属花生甘蓝物种基因组大小和开放阅读框文献Sesamum indicum L. Sesame 芝麻(2n = 26)293.7 Mb, 10,656 orfs 1Oryza brachyantha短药野生稻261 Mb, 32,038 orfs 2Chondrus crispus Red seaweed爱尔兰海藻105 Mb, 9,606 orfs 3Pyropia yezoensis susabi-nori海苔43 Mb, 10,327 orfs 4Prunus persica Peach 桃226.6 of 265 Mb 27,852 orfs 5Aegilops tauschii 山羊草(DD)4.23 Gb (97% of the 4.36), 43,150 orfs 6 Triticum urartu 乌拉尔图小麦(AA)4.66 Gb (94.3 % of 4.94 Gb, 34,879 orfs 7 moso bamboo (Phyllostachys heterocycla) 毛竹2.05 Gb (95%) 31,987 orfs 8Cicer arietinum Chickpea鹰嘴豆~738-Mb,28,269 orfs 9 520 Mb (70% of 740 Mb), 27,571 orfs 10Prunus mume 梅280 Mb, 31,390 orfs 11Gossypium hirsutum L.陆地棉2.425 Gb 12Gossypium hirsutum L. 雷蒙德氏棉761.8 Mb 13Citrus sinensis甜橙87.3% of ~367 Mb, 29,445 orfs 14甜橙367 Mb 15Citrullus lanatus watermelon 西瓜353.5 of ~425 Mb (83.2%) 23,440 orfs 16 Betula nana dwarf birch,矮桦450 Mb 17Nannochloropsis oceanica CCMP1779微绿球藻(产油藻类之一)28.7 Mb,11,973 orfs 18Triticum aestivum bread wheat普通小麦17 Gb, 94,000 and 96,000 orfs 19 Hordeum vulgare L. barley 大麦1.13 Gb of 5.1 Gb,26,159 high confidence orfs,53,000 low confidence orfs 20Gossypium raimondii cotton 雷蒙德氏棉D subgenome,88% of 880 Mb 40,976 orfs 21Linum usitatissimum flax 亚麻302 mb (81%), 43,384 orfs 22Musa acuminata banana 香蕉472.2 of 523 Mb, 36,542 orfs 23Cucumis melo L. melon 甜瓜375 Mb(83.3%)27,427 orfs 24Pyrus bretschneideri Rehd. cv. Dangshansuli 梨(砀山酥梨)512.0 Mb (97.1%), 42,812 orfs 25,26Solanum lycopersicum 番茄760/900 Mb,34727 orfs 27S. pimpinellifolium LA1589野生番茄739 MbSetaria 狗尾草属(谷子、青狗尾草)400 Mb,25000-29000 orfs 28,29 Cajanus cajan pigeonpea木豆833 Mb,48,680 orfs 30Nannochloropis gaditana 一种海藻~29 Mb, 9,052 orfs 31Medicago truncatula蒺藜苜蓿350.2 Mb, 62,388 orfs 32Brassica rapa 白菜485 Mb 33Solanum tuberosum 马铃薯0.73 Mb,39031 orfs 34Thellungiella parvula条叶蓝芥13.08 Mb 29,338 orfs 35Arabidopsis lyrata lyrata 玉山筷子芥? 183.7 Mb, 32670 orfs 36Fragaria vesca 野草莓240 Mb,34,809 orfs 37Theobroma cacao 可可76% of 430 Mb, 28,798 orfs 38Aureococcus anophagefferens褐潮藻32 Mb, 11501 orfs 39Selaginella moellendorfii江南卷柏208.5 Mb, 34782 orfs 40Jatropha curcas Palawan麻疯树285.9 Mb, 40929 orfs 41Oryza glaberrima 光稃稻(非洲栽培稻)206.3 Mb (0.6x), 10 080 orfs (>70% coverage) 42Phoenix dactylifera 棕枣380 Mb of 658 Mb, 25,059 orfs 43Chlorella sp. NC64A小球藻属40000 Kb, 9791 orfs 44Ricinus communis蓖麻325 Mb, 31,237 orfs 45Malus domestica (Malus x domestica)苹果742.3 Mb 46Volvox carteri f. nagariensis 69-1b一种团藻120 Mb, 14437 orfs 47 Brachypodium distachyon 短柄草272 Mb,25,532 orfs 48Glycine max cultivar Williams 82栽培大豆1.1 Gb, 46430 orfs 49Zea mays ssp. Mays Zea mays ssp. Parviglumis Zea mays ssp. Mexicana Tripsacum dactyloides var. meridionale 无法下载附表50Zea mays mays cv. B73玉米2.06 Gb, 106046 orfs 51Cucumis sativus 9930 黄瓜243.5 Mb, 63312 orfs 52Micromonas pusilla金藻21.7 Mb, 10248 orfs 53Sorghum bicolor 高粱697.6 Mb, 32886 orfs 54Phaeodactylum tricornutum 三角褐指藻24.6 Mb, 9479 orfs 55Carica papaya L. papaya 番木瓜271 Mb (75%), 28,629 orfs 56 Physcomitrella patens patens小立碗藓454 Mb, 35805 orfs 57Vitis vinifera L. Pinot Noir, clone ENTAV 115葡萄504.6 Mb, 29585 orfs 58 Vitis vinifera PN40024葡萄475 Mb 59Ostreococcus lucimarinus绿色鞭毛藻13.2 Mb, 7640 orfs 60 Chlamydomonas reinhardtii 莱茵衣藻100 Mb, 15256 orfs 61Populus trichocarpa黑三角叶杨550 Mb, 45000 orfs 62Ostreococcus tauri 绿藻12.6 Mb, 7892 orfs 63Oryza sativa ssp. japonica 粳稻360.8 Mb, 37544 orfs 64Thalassiosira pseudonana 硅藻25 Mb, 11242 orfs 65Cyanidioschyzon merolae 10D红藻16.5 Mb, 5331 orfs 66Oryza sativa ssp. japonica粳稻420 Mb, 50000 orfs 67Oryza sativa L. ssp. Indica籼稻420 Mb, 59855 orfs 68Guillardia theta -蓝隐藻,551 Kb, 553 orfs 69Arabidopsis thaliana Columbia拟南芥119.7 Mb, 31392 orfs 70参考文献1 Zhang, H. et al. Genome sequencing of the important oilseed crop Sesamum indicum L. Genome Biology 14, 401 (2013).2 Chen, J. et al. Whole-genome sequencing of Oryza brachyantha reveals mechanisms underlying Oryza genome evolution. Nat Commun 4, 1595 (2013).3 Collén, J. et al. Genome structure and metabolic features in the red seaweed Chondrus crispus shed light on evolution of the Archaeplastida. Proceedings of the National Academy of Sciences 110, 5247-5252 (2013).4 Nakamura, Y. et al. The first symbiont-free genome sequence of marine red alga, susabi-nori Pyropia yezoensis. PLoS ONE 8, e57122 (2013).5 Verde, I. et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nature Genetics advance online publication (2013).6 Jia, J. et al. Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature 496, 91-95 (2013).7 Ling, H.-Q. et al. Draft genome of the wheat A-genome progenitor Triticum urartu. Nature 496, 87-90 (2013).8 Peng, Z. et al. The draft genome of the fast-growing non-timber forest species moso bamboo (Phyllostachys heterocycla). Nature Genetics 45, 456-461 (2013).9 Jain, M. et al. A draft genome sequence of the pulse crop chickpea (Cicer arietinum L.). Plant Journal, DOI: 10.1111/tpj.12173 (2013).10 Varshney, R. K. et al. Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat Biotech 31, 240-246 (2013).11 Zhang, Q. et al. The genome of Prunus mume. Nat Commun 3, 1318 (2012).12 Lee, M.-K. et al. Construction of a plant-transformation-competent BIBAC library and genome sequence analysis of polyploid Upland cotton (Gossypium hirsutum L.). BMC Genomics 14, 208 (2013).13 Paterson, A. H. et al. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 492, 423-427 (2012).14 Xu, Q. et al. The draft genome of sweet orange (Citrus sinensis). Nat Genet 45,59–66 (2013).15 Belknap, W. R. et al. Characterizing the citrus cultivar Carrizo genome through 454 shotgun sequencing. Genome 54, 1005-1015 (2011).16 Guo, S. et al. The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat Genet 45, 51–58 (2013).17 Wang, N. et al. Genome sequence of dwarf birch (Betula nana) and cross-species RAD markers. Mol Ecol Article first published online: 21 NOV 2012 DOI:10.1111/mec.12131 (2012).18 Vieler, A. et al. Genome, functional gene annotation, and nuclear transformation of the heterokont oleaginous alga Nannochloropsis oceanica CCMP1779. PLoS Genet 8, e1003064 (2012).19 Brenchley, R. et al. Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 491, 705-710 (2012).20 Consortium, T. I. B. G. S. A physical, genetic and functional sequence assembly of the barley genome. Nature 491, 711–716 (2012).21 Wang, K. et al. The draft genome of a diploid cotton Gossypium raimondii. Nature Genetics 44, 1098–1103 (2012).22 Wang, Z. et al. The genome of flax (Linum usitatissimum) assembled de novo from short shotgun sequence reads. The Plant Journal 72, 461-473 (2012).23 D'Hont, A. et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 488, 213–217 (2012).24 Garcia-Mas, J. et al. The genome of melon (Cucumis melo L.). PNAS 109, 11872-11877 (2012).25 reporter, A. G. s. Consortium releases pear genome data. GenomeWeb Daily News (2012).26 Wu, J. et al. The genome of pear (Pyrus bretschneideri Rehd.). GenomeRes.Published in Advance November 13, 2012, doi:10.1101/gr.144311.112 (2012).27 Consortium, T. T. G. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485, 635–641 (2012).28 Bennetzen, J. L. et al. Reference genome sequence of the model plant Setaria. Nat Biotech 30, 555-561 (2012).29 Zhang, G. et al. Genome sequence of foxtail millet (Setaria italica) provides insights into grass evolution and biofuel potential. Nat Biotech 30, 549-554 (2012).30 Varshney, R. K. et al. Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat Biotech 30, 83-89 (2012).31 Radakovits, R. et al. Draft genome sequence and genetic transformation of the oleaginous alga Nannochloropis gaditana. Nat Commun 3, 686 (2012).32 Young, N. D. et al. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 480, 520–524 (2011).33 Wang, X. et al. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 43, 1035-1039 (2011).34 Consortium, T. P. G. S. Genome sequence and analysis of the tuber crop potato. Nature 475, 189-195 (2011).35 Dassanayake, M. et al. The genome of the extremophile crucifer Thellungiella parvula. Nat. Genet. 43, 913-918 (2011).36 Hu, T. T. et al. The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat. Genet. 43, 476-481 (2011).37 Shulaev, V. et al. The genome of woodland strawberry (Fragaria vesca). Nat. Genet. 43, 109-116 (2011).38 Argout, X. et al. The genome of Theobroma cacao. Nat. Genet. 43, 101-108 (2011).39 Gobler, C. J. et al. Niche of harmful alga Aureococcus anophagefferens revealed through ecogenomics. PNAS 108, 4352-4357 (2011).40 Banks, J. A. et al. The selaginella genome identifies genetic changes associated with the evolution of vascular plants. Science 332, 960-963 (2011).41 Sato, S. et al. Sequence analysis of the genome of an oil-bearing tree, Jatropha curcas L. DNA Res. 18, 65-76 (2011).42 Sakai, H. et al. Distinct evolutionary patterns of Oryza glaberrima deciphered by genome sequencing and comparative analysis. Plant Journal 66, 796-805 (2011).43 Al-Dous, E. K. et al. De novo genome sequencing and comparative genomics of date palm (Phoenix dactylifera). Nat Biotech 29, 521-527 (2011).44 Blanc, G. et al. The Chlorella variabilis NC64A genome reveals adaptation to photosymbiosis, coevolution with viruses, and cryptic sex. Plant Cell 22, 2943-2955 (2010).45 Chan, A. P. et al. Draft genome sequence of the oilseed species Ricinus communis. Nat Biotech 28(951-956 (2010).46 Velasco, R. et al. The genome of the domesticated apple (Malus x domestica Borkh.). Nat. Genet. 42, 833-839 (2010).47 Prochnik, S. E. et al. Genomic analysis of organismal complexity in the multicellular green alga Volvox carteri. Science 329, 223-226 (2010).48 Initiative, T. I. B. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 463, 763-768 (2010).49 Schmutz, J. et al. Genome sequence of the palaeopolyploid soybean. Nature 463, 178-183 (2010).50 Hufford, M. B. et al. Comparative population genomics of maize domestication and improvement. Nat Genet 44, 808-811 (2012).51 Wei, F. et al. The physical and genetic framework of the maize B73 genome. PLoS Genet 5, e1000715 (2009).52 Huang, S. et al. The genome of the cucumber, Cucumis sativus L. Nat. Genet. 41, 1275-1281 (2009).53 Worden, A. Z. et al. Green evolution and dynamic adaptations revealed by genomes of the marine picoeukaryotes Micromonas. Science 324, 268-272 (2009).54 Paterson, A. H. et al. The Sorghum bicolor genome and the diversification of grasses. Nature 457, 551-556 (2009).55 Bowler, C. et al. The Phaeodactylum genome reveals the evolutionary history of diatom genomes. Nature 456, 239-244 (2008).56 Ming, R. et al. The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature 452, 991-996 (2008).57 Rensing, S. A. et al. The Physcomitrella genome reveals evolutionary insights into the conquest of land by plants. Science 319, 64-69 (2008).58 Velasco, R. et al. A high quality draft consensus sequence of the genome of a heterozygous grapevine variety. PLoS One 2, e1326 (2007).59 Jaillon, O. et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463-467 (2007).60 Palenik, B. et al. The tiny eukaryote Ostreococcus provides genomic insights into the paradox of plankton speciation. PNAS 104, 7705-7710 (2007).61 Merchant, S. S. et al. The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 318, 245-250 (2007).62 Tuskan, G. A. et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313, 1596-1604 (2006).63 Derelle, E. et al. Genome analysis of the smallest free-living eukaryote Ostreococcus tauri unveils many unique features. PNAS 103, 11647-11652 (2006). 64 Project, I. R. G. S. The map-based sequence of the rice genome. Nature 436,793-800 (2005).65 Armbrust, E. V. et al. The genome of the diatom Thalassiosira Pseudonana: ecology, evolution, and metabolism. Science 306, 79-86 (2004).66 Matsuzaki, M. et al. Genome sequence of the ultrasmall unicellular red alga Cyanidioschyzon merolae 10D. Nature 428, 653-657 (2004).67 Goff, S. A. et al. A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296, 92-100 (2002).68 Yu, J. et al. A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 296, 79-92 (2002).69 Douglas, S. et al. The highly reduced genome of an enslaved algal nucleus. Nature 410, 1091-1096 (2001).70 Kaul, S. et al. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796-815 (2000).。
菌株全基因组测序测目的

菌株全基因组测序测目的随着科技的飞速发展,基因组测序技术也得到了热烈关注。
全基因组测序作为一种高通量测序技术,被广泛应用于生物学研究中。
对于微生物,全基因组测序技术在分子生物学、生态学、农业以及医学等领域都有着广泛的应用。
本文将介绍菌株全基因组测序的目的、意义及运用等方面的相关信息。
一、菌株全基因组测序的目的菌株全基因组测序的主要目的涉及以下三个方面:1.识别基因组中的基因和功能。
首先,菌株全基因组测序可以帮助我们准确、快速地识别菌株中的基因和功能。
通过测序,我们可以确定菌株整个基因组的序列信息,并通过生物信息学方法将其转化为可识别的DNA序列。
基于这些序列信息,我们可以确定菌株基因组中的基因和其对应的功能,从而揭示菌株的生物学特征。
2.深入了解菌株的演化历史和遗传变异。
其次,菌株全基因组测序可以帮助我们深入了解菌株的演化历史和遗传变异。
对于微生物而言,基因组具有广泛的可塑性和适应性。
通过测序和比较不同菌株基因组序列,我们可以了解菌株之间存在的遗传差异和基因演化历史,为后续科学研究提供重要的参考数据。
3.为微生物学研究提供基础数据和资源。
最后,菌株全基因组测序不仅可以在直接应用方面得到应用,同时也为研究社区提供了重要的数据和资源。
这些数据和资源可以用于虚拟基因组计划、菌群组学和转录组学等领域,可以为微生物学、生态学、医学等各个领域的研究提供强有力的支持。
二、菌株全基因组测序的意义1.准确分析菌株的生物学特征。
菌株全基因组测序可以帮助我们准确分析菌株的生物学特征。
在微生物学领域,全基因组测序成为了研究微生物多样性、环境适应性和致病性等方面的强有力工具。
通过对菌株基因组序列的分析,我们可以了解其生态环境、代谢途径、转录调控等方面的生物学特征。
2.揭示微生物的进化关系。
全基因组测序可以揭示微生物之间的进化关系。
通过比较不同菌株的基因组序列,我们可以了解它们之间的遗传关系和进化轨迹。
同时,全基因组测序还可以检测相关基因的变异和表达水平的变化,对了解微生物的丰富性和多样性提供了重要的支持。
动植物全基因组测序

2013.072013.102014.092014.112015.04图1 异源多倍体棉花基因组共线性分析与非对称进化分析图2 MYB基因家族表达模式分析, Jiang W, et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement [J]. Nature Biotechnology, 2015, 33(5): 531-537.图3 金丝猴植食性机制的分析图4 金丝猴有效群体大小分析参考文献Zhou X, Wang B, Pan Q, Li R, Li M. Whole-genome sequencing of the nub-nosed monkey provides insights into folivory and evolutionary history [J]. Nature Genetics, 2014, 46(12):1303-1310.图5 藏猪及其它猪种的群体遗传结构分析参考文献Li M, Tian S, Jin L, et al. Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars [J]. Nature genetics, 2013, 45(12): 1431-1438.图6 进化分析结果图7 脂肪酸能量代谢途径蓝色表示正选择基因;红色表示特异性基因参考文献Qu Y, Zhao H, Han N, et al. Ground tit genome reveals avian adaptation to living at high altitudes in the Tibetan plateau [J]. Nature communications, 2013, 4.图1 7株野生大豆共有和特有基因集图2 野生大豆开花时间调控基因SNP和InDel变异参考文献Li Y, Zhou G, Ma J, Jiang W, Li R#, et al. De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits [J]. Nature biotechnology, 2014.32(10):1045-1052.图3 部分novel sequence在世界人群中的分布参考文献Li R, Li Y, Zheng H, et al. Building the sequence map of the human pan-genome [J]. Nature biotechnology, 2010, 28(1): 57-63.。
动植物重测序
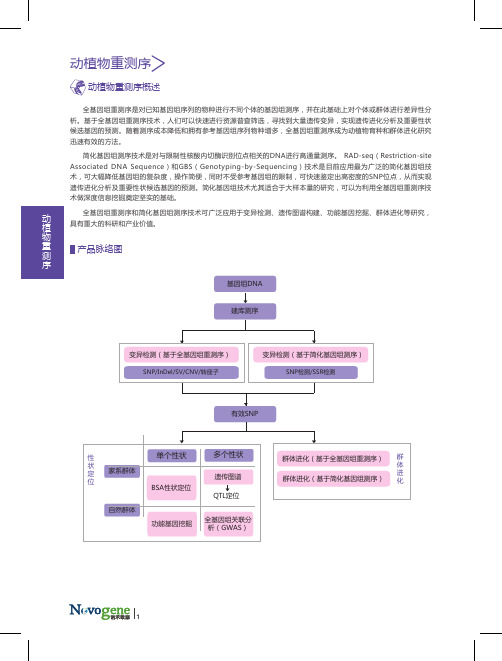
全基因组重测序是对已知基因组序列的物种进行不同个体的基因组测序,并在此基础上对个体或群体进行差异性分析。
基于全基因组重测序技术,人们可以快速进行资源普查筛选,寻找到大量遗传变异,实现遗传进化分析及重要性状候选基因的预测。
随着测序成本降低和拥有参考基因组序列物种增多,全基因组重测序成为动植物育种和群体进化研究迅速有效的方法。
简化基因组测序技术是对与限制性核酸内切酶识别位点相关的DNA进行高通量测序。
RAD-seq(Restriction-site Associated DNA Sequence)和GBS(Genotyping-by-Sequencing)技术是目前应用最为广泛的简化基因组技术,可大幅降低基因组的复杂度,操作简便,同时不受参考基因组的限制,可快速鉴定出高密度的SNP位点,从而实现遗传进化分析及重要性状候选基因的预测。
简化基因组技术尤其适合于大样本量的研究,可以为利用全基因组重测序技术做深度信息挖掘奠定坚实的基础。
全基因组重测序和简化基因组测序技术可广泛应用于变异检测、遗传图谱构建、功能基因挖掘、群体进化等研究,具有重大的科研和产业价值。
产品脉络图动植物重测序建库测序单个性状家系群体自然群体SNP/InDel/SV/CNV/转座子基因组DNA有效SNP性状定位群体进化群体进化(基于简化基因组测序) 群体进化(基于全基因组重测序) 变异检测(基于简化基因组测序)SNP检测/SSR检测遗传图谱全基因组关联分析(GWAS)功能基因挖掘变异检测(基于全基因组重测序) QTL定位BSA性状定位多个性状动植物重测序动植物重测序概述SNP检测、注释及统计基因组DNA350 bp小片段文库HiSeq PE150测序数据质控与参考基因组比对利用全基因组重测序技术对某一物种个体或群体的基因组进行测序及差异分析,可获得SNP、InDel、SV、CNV、PAV、转座子等大量的遗传多态性信息,建立遗传多态性数据库,为后续揭示进化关系、功能基因挖掘等奠定基础。
全基因组重测序技术在研究微生物物种中的应用

全基因组重测序技术在研究微生物物种中的应用随着科技的不断发展,生物学领域的研究也发生了巨大的变化。
全基因组重测序技术是其中的一个重要工具,它已经在微生物学研究中得到广泛的应用。
全基因组重测序技术可以对微生物物种进行深入的研究,有助于我们深入了解微生物群落的组成、演化和功能。
1. 全基因组重测序技术的原理全基因组重测序技术是一个高通量的DNA测序技术,它可以有效地对DNA序列进行快速、准确和高效的测定。
具体来说,这项技术是通过将微生物DNA分散到许多小碎片,并将这些碎片扩增、序列化和定位回原始位置来实现的。
通过重复这个过程,我们可以构建出完整的基因组序列,从而对微生物物种进行深入的研究和分析。
2. 全基因组重测序技术在微生物学研究中的应用全基因组重测序技术可以解决许多微生物学领域的研究问题。
例如,在微生物的功能研究中,通过全基因组重测序技术可以发现微生物环境中的微生物种类和数量,并确定它们在特定功能的发挥中的作用。
此外,通过对微生物中个体突变的分析,可以检测到微生物中与疾病相关的突变,并进一步阐明疾病的病理生理机制。
在微生物物种的系统发育和分类研究中,全基因组重测序技术同样具有重要的作用。
利用序列数据进行分析,可以得到微生物物种的系统分类树,研究微生物群落中物种的构成和演化关系。
此外,在微生物种群的遗传多样性研究中,全基因组重测序技术几乎已经成为了标准的研究工具,对于不同微生物物种之间的遗传多样性进行深入的比较和分析。
3. 全基因组重测序技术的优缺点全基因组重测序技术的优点在于快速、准确、灵敏和可重复性高,可以为我们提供比较全面的微生物物种信息。
此外,全基因组重测序技术对于检测微生物中新的基因和功能也有很大的帮助,有助于进一步挖掘微生物生物学的潜力。
然而,全基因组重测序技术也存在一些局限性。
其中最明显的问题是重测序过程中的DNA损失和断裂,这可能导致测序结果的不准确性。
此外,全基因组重测序技术对于不同物种之间的比较和分析存在一定的局限性,需要结合其他分析方法来解决。
基于全基因组序列的物种分类与进化研究

基于全基因组序列的物种分类与进化研究随着科技的进步和高通量测序技术的发展,全基因组测序已成为物种分类和进化研究中的重要手段。
全基因组测序是指对某一物种的基因组DNA的大部分区域进行DNA序列测定,并在其中识别基因和其它功能性区域,通过分析这些信息来了解基因组的结构和功能。
全基因组测序可以在分子水平上揭示物种的演化历史,从而使科学家更好地理解生命的起源和进化。
物种分类和进化研究是生物学的两个重要领域,早在达尔文时代,人们就开始对物种的进化关系进行研究。
传统的物种分类主要依据形态特征和比较遗传信息进行,而全基因组测序则提供了更为全面和准确的遗传信息来源,使得物种分类和进化研究进入了一个全新的阶段。
通过全基因组测序,科学家可以对不同种类的基因组进行比较,从而了解它们之间的相似性和差异性,进而对它们进行分类。
最近25年来,17个鸟纲类、17个哺乳纲类和3个爬行纲类的全基因组测序数据已经获得。
这些数据使得科学家能够更好地理解不同物种之间的演化历史,包括它们是如何分化成各自的物种以及这些物种之间的亲缘关系。
基于全基因组测序的物种分类和进化研究中的一个重要问题是如何将众多基因组序列相互进行比较。
研究人员通常会对物种的基因组序列进行聚类分析,统计相似性指数,使用分子进化树等技术,以识别物种之间的关系。
分子进化树是基于分子序列的邻接法,通过计算基因序列之间的差异性来推断物种的演化历史。
此外,还有许多其他技术,例如计算基因组大小和对基因组中各个区段的编码密度进行比较等。
基于全基因组测序的物种分类和进化研究所产生的数据也带来了许多挑战。
例如,全基因组测序会收集大量的基因组数据,但这些数据也会带来处理数据的困难。
此外,研究人员需要对不同物种之间的形态特征和分子信息进行结合,从而确定各物种之间的分类关系。
总体来说,全基因组序列的遗传信息有助于科学家了解生物的演化历史,从而对其亲缘关系进行分类和进化研究。
虽然全基因组测序可能带来一些技术挑战,但这项技术仍然为科学家提供了一种更为全面、准确和深入的研究动植物进化关系的方法。
基因组测序的概念

基因组测序的概念1. 概念定义基因组测序是指对生物体的全部基因组进行测序的过程。
基因组是一个生物体所有基因的集合,包括编码蛋白质的基因和调控基因等。
基因组测序可以揭示生物体的遗传信息,帮助我们了解生物体的遗传特征、进化历史和疾病相关性等。
2. 重要性2.1 揭示生物多样性和进化关系通过对不同物种的基因组进行测序比较,可以揭示不同物种之间的遗传差异和相似性,进而推断它们之间的进化关系。
例如,通过对人类与其他灵长类动物(如黑猩猩、大猩猩)基因组进行比较,可以揭示人类与这些动物之间的亲缘关系。
2.2 揭示疾病相关基因和个体风险通过对人类个体或群体的基因组进行测序分析,可以发现与特定疾病相关的遗传变异。
这些变异可能涉及某个单一基因或多个基因之间的相互作用。
对于常见疾病(如癌症、心血管疾病)的遗传风险因素的了解,有助于早期预防、诊断和治疗。
2.3 促进个性化医学发展基因组测序可以揭示个体的遗传变异,包括药物代谢能力、易感基因等。
这些信息对于制定个性化治疗方案非常重要。
通过基因组测序,可以预测某些药物在患者体内的代谢速度和效果,从而指导选择最合适的药物和剂量。
2.4 推动科学研究进展基因组测序为科学家提供了大量的遗传信息,有助于推动生物学、医学等领域的科学研究。
通过对不同物种或群体进行大规模基因组测序,可以发现新基因、新功能以及新的生物过程,并深入理解它们在生物系统中的作用。
3. 测序技术目前常用的基因组测序技术主要包括以下几种:3.1 Sanger测序Sanger测序是一种经典且可靠的DNA测序方法。
它通过利用DNA聚合酶合成DNA链的特性,以及加入带有荧光标记的二进制链终止核苷酸(ddNTPs),从而实现对DNA序列的测定。
Sanger测序具有高准确性和较长读长的优点,但其测序速度较慢,且成本较高。
3.2 下一代测序(NGS)下一代测序是指一系列新型高通量测序技术,包括 Illumina HiSeq、Ion Torrent PGM、Pacific Biosciences等。
全基因组测序原理

全基因组测序原理
全基因组测序是一种基因组大规模测序技术,其目的是通过检测某个物种的所有基因组,完成基因组的完全测序和某个物种基因组的细节分析,也就是某个物种全部基因组序列的测序和分析过程。
全基因组测序首先利用酶切技术,将一个基因组长度的DNA片段或单个染色体长的DNA片段分割成较小的片段,避免整个基因组的同时测序产生的种种技术困难。
接着,由于片段的大小大约在200bp-1000bp,所以可以利用多种方法将此片段分叉成若干个物种,然后放在microarray上,实现物种的大规模测序。
整个过程往往利用生物信息技术、定点突变、拷贝数变异等分析技术,以及计算机技术对组学数据进行分析,有效地筛选出有价值的基因序列。
植物基因组测序项目揭示遗传多样性和进化关系

植物基因组测序项目揭示遗传多样性和进化关系【引言】近年来,随着基因组测序技术的飞速发展,人类对植物遗传多样性和进化关系的研究取得了显著的进展。
植物基因组测序项目在揭示遗传多样性和进化关系方面发挥着重要作用。
本文将就此话题展开讨论,探讨植物基因组测序对于我们了解植物遗传多样性和进化关系的重要性以及相关研究成果和未来的发展方向。
【植物基因组测序的重要性】植物基因组测序是一种通过测定植物基因组的DNA序列来研究植物的遗传多样性和进化关系的方法。
它可以提供大量的遗传信息,揭示植物物种的遗传多样性和进化关系,对于我们判断植物物种间的亲缘关系、了解植物的进化历史以及保护和利用自然资源具有重要价值。
【植物遗传多样性的揭示】植物基因组测序项目可通过分析大量植物基因组的DNA序列来揭示植物遗传多样性。
不同植物物种间的基因组序列差异可以用来衡量它们之间的亲缘关系。
通过对不同物种的基因组序列进行比对和分析,可以构建植物物种间的遗传多样性树,确定物种的来源和演化关系。
这不仅有助于我们系统地了解植物的分类演化,还可以为植物育种和保护提供理论依据。
【植物进化关系的揭示】植物基因组测序项目还可以揭示植物的进化关系。
植物基因组测序可以提供丰富的遗传信息,包括基因的结构、功能和调控等。
通过对植物基因组的比较研究,可以了解植物群体的遗传变异和进化过程。
例如,通过比较不同物种的基因组序列,我们可以发现基因的保守和变异部分,这有助于我们研究植物物种的起源、演化路径和适应性进化等重要问题。
【相关研究成果】植物基因组测序项目已经在揭示植物遗传多样性和进化关系方面取得了重要的研究成果。
例如,通过对拟南芥(Arabidopsis thaliana)基因组的测序和分析,我们了解了拟南芥的基因组结构和调控网络,以及它在进化中的起源和演化途径。
类似地,对水稻(Oryza sativa)基因组的测序和分析揭示了水稻的起源和适应性进化,为水稻育种提供了重要的线索。
银杏全基因组测序及生物信息学分析

银杏全基因组测序及生物信息学分析1. 本文概述随着生物科学技术的飞速发展,基因组测序已成为解析生物种类遗传特征、生长发育机制及进化历史的重要手段。
银杏(Ginkgo biloba L.),作为一种古老的植物,具有极高的科学研究价值。
银杏全基因组测序及生物信息学分析的研究,不仅有助于揭示银杏独特的生物学特性,而且对于理解植物进化历程具有重要意义。
本文通过对银杏全基因组进行测序,并运用生物信息学方法进行深入分析,旨在为银杏的遗传改良、种质资源保护以及相关药物开发等领域提供科学依据。
本文首先介绍了银杏全基因组测序的方法和结果,然后对银杏基因组的结构特征进行了详细分析,最后探讨了银杏基因在生长发育、逆境响应等方面的功能。
本研究不仅丰富了我们对银杏这一古老植物的了解,也为植物基因组学研究提供了新的视角和数据资源。
2. 材料与方法银杏样本来源:本研究选取成年银杏植株作为实验材料,所有样本均来自我国某银杏种植基地。
样本采集:在银杏生长期,采集健康叶片样本,立即冻存于液氮中,并转移至80C冰箱保存,以备后续基因组DNA提取。
基因组DNA提取:采用改良的CTAB法提取银杏基因组DNA,并通过琼脂糖凝胶电泳和紫外分光光度计对DNA的质量和浓度进行评估。
测序策略:采用高通量测序技术,包括Illumina HiSeq Ten平台和PacBio SMRT技术,进行银杏全基因组测序。
文库构建与测序:将提取的基因组DNA进行片段化、末端修复、加A尾,然后连接测序接头,构建测序文库。
通过Illumina HiSeq Ten 平台进行双端测序,利用PacBio SMRT技术进行长片段测序。
质量控制:对原始测序数据进行质量控制,包括去除接头序列、低质量序列等,确保后续分析的准确性。
组装策略:采用从头组装和辅助组装相结合的策略,利用Illumina短读序列和PacBio长读序列进行组装。
组装软件:使用如Canu、Flye等软件进行初步组装,然后利用Pilon、NextPolish等进行优化。
利用基因测序技术研究物种起源与进化关系

利用基因测序技术研究物种起源与进化关系标题:基因测序技术揭示物种起源与进化关系引言:物种起源与进化一直是生物学研究的核心问题之一。
随着DNA测序和分析技术的快速发展,基因测序技术已经成为解答这一问题的重要工具。
本文将探讨基因测序技术如何揭示物种起源与进化关系,并展示一些相关研究的最新发现。
一、DNA测序技术的发展:DNA测序技术经过数十年的发展,从最早的Sanger测序方法到如今的高通量测序技术,不断提高了测序速度和准确性。
高通量测序技术,如Illumina测序平台,使得科学家们能够快速测序大量DNA样品,开启了大规模基因组测序的时代。
二、物种起源与基因测序关系的解析:1. 比较基因组学:通过测序多个物种的基因组,并进行比较分析,科学家们可以揭示出物种间的共享基因和基因组差异。
例如,通过比较人类和其他灵长类动物的基因组,可以了解到人类与大猩猩之间的进化关系。
2. 进化树分析:基因测序技术可以帮助构建进化树,揭示物种起源和进化的关系。
通过比较物种基因组的遗传差异,科学家们可以推断各种生物的演化路径。
例如,通过测序多个哺乳动物的基因组,可以构建哺乳动物的进化树,了解不同物种之间的演化关系。
三、基因测序技术揭示的最新发现:1. 人类起源:通过测序人类和其他人类近亲的基因组,科学家们发现人类与尼安德特人和丹尼索瓦人有共享基因,表明这些物种在某种程度上有基因交流和远古基因交配。
2. 物种演化:基因测序技术也揭示了许多物种的演化关系。
例如,通过测序鸟类基因组,研究者发现红色视觉基因在鸟类中的不同程度的退化,这解释了为何鸟类中不同物种的红色视觉能力存在差异。
结论:基因测序技术在揭示物种起源与进化关系方面发挥着重要作用。
通过密集测序各个物种的基因组,并进行比较分析,我们能够了解到物种间的遗传差异和演化历程。
未来,随着测序技术的不断进步,我们可以预期,更多关于物种起源与进化的谜团将会被揭开。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
已完成植物基因组测序情况(更新至2014年11月)中文名拉丁名发表时间刊物科、属基因组大小拟南芥Arabidopsis thaliana 2000.12 Nature 十字花科、鼠耳芥属125M水稻Oryza sativa. ssp. indica 2002.04 Science 禾本科、稻属466M水稻Oryza sativa. ssp.japonica2002.04 Science 禾本科、稻属466M杨树Populus trichocarpa 2006.09 Science 杨柳科、杨属480M 葡萄Vitis vinifera 2007.09 Nature 葡萄科、葡萄属490M衣藻Chlamydomonasreinhardtii2007.01 Science 衣藻科、衣藻属130 M小立碗藓Physcomitrella pattens 2008.01 Science 葫芦藓科、小立碗藓属480M 番木瓜Carica papaya 2008.04 Nature 番木瓜科、番木瓜属370M 百脉根Lotus japonicus 2008.05 DNA Res. 豆科472 Mb三角褐指藻Phaeodactylumtricornutum2008.11 Nature 褐指藻属27.4M高粱Sorghum bicolor 2009.01 Nature 禾本科、高粱属730M 玉米Zea mays ssp. mays 2009.11 Science 禾本科、玉米属2300M 黄瓜Cucumis sativus 2009.11 Nature Genetics 葫芦科、黄瓜属350M 大豆Glycine max 2010.01 Nature 豆科、大豆属1100M二穗短柄草Brachypodiumdistachyon2010.02 Nature 禾本科、短柄草属260M褐藻Ectocarpus 2010.06 Nature 水云属196M 团藻Volvox carteri 2010.07 Science 团藻属138M蓖麻Ricinus communis 2010.08 NatureBiotechnology大戟科、蓖麻属350M小球藻Chlorella variabilis 2010.09 Plant Cell 小球藻科46M苹果Malus × domestica 2010.09 Nature Genetics 蔷薇科、苹果属742M森林草莓Fragaria vesca 2010.12 Nature Genetics 蔷薇科、草莓属240M可可树Theobroma cacao 2010.12 Nature Genetics 梧桐科、可可属430-Mb 野生大豆Glycine soja 2010.12 PNAS 豆科、大豆属915.4 Mb褐潮藻类Aureococcusanophagefferens2011.02 PNAS 57M麻风树Jatropha curcas 2010.12 DNA Res. 大戟科、麻风树属410M 卷柏Selaginella moellendorffii 2011.05 Science 卷柏属212M枣椰树Phoenix dactylifera 2011.05 Naturebiotechnology棕榈科685M琴叶拟南芥Arabidopsis lyrata 2011.05 Nature Genetics 十字花科、鼠耳芥属206.7 Mb 马铃薯Solanum tuberosum 2011.07 Nature 茄目、茄科、茄属844M条叶蓝芥Thellugiella parvula 2011.08 Nature Genetics 盐芥属140M白菜Brassica rapa 2011.08 Nature Genetics 十字花科、芸薹属485M 印度大麻Cannabis sativa 2011.1 Genome biology 大麻属534M木豆Cajanus cajan 2011.11 Naturebiotechnology豆科、木豆属833M蒺藜苜蓿Medicago truncatula 2011.11 Nature 豆科苜蓿属500M 蓝载藻Cyanophora paradoxa 2012.02 Science 灰胞藻门70M谷子Setaria italica 2012.05 Naturebiotechnology禾本科、狗尾草属490M谷子Setaria italica 2012.05 Naturebiotechnology禾本科、狗尾草属预估510M,组装出400M番茄Solanum lycopersicum 2012.05 Nature 茄科、茄属900Mb 甜瓜Cucumis melo 2012.07 PNAS 葫芦科、甜瓜属450Mb 亚麻Linum usitatissimum 2012.07 Plant Journal 亚麻科、亚麻属373Mb 盐芥Thellungiella salsuginea 2012.07 PNAS 十字花科、盐芥属260Mb 香蕉Musa acuminata 2012.07 Nature 芭蕉科、芭蕉属523Mb 雷蒙德氏棉Gossypium raimondii 2012.08 Nature Genetics 锦葵科、棉属775.2Mb 大麦Hordeum vulgare 2012.1 Nature 禾本科、大麦属 5.1Gb梨Pyrus bretschneideri 2012.11 Genome Research 蔷薇科、梨属527Mb 西瓜Citrullus lanatus 2012.11 Nature Genetics 葫芦科、西瓜属425 Mb 甜橙Citrus sinensis 2012.11 Nature Genetics 芸香科、柑橘属367 Mb 小麦Triticum aestivum 2012.11 Nature 禾本科、小麦属17Gb两种小型藻Bigelowiella natans,Guillardia theta2012.11 Nature 95Mb 87Mb棉花(雷蒙德氏棉)Gossypium raimondii 2012.12 Nature 锦葵科、棉属761.4Mb梅花Prunus mume 2012.12 NatureCommunications蔷薇科、梨属280M鹰嘴豆Cicer arietinum 2013.01 Naturebiotechnology豆科、鹰嘴豆属738Mb橡胶树Hevea brasiliensis 2013.02 BMC Genomics 大戟科、橡胶树属 2.15Gb 毛竹Phyllostachys heterocycla 2013.02 Nature Genetics 竹科、钢竹属 2.075 Gb短花药野生稻Oryza brachyantha 2013.03NatureCommunications禾本科稻属342Mb-362Mb小麦A Triticum urartu 2013.03 Nature 禾本科、小麦属 4.94 Gb 小麦D grassAegilops tauschii 2013.03 Nature 禾本科、小麦属 4.36Gb 桃树Prunus persica 2013.03 Nature Genetics 蔷薇科、梨属265 Mb 丝叶狸藻Utricularia gibba 2013.05 Nature 狸藻科、狸藻属82Mb中国莲Nelumbo nucifera Gaertn 2013.05 Genome biology 睡莲科、莲属929 Mb 挪威云杉Picea abies 2013.05 Nature 松科、云杉属19.6G海洋球石Emiliania huxleyi 2013.06 Nature 定鞭藻纲141.7Mb藻虫黄藻Symbiodinium minutum 2013.07 Current Biology 甲藻门 1.5G 油棕榈Elaeis guineensis 2013.07 Nature 棕榈科、油棕榈属 1.8G枣椰树Phoenix dactylifera 2013.08 NatureCommunications棕榈科、刺葵属671 Mb醉蝶花Tarenaya hassleriana 2013.08 Plant Cell 醉蝶花科、醉蝶花属290 Mb 莲Nelumbo nucifera 2013.08 Plant Journal 睡莲科、莲属879 Mb桑树Morus notabilis 2013.09 NatureCommunications桑科、桑属357 Mb猕猴桃Actinidia chinensis 2013.10 NatureCommunications猕猴桃属616.1 Mb胡杨Populus euphratica 2013.11 NatureCommunications杨属496.5 Mb八倍体草莓F. x ananassa 2013.12 DNA Research 草莓属698 Mb 康乃馨Dianthus caryophyllus L. 2013.12 DNA Research 石竹属622 Mb 甜菜Beta vulgaris ssp. vulgaris 2013.12 Nature 藜科甜菜属566.6 Mb 无油樟(互叶梅)Amborella trichopoda 2013.12 Science 无油樟属748 Mb辣椒Capsicum annuum(Criolo de Morelos334)2014.1 Nature Genetics 辣椒属 3.48G芝麻Sesamum indicum 2014.2 Genome Biology 胡麻科胡麻属274 Mb辣椒Capsicum annuum(Zunla-1)2014.3 PNAS 辣椒属 3.48G火炬松Pinus taeda(Loblollypine)2014.3 Genome Biology 松属23.2G棉花(亚洲棉)Gossypium arboreum 2014.5 Nature Genetics 锦葵科、棉属1694Mb 萝卜Raphanus sativus L. 2014.5 DNA Research 十字花科、萝卜属402Mb甘蓝Brassica oleracea 2014.5 Naturecommunications十字花科、芸薹属630Mb菜豆Phaseolus vulgaris L.2014.6 Nature Genetics 豆科,菜豆属587Mb野生大豆Glycine soja2014.7Naturecommunications豆科、大豆属868 Mb普通小麦Triticum aestivum 2014.7 Science 禾本科17Gb野生西红柿Solanum pennellii 2014.7 Nature Genetics茄科942 Mb非洲野生稻Oryza glaberrima2014.8 Nature Genetics禾本科316 Mb油菜Brassica napus2014.8 Science十字花科630 Mb中果咖啡Coffea canephora 2014.9 Science 茜草科,咖啡属710 Mb茄子Solanum melongena 2014.9 DNA Research 茄科、茄属1093 Mb多个野生大豆Glycine soja 2014.9Naturebiotechnology豆科、大豆属889.33~1,118.34Mb绿豆Vigna radiata 2014.10 Naturecommunications豆科、豇豆属543 Mb啤酒花Humulus lupulus 2014.11 Plant and CellPhysiology大麻科、葎草属 2.57 Gb蝴蝶兰Phalaenopsis equestris2014.11 Nature Genetics 兰科、蝴蝶兰属 1.16 Gb。