医疗器械标签及CE标志
医疗器械CE

分类大的分类上,CE认证中医疗器械的分类有Ⅰ类、Ⅱa类以及Ⅱb类,基础是根据适用范围来确定的。
Ⅰ类是指非侵入式,即只和病人的皮肤接触,不与病人内部的器官或者组织接触类型的医疗器械。
但是,某些虽然不会和人体内部发生接触,但是却对人的体内循环或者代谢有影响的就不算归在这一类了。
一般来说像手术中会用到的导尿管、手术巾这种把病人不能再回到体内的体液收集起来的医疗器械,还有理疗类的石膏、牵引器、还有近几年比较火的静脉曲张袜这种外部作用的医疗器械,还有就是帮助病人移动的各种器械,比如轮椅等都属于Ⅰ类医疗器械的范围。
不过手机血液的血袋就是在另外特别规定一种医疗器械了,属于Ⅱb类。
但是要注意的就是,有一些会参与到体液改变的器械,像是各种滤网、还有血液透析机中分离有害物质的部件等都不是属于Ⅱb类医疗器械的,而是属于Ⅱa类。
普通的敷料,也就是用于皮肤表面的破损和溃烂清洁、保护作用的棉球、棉签、绷带、纱布等,都是算在Ⅰ类医疗器械的。
因为伤口不深,只是表面的问题,不会和真皮层接触。
而用于手术中的那些,回个人体的内部组织、真皮接触的,就是Ⅱb类医疗器械了。
上次说的和人体发生接触,但是只起到连接作用的医疗器械是Ⅱa类,那么相对的,就会有CE认证Ⅱb类医疗器械。
Ⅱb类医疗器械是指,听过这种器械,会改变人体内部的各种液体成分和容量的生物、化学类型医疗器械。
最简单的例子就是在治疗尿毒症中常用的血液透析机,帮助清除人体血液内的有毒物质,造成血液改变的机器。
还有就是通过帮助细胞分离的器械。
还有就是在比较深的伤口在处理的时候必须要用的各种敷料,也属于Ⅱb类医疗器械.用于引导各类型液体或者是人体的血液、体液的注入、输出、储存的,不进入人体的这类器械为定义下的Ⅱa类医疗器械。
各种引流管,氧气输送的管道、手术麻醉时用的指示器、输液器、鼻饲管、用于临时存放人体某器官或者组织的瓶子等,在CE认证医疗器械指令中,都是属于Ⅱa类医疗器械。
但如果厂家制造的某种管道,是不和这些器械连接发生作用的,就要是Ⅱb类了。
关于体外诊断医疗器械的9879EC指令

第6条 标准与技术法规常设委员会
❖ 1.欧洲联盟委员会应得到常设委员会的帮助。
❖ 2.对于法规的建立,首先,欧洲联盟委员会的代表 应向常设委员会提交一份所采取措施的草案。常设 委员会应在主席根据事情的紧急程度规定的期限内 对本草案提出意见,必要时可采取投票方式决定。
第7条 医疗器械常设委员会
❖ 常设委员会中成员国代表的票数应按规定的方式加权,主席 不参加投票。如果设想的措施与常设委员会的意见一致,欧 盟委员会应批准该措施。若不一致,或常设委员会没有提出 意见,欧盟委员会应立即向欧盟理事会提交一项拟采取的措 施提案。欧盟理事会应根据特定多数的原则采取行动。
(d) “自我检测器械”是指制造商规定能够由非专业 人员在家庭环境中使用的任何器械;
eg:易知欣、易知盖
第1条 适用范围、定义
(e) “性能评定器械”是指制造商规定用在医学分析 实验室中或用在其所在场所以外的其他适当环境中 进行一项或多项性能评定研究的任何器械;
第1条 适用范围、定义
(f) “制造商”是指在以其名义将器械投放市场之前 对器械的设计、制造、包装和标志负有责任的自然 人或法人,不管这些工作是制造商完成的还是由第 三方完成的;
2.欧洲联盟委员会应尽快地与有关方面磋商。
3.如果不符合要求的器械加贴了CE标志,主管成员国应对加 贴该标志的任何人采取适当的行动,并应通知欧洲联盟委员会 和其他成员国。
4.欧洲联盟委员会应保证成员国了解该程序的进展情况及结果。
第9条 合格评定程序
所有器械在加贴EC标志时,制造商应实施本指令 所述的程序,并应在将器械投放市场前编写EC合格 声明。
第10条 制造商和器械的注册
❖ 1. 制造商应向其营业注册地点所在的成员国主管当 局通报:营业注册地点的地址、有关试剂、试剂产 品的信息以及任何重大改变(停止投放市场);对 其他器械适当的说明;
CE分类规则
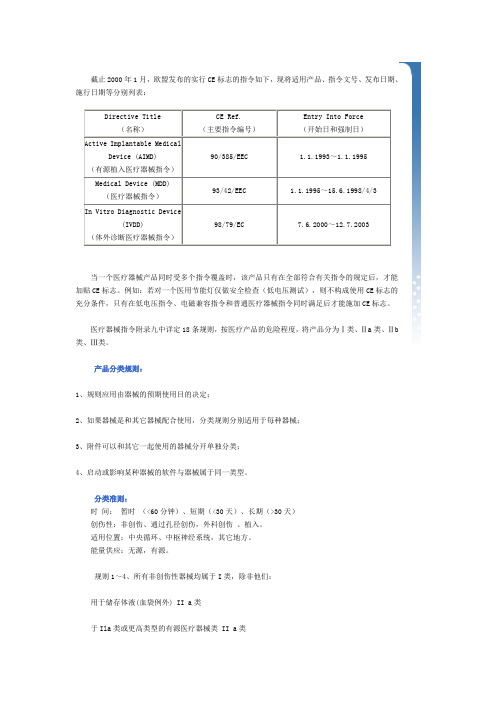
截止2000年1月,欧盟发布的实行CE标志的指令如下,现将适用产品、指令文号、发布日期、施行日期等分别列表:Directive Title (名称)CE Ref.(主要指令编号)Entry Into Force(开始日和强制日)Active Implantable MedicalDevice (AIMD)(有源植入医疗器械指令)90/385/EEC 1.1.1993~1.1.1995Medical Device (MDD)(医疗器械指令)93/42/EEC 1.1.1995~15.6.1998/4/3 In Vitro Diagnostic Device(IVDD)(体外诊断医疗器械指令)98/79/EC 7.6.2000~12.7.2003当一个医疗器械产品同时受多个指令覆盖时,该产品只有在全部符合有关指令的规定后,才能加贴CE标志。
例如:若对一个医用节能灯仅做安全检查(低电压测试),则不构成使用CE标志的充分条件,只有在低电压指令、电磁兼容指令和普通医疗器械指令同时满足后才能施加CE标志。
医疗器械指令附录九中详定18条规则,按医疗产品的危险程度,将产品分为Ⅰ类、Ⅱa类、Ⅱb 类、Ⅲ类。
产品分类规则:1、规则应用由器械的预期使用目的决定;2、如果器械是和其它器械配合使用,分类规则分别适用于每种器械;3、附件可以和其它一起使用的器械分开单独分类;4、启动或影响某种器械的软件与器械属于同一类型。
分类准则:时间:暂时(<60分钟)、短期(<30天)、长期(>30天)创伤性:非创伤、通过孔径创伤,外科创伤、植入。
适用位置:中央循环、中枢神经系统,其它地方。
能量供应:无源,有源。
规则1~4、所有非创伤性器械均属于I类,除非他们:用于储存体液(血袋例外) II a类于Ila类或更高类型的有源医疗器械类 II a类改变体液成分 II a/II b类一些伤口敷料 II a/II b类规则5、侵入人体孔径的医疗器械暂时使用(牙科压缩材料、检查手套) I类短期使用(导管、隐形眼镜) II a类长期使用(正常牙线) II b类规则6-8、外科创伤性器械再使用的外科器械(钳子,斧子) I类暂时或短期使用(缝合针。
医疗器械标签及CE标志

CE标志的意义
进入欧洲市场的通行证
加贴CE标志的医疗器械可以合法地进入欧洲 市场。
提高企业形象
加贴CE标志的医疗器械可以提升企业的形象 和知名度。
提高产品可信度
加贴CE标志的医疗器械可以增加消费者对产 品的信任度。
避免市场风险
加贴CE标志的医疗器械可以避免因不符合相 关指令和标准而产生的市场风险。
。
使用说明
提供简明扼要的说明,指导用 户正确使用医疗器械。
警告和注意事项
对可能存在的风险和注意事项 进行明确标注,确保用户安全
使用。
标签位置要求
01
02
03
明显可见
标签应放置在医疗器械的 明显位置,便于用户查看 。
不影响使用
标签的位置不应妨碍医疗 器械的正常使用和操作。
持久性
标签应具有较好的持久性 ,不易脱落或损坏。
CE标志的要求
符合相关指令要求
医疗器械必须符合相关的欧洲指令要 求,才能加贴CE标志。
符合安全标准
医疗器械必须符合相关的欧洲安全标 准,才能加贴CE标志。
符合质量管理体系要求
医疗器械必须符合ISO 13485质量管 理体系要求,才能加贴CE标志。
符合公告机构要求
医疗器械必须经过公告机构的认证, 才能加贴CE标志。
标签材质要求
防水防腐
标签应具备防水和防腐性 能,以确保在医疗器械的 整个生命周期内保持清晰 可见。
耐磨耐久
标签应具备耐磨和耐久性 ,能够承受医疗器械在使 用过程中的摩擦和碰撞。
可读性强
标签应选用清晰易读的字 体和颜色,确保用户在各 种环境下都能够轻松查看 和理解标签内容。
03
CE标志的要求和意义
英国药品和医疗器械认证标识

英国药品和医疗器械认证标识
英国药品和医疗器械认证标识包括UKCA认证和CE认证。
UKCA认证是医疗器械在英国获得市场准入的合规程序,取代了之前的CE 认证。
对于确保医疗器械符合英国《医疗器械法规》(UK MDR)的要求,该认证非常重要。
原则上,所有要在英国市场销售的较高分类级别的医疗器械和体外诊断医疗器械都需要获得UKCA认证,包括新推出的产品和已经获得批准的产品。
要获得医疗器械UKCA认证,制造商需要委托一家被称作英国认可机构(UK Approved Body)的第三方机构进行认证,认证过程包括产品分类、技术文件评审、质量体系评估和最终认证,必须满足所有相关要求以证实合规性。
CE认证在英国脱欧前是欧盟的产品安全认证标志,英国脱欧后CE认证在北爱尔兰地区仍然有效。
在英格兰、威尔士和苏格兰地区,CE认证标志或带有CE认证的医疗器械产品仍可被接受。
以上信息仅供参考,如需获取更多详细信息,建议咨询专业法律人士。
医疗器械标签及ce标志

目录
• 引言 • 医疗器械标签的重要性 • CE标志的介绍 • 医疗器械标签和CE标志的关联 • 医疗器械标签和CE标志的实例分析 • 总结与展望
01
引言
主题简介
医疗器械标签
医疗器械的标签是向用户提供产品信息的必要手段,包 括产品名称、生产商、使用说明等。
CE标志
CE标志是欧盟对符合相关指令要求的产品的安全认证 标志,医疗器械产品必须加贴CE标志才能在欧盟市场 销售。
够正确使用产品。
标签位置
标签应贴在显眼的位置,方便用 户查看,同时也要考虑不影响产
品的正常使用。
字体和颜色
标签的字体和颜色应清晰易读, 符合相关法规要求,以确保用户
能够准确获取标签信息。
CE标志实例分析
标志位置
CE标志应清晰可见地贴在产品上,以便用户和监管机构识别。
尺寸和颜色
CE标志的大小和颜色应符合相关法规要求,以确保其具有足够的 辨识度。
认证信息
CE标志应包含认证机构名称、认证编号等信息,以便用户和监管 机构核实产品是否符合相关法规要求。
标签与CE标志关联的实例分析
01
02
03
关联方式
医疗器械标签和CE标志应 相互关联,确保用户能够 通过标签了解产品符合CE 认证的情况。
信息一致性
标签和CE标志上的信息应 保持一致,避免出现矛盾 或误导用户的情况。
标签也是医疗器械产品在市场上的宣 传和推广的重要手段,符合要求的标 签可以提高产品的市场竞争力。
标签可以向用户传达产品的基本安全 性和性能信息,帮助用户正确使用产 品,避免因误用而导致的风险。
医疗器械标签和CE标志的实
05
例分析
医疗器械标签及CE标志

93/42/EEC,MDD指令不适用于:
-- A 98/79/EC指令涉及的体外诊断器械 -- B 90/385/EEC 指令涉及的有源植入式器 械 -- C 65/65/EEC指令涉及的药品,包括涉及 的89/381/EEC 指令的来源于血液的药品 -- D 76/768/EEC 指令涉及的化妆品 -- E 人血、人血制品、人血浆或人血细胞 -- F 人体移植物、人体组织或细胞,或含有 人体组织或细胞或由其衍生的制品
特殊的条款(附录VIII和X)允许专用器械和临床 研究用器械被使用而无需带有CE标志。
条款5:符合协调标准的医疗器械被认为满 足基本要求
条款6:标准和技术法规委员会 条款7:医疗器械委员会 条款8:如果发现医疗器械不安全的话,保
护条款允许某个成员国采取行动,召回已 上市产品,或禁止,限制其投放市场
Requirements
Informative notes
Authorized representative in the European Community
欧盟授权 代表
NOTE 1 This symbol is
This symbol shall be accompanied by the name and address of the authorized representative in the European Community, adjacent to the symbol. 该符号应附有欧盟授权代表的名称和地址, 并紧靠该符号
• 附录1:基本要求 • 附录2:完整的质量保证体系 • 附录3:产品型式试验 • 附录4:产品验证 • 附录5:生产质量保证体系 • 附录6:最终产品质量保证体系 • 附录7:自我符合性声明 • 附录8:特殊用途的器械声明 • 附录9:分类规则 • 附录10:临床评估 • 附录11:选择公告机构准则 • 附录12:合格的CE标志
医疗器械指令及CE标志 PPT课件

CE 标志
CE标志的意义 • 表明该器械在欧盟内满足相关指令的基本要求 • 表明该器械在欧盟内被合法地投放市场 • 表明该器械已进行了一个合格评定程序
ppt课件
24
CE 标志
CE标志加贴的地方
• 尽可能产品本身或其标牌上
• 无菌包装上 • 使用说明书上 • 外包装上 • 其他任何地方
ppt课件
17
医疗器械指令:基本框架
条款17:符合基本要求并通过了相应的符合性评价程序的医疗器械必 须带有CE标志,参照附录XII
条款18:误用CE标志
条款19:禁止或限制的决定
条款20:保密性
条款21:指令的废止和修改 条款22:执行和过渡期 条款23:本规定向各成员国发送
ppt课件
4
医疗器械指令:定义、范围
• 医疗器械:是指制造商预定用于人体以下目的的任何仪器、 装置、器具或其他物品,无论它们是单使用还是组合使用, 包括独立用于诊断和治疗的软件: — 疾病的诊断、预防、监视、治疗或减轻 — 操作或残障的诊断、监视、治疗、减轻或修补 — 解剖学和生理过程的探查,替换或变更 — 妊娠的控制 其作用于人体体表或体内的主要设计作用不是用药理学, 免疫学或代谢的手段获得,但可能有这些手段参与并起一 定辅助作用。 . 附件:本身虽然不是器械,但由其制造商专门指定与器械 一起使用,使其能按照器械制造商预定的器械用途来使用 5 ppt课件 的物品。
ppt课件
28
IVDD产品分类.依据附录2-ListA
Virology: 病毒学 • — HIV Infections (I and II) • — HIV (I+II) • — HTLV I and II Infections • — HTLV (I+II) • — Hepatitis Infections (HBV, HCV, HDV, not HAV)
什么是CE Marking

什么是CE Marking(CE标示)CE Marking(CE标示)是产品进入欧盟境内销售的通行证。
欧盟为了保障其会员国内人民生命与财产安全,陆续订出了许多安全指令,规定出许多需要粘贴CE标志的产品,如机械、低电压电气产品、电磁兼容性产品等。
有些产品更强制规定须由核可之验证机构执行验证,取得证明后一律贴上CE标志,始得于欧盟各国销售。
“CE”标志是一种安全认证标志,被视为制造商打开并进入欧洲市场的护照。
凡是贴有“CE”标志的产品就可在欧盟各成员国内销售,无须符合每个成员国的要求,从而实现了商品在欧盟成员国范围内的自由流通。
在欧盟市场“CE”标志属强制性认证标志,不论是欧盟内部企业生产的产品,还是其他国家生产的产品,要想在欧盟市场上自由流通,就必须加贴“CE”标志,以表明产品符合欧盟《技术协调与标准化新方法》指令的基本要求。
这是欧盟法律对产品提出的一种强制性要求。
CE两字,是从法语“Communate Europpene”缩写而成,是欧洲共同体的意思。
欧洲共同体后来演变成了欧洲联盟(简称欧盟)。
近年来,在欧洲经济区(欧洲联盟、欧洲自由贸易协会成员国,瑞士除外)市场上销售的商品中,CE标志的使用越来越多,CE标志加贴的商品表示其符合安全、卫生、环保和消费者保护等一系列欧洲指令所要表达的要求。
在过去,欧共体国家对进口和销售的产品要求各异,根据一国标准制造的商品到别国极可能不能上市,作为消除贸易壁垒之努力的一部分,CE应运而生。
因此,CE代表欧洲统一(CONFORMITE EUROPEENNE)。
事实上,CE还是欧共体许多国家语种中的"欧共体"这一词组的缩写,原来用英语词组EUROPEAN COMMUNITY缩写为EC,后因欧共体在法文是COMMUNATE EUROPEIA,意大利文为COMUNITA EUROPEA,葡萄牙文为COMUNIDADE EUROPEIA,西班牙文为COMUNIDADE EUROPE等,故改EC为CE。
医疗器械的标志和标签解读

标志与标签关系
01
互补关系
标志和标签在医疗器械的标识体系中相互补充,共同构成了完整的信息
传递系统。标志以图形或文字形式提供简洁明了的信息,而标签则提供
更为详细的说明和解释。
02
层级关系
在信息传递的层级上,标志通常位于较高的层级,用于快速识别和传达
关键信息;而标签则位于较低的层级,提供更为详尽的信息供使用者深
若发现标志和标签模糊不清或损坏严重,应及时更换新 的标志和标签。
在医疗器械维修、保养过程中,应注意保护标志和标签 的完整性,避免损坏或污染。
医疗器械生产企业应建立标志和标签管理制度,明确更 新和维护的流程和责任人。
06
实例分析:某品牌医疗器械标志和标签解 读
实例背景介绍
医疗器械品牌:某国际知名品 牌
FDA认证标志
表示该医疗器械已经通过美国食品药品监督管理局(FDA)的审核和认证,可以在美国市场销售和使用。FDA认 证标志是进入美国市场的必要条件之一。
04
医疗器械常见标签解读
产品名称及型号规格标签
产品名称
标识符号
标明医疗器械的通用名称,应与注册 证或备案凭证上的产品名称一致。
部分医疗器械会有特定的标识符号, 如无菌标识、一次性使用标识等。
01
02
03
04
标志和标签应清晰、易读、持 久,确保在医疗器械的整个生
命周期内保持清晰可见。
设计应简洁明了,避免使用容 易产生误解的图形和文字。
应使用对比强烈的颜色,以便 在各种光线条件下都能清晰识
别。
对于小型医疗器械,标志和标 签应适当缩小,但仍需确保内
容的清晰度和可读性。
粘贴位置及方式要求
标志和标签应粘贴在医疗器械的显著 位置,方便用户和使用者查看。
医疗器械ce认证标准详细解读

医疗器械ce认证标准详细解读一.医疗器械CE认证标准1.1 概述在欧洲市场销售的医疗器械需要符合欧盟制定的CE认证标准。
CE认证标准是指医疗器械必须符合欧洲经济区(EEA)的法律要求,以确保其安全性、有效性和符合环境保护要求。
1.2 CE认证流程医疗器械CE认证的流程包括以下步骤:- 提交申请:制造商或其授权代表填写申请表格并提交相关文件。
- 技术文件评审:CE认证机构对技术文件进行评审,包括设计文件、制造文件、验收文件等。
- 制造商评估:CE认证机构会进行现场审核,确认制造商是否符合相关要求。
- 认证决策:CE认证机构根据评审和审核结果,作出认证决策。
- 发放证书:认证通过后,CE认证机构会发放CE认证证书。
1.3 CE标志通过CE认证的医疗器械可以在欧洲市场上销售,并需要在产品上贴上CE标志。
CE标志是制造商声明其产品符合欧洲法律要求的标志。
二.CE认证标准的详细解读2.1 相关法规CE认证标准的基础是欧盟制定的相关法规,包括以下几个主要法规:- 医疗器械指令(Medical Device Directive,MDD) - 医疗器械法规(Medical Device Regulation,MDR) - 原材料法规(Material Regulations)- 环境法规(Environmental Regulations)2.2 技术文件要求制造商需要提交完整的技术文件,以证明其医疗器械符合CE认证标准。
技术文件的要求包括以下内容:- 设计文件:包括产品规格、设计图纸、工艺流程等。
- 制造文件:包括材料清单、组装过程、质量控制流程等。
- 验收文件:包括测试报告、检验记录、验证文件等。
2.3 环境评估要求制造商需要对其医疗器械的环境影响进行评估,并采取相应的环境保护措施。
环境评估要求包括以下内容:- 原材料选择:选择符合环境要求的原材料。
- 废弃物处理:制定废弃物处理方案,确保符合环保要求。
- 能源消耗控制:尽量减少能源消耗,减少对环境的影响。
医疗器械产品标签要求详解

医疗器械产品标签要求详解随着医疗技术的发展与进步,医疗器械产品在我们日常生活中的应用越来越广泛。
而医疗器械产品标签作为产品的重要组成部分,对于产品的质量、安全性和使用说明起到了至关重要的作用。
本文将详细探讨医疗器械产品标签的要求和规范。
一、标签的基本要求医疗器械产品标签是提供给使用者、患者以及医疗机构等相关方的重要信息载体,其信息准确、明确且易于理解是至关重要的。
因此,医疗器械产品标签应符合以下基本要求:1. 信息完整性:标签应包含产品名称、型号规格、批号、注册证号、生产日期、有效期、生产厂家、使用方法、注意事项等关键信息。
这些信息旨在帮助使用者正确使用产品,并在必要时追溯产品的生产和使用情况。
2. 清晰易读:标签上的文字和数字应清晰可辨,字号大小适中,以确保使用者能够方便地阅读和理解标签上的信息。
此外,适当的行距、段落分隔和标点符号的使用也有助于提高标签的可读性。
3. 物理耐久性:医疗器械产品常常需要在特殊环境下使用,因此标签应具备较强的耐磨、耐腐蚀和耐高温等物理性能。
这样可以确保标签在产品使用过程中不易脱落、模糊或变形。
4. 标签材质选择:医疗器械产品标签的材质应符合相关的法规和标准要求。
常见的标签材质包括纸质标签和塑料标签,其选择应根据产品的特性、用途和环境条件来确定。
5. 标签附着方式:医疗器械产品标签应能够牢固地附着在产品上,并且不易被人为或环境因素所破坏。
常见的标签附着方式包括胶粘剂、热熔胶和螺纹等方式,使用时应根据产品的特性和使用环境来选择合适的附着方式。
二、标签内容要求医疗器械产品标签的内容要求与产品的特性和用途密切相关。
除了基本要求中提到的必要信息外,医疗器械产品标签还应包含以下内容:1. 使用说明:详细描述产品的使用方法、操作步骤和注意事项,并提供示意图、图表或配套说明书等以帮助使用者正确操作和使用产品。
2. 成分和材料:如适用,标签上应清楚地标注产品的成分和材料,以便患者或使用者能够了解产品的物质组成,避免对过敏源造成不良反应。
医疗器械CE认证及MDD指令介绍

医疗器械CE认证及MDD指令介绍CE认证CE是Conformité Européene的缩写,意为“符合欧洲要求”。
CE认证是欧洲共同市场的准入标志,表示产品符合欧洲对质量、安全、环保等要求的标准。
对于医疗器械来说,获得CE认证意味着该产品已经通过了相应的评估程序,符合欧盟的相关法规要求。
医疗器械CE认证是基于欧洲理事会93/42/EEC指令(MDD指令)的要求进行的。
该指令规定了医疗器械制造商需要适用的技术要求、质量规范、性能评价和临床实验等方面的要求。
只有通过了符合相应技术要求的医疗器械,才能够获得CE认证,并且在欧洲市场上销售和使用。
MDD指令MDD指令全称为Council Directive 93/42/EEC,是欧盟于1993年制定的一项关于医疗器械的指令。
MDD指令的主要目的是确保医疗器械在欧洲市场销售和使用时的质量、安全和性能等方面符合一定的标准。
MDD指令规定了医疗器械的分类、CE认证程序、质量管理体系要求等方面的内容。
研发和制造医疗器械的企业需要根据指令的要求完成技术文件的准备、产品设计和开发、质量管理体系的建立等工作,并申请CE认证。
根据MDD指令的要求,医疗器械被分为四个类别,从I类到IV类,级别根据其潜在风险的程度逐级增高,从低风险到高风险。
对于不同类别的医疗器械,制造商需要提供不同程度的技术文件和相应的审核程序。
MDD指令还规定了医疗器械质量管理体系的要求,制造商需要建立适当的质量体系,包括文件控制、风险管理、验收测试等环节,以确保产品的质量和安全性。
总结医疗器械CE认证及MDD指令是欧洲对医疗器械质量和安全管理的法规要求。
通过获得CE认证,医疗器械制造商可以在欧洲市场销售产品。
而MDD指令对于医疗器械的分类、CE认证程序、质量管理体系要求等进行了规定。
通过遵循MDD指令的要求,制造商可以确保自己的产品符合欧洲市场的标准。
这些法规体系的建立和遵守,可以保证医疗器械的质量、安全和性能,保护患者和使用者的权益。
ce标志尺寸要求

CE标志尺寸要求概述CE标志是欧洲共同市场认证的标志,用于证明产品符合欧洲法规的关于安全、健康、环保以及消费者保护方面的要求。
CE标志尺寸要求是指在产品上打印或附着CE标志时,需要满足的标志大小尺寸规定。
本文将就CE标志尺寸要求进行全面、详细、完整且深入的探讨。
CE标志的重要性CE标志是欧洲市场准入的重要条件之一,几乎所有希望在欧洲市场销售的产品都需要取得CE认证并贴上CE标志。
CE认证证明产品符合欧洲的相关法规和标准,消费者在购买产品时可以通过CE标志来判断产品的合法性和质量可靠性。
因此,CE标志在欧洲市场中具有重要的指导和信任作用。
CE标志的尺寸要求CE标志的尺寸要求是指在产品上打印或附着CE标志时,CE标志的大小和比例应满足一定的规定。
根据欧洲立法规定,CE标志的尺寸应不小于5毫米,以确保标志清晰可见。
CE标志的最小尺寸根据欧洲指令,CE标志在产品上的最小尺寸为5毫米。
这意味着CE标志的任何一个边长都不能小于5毫米。
CE标志的比例要求CE标志的宽度与高度之比应保持为5:4。
也就是说,CE标志的高度应为宽度的四分之五。
CE标志尺寸的适用范围CE标志尺寸要求适用于几乎所有需要进行CE认证并在欧洲市场销售的产品。
无论是机械设备、电气设备、医疗器械、建筑材料还是消费类产品,只要需要CE认证,都需要在产品上正确地打印或附着CE标志,并满足CE标志尺寸要求。
CE标志尺寸要求的例外情况尽管大多数产品都需要满足CE标志尺寸要求,但也有一些例外情况。
例如,对于比较小的产品或包装较小的产品,如果由于技术原因无法满足CE标志的5毫米尺寸要求,允许使用比5毫米更小的尺寸,但仍然需要保持良好的可见性。
如何正确使用CE标志除了满足CE标志的尺寸要求外,还需要注意以下几点以确保正确使用CE标志:1.只有通过了相应的CE认证的产品才能使用CE标志。
未经CE认证的产品禁止使用CE标志,否则将承担法律责任。
2.CE标志应放置在产品上易于识别的位置,并不能被误解为其他标志。
带ce标志的产品的标签说明及语言的控制程序ok

程序文件XHM -B -04.25-2011 7.5.3带CE 标志的产品的标签、说明及语言控制程序版本号:C 修改状态:0 共2页 第1页1 目的为确保带有CE 标志产品的标签、说明及语言符合93/94/EEC —1993《欧洲共同体理事会法令》的要求。
2 适用范围本程序适用于带CE 标志产品的标签、说明及语言的编制、使用、检验过程的控制。
3 职责3.1 技术科负责设计带CE 标志产品的标签、说明及语言的文件。
3.2 技术科负责带CE 标志产品的标签、说明及语言设计文件的审核。
3.3 质检科负责带CE 标志产品的标签、说明及语言的检验。
3.4 生产车间负责带CE 标志产品的标签、说明及语言的使用。
4 程序4.1 技术科在设计带CE 标准产品的标签、说明及语言的文件时,必须满足EN 980:2008《医疗器械标签中使用的图形符号》的要求,并参照出口供应合同的要求及附录中关于语言的要求。
4.2 技术科对带CE 标志产品的标签、说明及语言的设计文件进行审核后,必须报欧盟代表确认,当技术科审核不能满足要求时,可以委托外部审核。
4.3 质检科对带CE 标志产品的标签,说明及语言的检验,必须依据经欧盟代表确认的关于标签,说明及文字的设计文件。
4.4 生产车间在使用带CE 标志产品的标签、说明及语言时,必须严格执行生产过程控制程序。
4.5 带CE 标志产品的标签、说明及语言文字,一般应包括如下内容和要求。
4.5.1标签的内容和要求:a)公司名称、地址、电话;b)欧盟授权代表名称、地址、电话;c)产品名称、规格、型号、数量、净重、毛重; d)不得重复使用的字样符号: ①字样:For sing usc only ;②符号:③符号表示一次性使用; e)产品经EO 灭菌字样符号: ①字样: Stefled dy EO②符号:③符号表示产品经EO 灭菌; f)产品无毒、无热原的字样; g)请阅读说明的字样或符号: ①字样:Caution ②符号:③符号表示提醒使用者用前阅读使用说明; h)生产批号: ①字样:LOT ②符号:③符号表示生产批号,符号后边紧接着生产批号、用6位数字表示。
医疗器械标签和语言控制程序

文件制修订记录1.0目的根据MDD(93/42/EEC)和EN980、EN1041的要求,本程序文件规定带有CE标志产品的标签和语言控制程序。
2.0范围适用于CE标志产品的标签和语言控制。
3.0职责3.1技术部负责制订标签和语言的有关技术文件;3.2综合部负责标签的采购;3.3生产部负责标签的执行;3.4质量部负责监督执行及确认。
4.0程序4.1标签包含的内容:4.1.1公司的名称和地址;4.1.2使得使用者确认产品及包装的内容所必须的资料(包含产品名称、规格型号、产品配置数量、产品包装资料);4.1.3对无菌医疗器械,须在标签上注明无菌的“STERILE”表示;4.1.4产品序列号,在其前面用符号“SN”表示产品的批号;适用时,产品批号在其前面用符号“LOT”表示;4.1.5适用时,产品应注明使用的期限,为安全起见应采用年月表示;4.1.6当器材为一次性使用,其使用符号“For Single Use”表示;4.1.7产品的生产日期采用年份加月份表示;例:生产日期2022.09表示产品的制造日期为2022年9月。
4.1.8适用时,产品若为特殊订制者(患者专用器械)使用,其使用符号“Custom-made device”表示;4.1.9适用时,产品若为临床调查使用,其使用符号“exclusively for clinicalinvestigations”表示;4.1.10适当时,产品有任何特殊要求贮存、搬运、包装条件;4.1.11产品具有的任何特殊操作说明;4.1.12产品具有的任何警告事项及/或小心事项;4.1.13适用时,代理商的名称和地址;4.1.14出口到欧盟的产品,其标签或外包装箱或使用说明书上必须有CE授权代表的名称和地址;4.1.15适用时,应具有消毒的方法;4.1.16取得CE认证证书后,产品应带有CE标志。
4.2使用说明书的内容主要包括以下内容:4.2.1产品的功能特性及预期用途;4.2.2使用说明书应具有上述标签提到的有关内容,其4.1.4,4.1.6点除外;4.2.3任何不好的副作用;4.2.4当产品在使用或治疗过程中,会造成与其他器械之间产生相互干扰的可能性方面的详细说明;4.2.5与其他器械的连接,必须包含足够详细的相关特性,便于操作者正确使用;4.2.6适当时,包含产品插入时会造成某些危险的资料;4.2.7产品的正确安装、正常使用及安全运作所需的详细资料,以保证产品能安全、持续运作所需的保养和校正说明;4.2.8无菌包装被损坏时所需要的资讯,适当时应提供重新杀菌的详细说明;4.2.9对于可再使用的器材应容许再使用的适当过程,必须包含清洁、消毒、包装的说明。
法规定医疗器材须经验证并附“CE”标志

法规定医疗器材须经验证并附“CE”标志
佚名
【期刊名称】《质量技术监督研究》
【年(卷),期】1995(000)006
【摘要】法国政府日前颁布一项法令(95—292号),规定医疗器材的制造及销售,须经过法国或EU认可的验证机构验证并挂上“CE”标志后方可在市场出售。
该法令实行的目的为使医疗器材挂上“CE”标志后,即可于EU各会员国市场内通【总页数】1页(P19-19)
【正文语种】中文
【中图分类】F203
【相关文献】
1.我国商标法对地理标志保护的探讨——兼评商标法修改稿第七章的规定 [J], 李祖明
2.POCT 法与 ACCESS化学发光法检测心肌标志物的比对 [J], 何啸;管华玲
3.安徽省规定八类收费项目须经集体审议 [J],
4.法国医疗器材产品“CE”标志规定 [J], 无
5.教育部拟规定国家级教学成果奖须经4年以上实践检验 [J], 无;
因版权原因,仅展示原文概要,查看原文内容请购买。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
• 附录1:基本要求 • 附录2:完整的质量保证体系 • 附录3:产品型式试验 • 附录4:产品验证 • 附录5:生产质量保证体系 • 附录6:最终产品质量保证体系 • 附录7:自我符合性声明 • 附录8:特殊用途的器械声明 • 附录9:分类规则 • 附录10:临床评估 • 附录11:选择公告机构准则 • 附录12:合格的CE标志
Title of symbol
Requirements
Informative notes
Manufacturer
制造商
This symbol shall be accompanied by the name and address of the manufacturer (i.e. the person placing the medical device on the market), adjacent to the symbol. 该符号应附有制造商(将医疗器械投放到 市场的人)的名称和地址,并紧靠该符号
93/42/EEC 医疗器械指令
范围
93/42/EEC,MDD指令不适用于:
-- A 98/79/EC指令涉及的体外诊断器械 -- B 90/385/EEC 指令涉及的有源植入式器 械 -- C 65/65/EEC指令涉及的药品,包括涉及 的89/381/EEC 指令的来源于血液的药品 -- D 76/768/EEC 指令涉及的化妆品 -- E 人血、人血制品、人血浆或人血细胞 -- F 人体移植物、人体组织或细胞,或含有 人体组织或细胞或由其衍生的制品
0123
公告机构代号
-- 如被缩小或放大,上述的比例关系应得到保证
-- 如指令没有规定特定的尺寸,CE标志两个字母必须具有 基本相同的垂直高度,且不得低于5mm
-- CE标志必须以清晰、不易擦掉的方式加贴
--小型医疗器械可免于此最低尺寸的限制
CE 标志
• CE符合标志须以明显,清晰、持久的方式 贴附在器械或其无菌包装的适当位置。也 可印附在使用说明书上。适当时,CE标志 也必须加附在销售包装上。
NOTE 1 This symbol is used to indicate information that is required in Europeb NOTE 2 The full definition of “manufacturer” is given in EU Directives 90/385/EEC, 93/42/EEC and 98/79/EC. NOTE 3 Guidance on the requirements for EU Directives 90/385/EEC and 93/42/EEC is given in EN 1041. NOTE 4 The date of manufacture, as well as the name and address of the manufacturer, can be combined in one symbol. 制造日期也可以和制造商的名称和地址 一样结合为一个符号
CE 标志
CE标志的意义 • 表明该器械在欧盟内满足MDD的基本要求 • 表明该器械在欧盟内被合法地投放市场 • 表明该器械已通过了一个相应的符合性评
价程序
CE 标志
CE标志加贴的地方
• 尽可能产品本身或其标牌上 • 无菌包装上 • 使用说明书上 • 外包装上
CE 标志
Conformity European
• 禁止加附任何有可能使第三方误解CE标志 含义或图案的标志或铭文,只有在不影响 CE标志的可见度和清晰度的情况下,其它 标志才可加附于器械、包装或随附器械的 活页说明上。
CE 标志
• 如果某个公告机构参与所进行的符合性评 价程序,则通常加贴有其标识号的CE标志 。由于公告机构需要参与除依据附录Ⅶ的 程序之外全部的符合性评价程序。因此没 有标识号的CE标志仅用于无需灭菌和没有 测量功能的Ⅰ类医疗器械。
基本框架
93/42/EEC指令共有23条款和12附录, 其内容和重点包括:
条款1:本指令适用于医疗器械和其附件 条款2:成员国必须保证医疗器械只有在保证人身
安全和健康时才能被投放市场并投入使用 条款3:一种能保证安全和健康的“器械”是指满
足附录I中基本要求的器械 条款4:带有CE标志的医疗器械可在欧盟内流通。
特殊的条款(附录VIII和X)允许专用器械和临床 研究用器械被使用而无需带有CE标志。
条款5:符合协调标准的医疗器械被认为满 足基本要求
条款6:标准和技术法规委员会 条款7:医疗器械委员会 条款8:如果发现医疗器械不安全的话,保
护条款允许某个成员国采取行动,召回已 上市产品,或禁止,限制其投放市场
条款13:分类改变和公告
条款14:负责将器械投放市场的人员的注 册,欧盟数据库
条款15:临床调查,参照附录X
条款16:公告机构
条款17:符合基本要求并通过了相应的符 合性评价程序的医疗器械必须带有CE标志 ,参照附录XII
条款18:误用CE标志 条款19:禁止或限制的决定 条款20:保密性 条款21:指令的废止和修改 条款22:执行和过渡期 条款23:本规定向各成员国发送
ISO 15223-1:2012)
Medical devices - Symbols to be used with medical device labels, labelling and information to be suppliedReference numb源自r of symbol基本要求
• 安全使用器械所必需的资料应尽量以可行 的和适当的方式显示在器械或其包装上, 必要时也可列于其销售包装上。如果有关 的信息不能显示在器械的包装上,则必须 包含在使用说明书中。每件器械的包装中 必须包括使用说明书。作为例外,如果第 Ⅰ和Ⅱa类器械在没有说明书的情况下可以 安全的使用,则对此两类器械,使用说明 书不是必需的。
条款9:符合性评价程序与产品的分类(附 录IX中给出了分类规则)
条款10:事故报告的要求,执业医师和医 疗机构,制造商和欧盟代表都应向主管当 局报告
条款11:医疗器械必须通过某一些程序( 附录II~VII中的所述)以便证明它们符合基 本要求
条款12:有关系统和打包的医疗器械上市 的特殊程序