2005药典药材含量测定归类
《中国药典》2005版成方制剂和单味制剂明

《中国药典》2005版成方制剂和单味制剂明明目地黄丸拼音名:Mingmu Dihuang Wan英文名:书页号:2005年版一部-492【处方】熟地黄160g山茱萸(制)80g牡丹皮60g 山药80g茯苓60g泽泻60g枸杞子60g菊花60g 当归60g白芍60g 蒺藜60g石决明(煅)80g【制法】以上十二味,粉碎成细粉,过筛,混匀。
每100g粉末用炼蜜35~50g 加适量的水泛丸,干燥,制成水蜜丸;或加炼蜜90~110g制成小蜜丸或大蜜丸,即得。
【性状】本品为黑褐色至黑色的水蜜丸、黑色的小蜜丸或大蜜丸;气微香,味先甜而后苦、涩。
【鉴别】(1) 取本品,置显微镜下观察:淀粉粒三角状卵形或矩圆形,直径24~40μm ,脐点短缝状或人字状。
不规则分枝状团块无色,遇水合氯醛液溶化;菌丝无色或淡棕色,直径4 ~6μm。
薄壁组织灰棕色至黑棕色,细胞多皱缩,内含棕色核状物。
果皮表皮细胞橙黄色,表面观类多角形,垂周壁略连珠状增厚。
薄壁细胞类圆形,有椭圆形纹孔,集成纹孔群。
内皮层细胞垂周壁波状弯曲,较厚,木化,有稀疏细孔沟。
种皮石细胞表面观不规则多角形,壁厚,波状弯曲,层纹清晰。
花粉粒类圆形,直径24~34μm ,外壁有刺,长3 ~5μm,具3 个萌发孔。
纤维直径15~35μm ,壁厚,微木化,有大的圆形纹孔。
果皮纤维木化,上下层纵横交错排列。
不规则团块暗灰色,不透明,加酸后产生气泡。
(2) 取本品水蜜丸18g,研碎;或取小蜜丸或大蜜丸24g,切碎。
加乙醚10ml使湿润,加石油醚(30~60℃)40ml,超声处理15分钟,静置过夜,滤过,滤液挥干,残渣加丙酮2ml 使溶解,作为供试品溶液。
另取丹皮酚对照品,加丙酮制成每1ml含1mg 的溶液,作为对照品溶液。
照薄层色谱法(附录ⅥB)试验,吸取供试品溶液6μl 、对照品溶液4μl,分别点于同一硅胶G薄层板上,以环己烷-乙酸乙酯(3:1)为展开剂,展开,取出,晾干,喷以盐酸酸性5 %三氯化铁乙醇溶液,在105 ℃加热约5 分钟。
《中国药典》2005版中药材自然分类索引编制及分析

在读 中药学博 士生, 博士学位 , 主要从事 中药品种、 品质 与药效研究 工作.
・
1 53 ・ 8
维普资讯
时珍 国医国药 20 0 6年第 1 7卷第 9期
续表 2 序号 科名
LSI H NM DCN N A E I E IA R S A C 06V L 1 N . I I E E IIEA DM T RA M DC E E R H20 O .7 O 9 IZ
《 中国药典) 0 5版 I ( )0 2 部 简称新 版药典 , 下同 ) 与历版药 典
一
表 2 双子 叶 植 物 药 材 分 类
Байду номын сангаас
样, 在编制时收集了药 材及其制品的 中文名 、 汉语拼音 、 拉丁名 和拉丁学名 , 在编制索引时采用 了笔 画索引 、 中文索引 、 汉语拼音 索引 以及拉丁名索引等 四种惯 用 的索引方 法。笔者按 照 自然分 类 系统 的原 则 , 根据 药 典 收 载 药 材 及 制 品 的 自然 属 性 , 其 中 的 对
《 国药典 ) 0 5版 中药 材 中 ) 0 2 自然 分 类 索 引 编 制 及 分 析
王 光 志 ,万德 光 ( 都 中医药大 学 , 成 四川 成 都 60 7 ) 10 5
摘 要 : 05 中国药典》I 对20 版《 部收我的药材及饮片、 植物油脂及提取物按照自然分类系统的方法进行 了分类编制和分
苦参含量测定

苦参含量测定出处:网络转载2009.08.17 16:16 来源:中药网责任编辑:信息部浏览2943次分享到:【导读】苦参注射液生物碱类成分指纹图谱的研究苦参为豆科植物苦参Sophora flavescens Ait的干燥根。
2005中国药典以苦参碱和氧化苦参碱为对照,采用高效液相色谱法测定其含量。
文献报道的苦参碱和氧化苦参碱的含量测定方法较多,主要.苦参注射液生物碱类成分指纹图谱的研究苦参为豆科植物苦参Sophora flavescens Ait的干燥根。
2005中国药典以苦参碱和氧化苦参碱为对照,采用高效液相色谱法测定其含量。
文献报道的苦参碱和氧化苦参碱的含量测定方法较多,主要为高效液相色谱法和薄层扫描法,另外还有GC、毛细管电泳法、离子交换法等。
一、高效液相色谱法中药网⑴2005版中国药典苦参含量测定项下:色谱条件:以氨基键合硅胶为填充剂;以乙腈-无水乙醇-3%磷酸溶液(80:10:10)为流动相;检测波长为220nm。
理论板数按氧化苦参碱峰计算应不得低于2000。
供试品溶液的制备:取本品粉末(过三号筛)约0.3g,精密称定,置具塞锥形瓶中,加浓氨试液0.5ml,精密加入三氯甲烷20ml,密塞,称定重量,超声处理(功率250W,频率33KHz)30分钟,放冷,再称定重量,用三氯甲烷补足减失的重量,摇匀,滤过。
精密量取续滤液5ml,通过中性氧化铝柱(100~200目,5g,内径1cm),依次以三氯甲烷、三氯甲烷-甲醇(7:3)各20ml洗脱,收集洗脱液,回收溶剂至干,残渣加无水乙醇适量使溶解,并转移至10ml量瓶中,加无水乙醇稀释至刻度,摇匀,即得。
⑵文献报道的HPLC法苦参药材的含量测定:张杰等用反相高效液相色谱法(RP-HPLC)测定苦参药材的含量:采用岛津LC10ATvp型高效液相色谱仪(日本岛津公司);Lambdal6紫外分光光度仪(美国PE公司);十八烷基硅烷键合硅胶柱(250mm?.6mm,5μm);乙腈-水(加0.04%三乙胺,以磷酸调pH7.0)(20:80)为流动相;流速:1.0mL/min;室温;紫外检测波长为215nm。
中国药典2005版

中国药典2005版中国药典2005版是我国药品管理局颁布的一部重要的药典标准,对我国临床医药工作起着至关重要的作用。
本文将对中国药典2005版进行全面介绍,包括其背景、内容和应用。
一、背景中国药典2005版是我国自1956年首次出版的中国药典以来的第十五版,于2005年正式颁布。
中国药典编委会负责统筹药典的编纂工作,其组成人员由国内知名的药学专家和学者组成。
二、内容中国药典2005版主要包括以下几个方面的内容:1. 药典编外部分:包括了药典的任务、药标核定原则、药品质量管理体系、词语的使用规范等。
2. 普通药材标准:对常用的草药、动植物组织、动物药材等进行了详细的检验和质量控制标准的规定。
3. 饮片标准:对饮片的质量、性状、理化指标以及微生物限度等方面进行了规定。
4. 中药制剂标准:包括了液体制剂、散剂、丸剂、软胶囊、糖衣片、注射剂等各种中药制剂的质量控制要求。
5. 化学药品标准:对化学药品的纯度、理化指标、微生物限度、重金属限度等方面进行了规定。
6. 辅料标准:对药品中常用的辅料如溶剂、稳定剂、吸附剂等的质量要求进行了规定。
7. 药材提取物标准:对药材提取物的成分测定、理化性质、微生物限度等方面进行了规定。
8. 理化检验:对药品的溶液浓度、酸度、含量测定、化学反应、物理性质等进行了详细的规定与分析方法。
三、应用中国药典2005版的应用范围非常广泛。
首先,医药生产企业必须严格按照中国药典2005版的标准进行生产,确保产品质量。
其次,药品监管机构会对生产企业进行药典标准的检查和抽查,对不符合标准的产品进行处罚。
此外,临床医生在用药过程中也需要参考中国药典2005版的相关标准,确保药物的质量和安全性。
对于研究人员和科研机构而言,中国药典2005版的标准也是他们研究和论文撰写的重要参考资料。
总结起来,中国药典2005版是我国药品管理工作的重要依据。
它对药品质量的控制起着重要作用,也为医药行业的发展提供了可靠的标准和依据。
中国药典2005年版一部标准凡例

一、本部正文分三部分排列:药材及饮片;植物油脂和提取物;成方制剂和单味制剂。
二、正文品种中文名称按笔画数顺序排列,同笔画数的字按起笔笔形─丨ノ丶フ顺序排列;单列的饮片排在相应药材的后面,制剂中同一品种凡因规格不同而臻主标准内容不可须单列者,在其名称后加括号注明规格;附录包括制剂通则、通用检测方法和指导原则,按分类编码;索引分别按中文索引、汉语拼音索引、拉丁名索引和拉丁学名索引顺序排列。
三、每一品种项下根据品种和剂型不同,按顺序可分别列有:⑴中文名称(必要时用括号加注副名),汉语拼音名与拉丁名;⑵来源;⑶处方;⑷制法;⑸性状;⑹鉴别;⑺检查;⑻浸出物;⑼含量测定;⑽性味与归经;⑾功能与主治;⑿用法与用量;⒀注意;⒁规格;⒂贮藏;⒃制剂等。
项目与要求四、药材的质量标准,一般按干品规定,特殊需用鲜品者,同时规定鲜品的标准,并规定鲜品用法与用量。
五、药材原植(动)物的科名、植(动)物名、学名、药用部位(矿物药注明类、族、矿石名或岩石名、主要成分)及采收季节和产地加工等,均属各该药材的来源范畴。
药用部位一般系指已除去非药用部分的商品药材。
采收(采挖等)和产地加工即对药用部位而言。
六、药材产地加工及炮制规定的干燥方法如下:(1) 烘干、晒干、阴干均可的,用“干燥”;(2) 不宜用较高温度烘干的,则用“晒干”或“低温干燥”(一般不超过60℃);(3) 烘干、晒干均不适宜的,用“阴干”或“晾干”;(4) 少数药材需要短时间干燥,则用“曝晒”或“及时干燥”。
制剂中的干燥系指烘干,不宜高温烘干的用“低温干燥”。
七、同一名称有多种来源的药材,其性状有明显区别的均分别描述。
先重点描述一种,其他仅分述其区别点。
分写品种的标题,一般采用习用的药材名。
没有习用名称者,采用植(动) 物名。
八、性状项下记载药品的外观、质地、横断面、臭、味、溶解度以及物理常数等。
(1) 外观性状是对药品的色泽和外表的感官描述。
(2) 溶解度是药品的一种物理性质。
中药材炮制通则(药典2005版)

中药材炮制通则(药典2005版)药材炮制系指将药材通过净制、切制、炮炙处理,制成一定规格的饮片,以适应医疗要求及调配、制剂的需要,保证用药安全和有效。
炮制药材的用水,应为饮用水。
炮制药材除另有规定外,应符合下列有关要求。
一、净制即净选加工。
经净制后的药材称为“净药材”。
凡供切制、炮炙或调配制剂的,均应使用净药材。
净制药材可根据其具体情况,分别选用挑选、风选、水选、筛选、剪、切、刮削、剔除、刷、擦、碾串火燎及泡洗等方法达到质量标准。
二、切制药材切制时,除鲜切、干切外,须经浸润使其柔软者,应少泡多润,防止有效成分流失。
软化处理方法有:喷淋、抢水洗、浸泡、润、漂、蒸。
并应按药材的大小、粗细、质地等分别处理。
注意掌握气温、水量、时间等条件。
切后应及时干燥,保证质量。
切制品有片、段、块、丝等。
其厚薄、长短、大小、宽窄通常为:片极薄片0.5mm以下,薄片1~2mm,厚片2~4mm;段短段5~10mm,长段10~15mm ;块 8~12mm的方块;丝细丝2~3mm,粗丝5~10mm。
其他不宜切制的药材,一般应捣碎用。
三、炮炙除另有规定外,常用的炮炙方法和要求如下。
1.炒炒制分清炒和加辅料炒。
炒时应火力均匀,不断翻动。
掌握加热温度、炒制时间及程度要求。
清炒取净药材置热锅中,用文火炒至规定程度时,取出,放凉。
需炒焦者,一般用中火炒至表面焦黄色,断面色加深为度,取出,放晾。
炒焦后易燃药材,可喷淋清水少许,再炒干或晒干。
麸炒取麸皮,撒在热锅中,加热至冒烟时,加入净药材,迅速翻动,炒至药材表面呈黄色或色变深时,取出,筛去麸皮,放凉。
除另有规定外,每100kg净药材,用麸皮10kg。
2.烫烫法常用的辅料为洁净河沙、蛤粉或滑石粉。
取河沙(蛤粉、滑石粉)置锅内,一般用武火炒热后,加入净药材,不断翻动,烫至表面鼓起、酥脆或规定的程度时,取出,筛去辅料,放凉。
如需醋淬时,筛去辅料后,趁热投入醋中淬酥。
3.煅煅制时应注意煅透,使酥脆易碎。
中华人民共和国药典2005年版

八、除另有规定外,片剂应密封贮存。 除正文品种另有规定外,片剂应进行以 下检查。
食品论坛 /
【重量差异】片剂重量差异的限度,应 符合下列有关规定。
平均片重或标示片重 0.30g以下
0.30g至0.30g以上
重量差异限度 ±7.5% ±5%
食品论坛 /
检查法 取供试品20片,精密称定总 重量,求得平均片重后,再分别精密称定 每片的重量,每片重量与平均片重相比较 (凡无含量测定的片剂,每片重量应与标 示片重比较),超出重量差异限度的不得 多于2片,并不得有1片超出限度1倍。
一、原料药与辅料混合均匀。含药量 小或含毒、剧药物的片剂,应采用适宜方法 使药物分散均匀。
食品论坛 /
二、凡属挥发性或对光、热不稳定 的药物,在制片过程中应遮光、避热,以 避免成分损失或失效。
三、压片前的物料或颗粒应控制水 分,以适应制片工艺的需要,防止片剂在 贮存期间发霉、变质。
咀嚼片不进行崩解时限检查。 凡规定检查溶出度、释放度的片剂, 不再进行崩解时限检查。
食品论坛 /
【发泡量】阴道泡腾片照下述方法 检查,应符合规定。
检查法 取25ml具塞刻度试管(内 径1.5cm)10支,各精密加水2ml,置 37℃±1℃水浴中5分钟后,各管中分别 投入供试品1片,密塞,20分钟内观察最 大发泡量的体积,平均发泡体积应不少 于6ml,且少于3ml的不得超过2片。
食品论坛 /
【分散均匀性】分散片照下述方法 检查,应符合规定。
检查法 取供试品2片,置 20℃±1℃的100ml水中,振摇3分钟,应 全部崩解并通过2号筛。
【微生物限度】口腔贴片、阴道片、 阴道泡腾片和外用可溶片照“微生物限度 检查法”(附录Ⅺ J)检查,应符合规定。
中药质量标准与05版药典

品种及其数据资料(包括标准起草依据、复核数据、 专家审定意见、历版药典及国外标准情况、国内生 产产品质量情况等):无
三、现行中药质量标准 若干问题讨论
1、关于中药质量标准的质量可控性
(1)中药与化学药品质量标准的区别
化学药品质量标准:是可以据以直接评价疗效的标准, 其含量高低直接与药效相关; 中药质量标准:还不是可以据以直接评价疗效的标准, 只能是药品生产与监督部门为保证不同批次药品质量的 一致和稳定,判断产品真伪优劣的尺度和依据。 中药标准控制的指标成分大多只能算作指标性成分。
• •
老药品管理法:包含国家药品标准和地方药品标准 新药品管理法:除中药饮片外,均执行国家药品标准。但实际上,中药 材也执行省级药品标准。
试行标准:新药经批准生产后,其药品标准为试行标准,试行期2年。 其它药品经批准后,需要进一步考察生产工艺及产品稳定性的,其药 品标准也可批准为试行标准 正式标准:试行期满转正后即为正式标准
一、国家药品标准管理-工作程序
4、国家药品标准的使用——药品标准的法定强制 性(药品必须符合国家药品标准) 药品经营、使用 药品监督检验
• •
药品检验机构需依据国家药品标准进行检验,出具 检验报告 标准增加检验内容:国家药品标准规定的检验方法 和检验项目不能检验时,药品检验机构可以补充检 验方法和检验项目进行药品检验,但需经国家局批 准后,才可作为认定药品质量的依据
(4)新药转正标准,现共出45册,其中中药约800种
二、中药质量标准现状-中药制剂 中药质量标准现状-
2、中药制剂国家药品标准-零散的约2000种 中药制剂国家药品标准-零散的约2000种 2000
(1)各种注册标准
中药新药试行标准 中药仿制药注册标准 中药保健药整顿试行标准 中药保护品种注册标准
《中国药典》2005年版Ⅰ部药材部分内容补正

《 国药 典 ) 0 5年版 I部 药 材 部分 内容 补 正 中 ) 0 2
沈 保 安 ( 安徽 省芜 湖市药 品 检验所 2 10 ) 4 0 1
关键 词 : 中国药典》 药材来源; 药材鉴定; 性味归经; 炮制; 内容补正 《 ;
中 标识 码 : C
或与《 药典》 采用 的“ 黑芝麻 ” 名相一致 。 13 香 附( . 莎草) 13 1 原 文 干 燥 根 茎 (8 .. 1 1页 ) 。
一
,
2 3 络石藤 . 2 3 1 原 文 茎呈圆柱形 , .. ……表 面红褐 色 , 点状 皮孔 及不 定 有 根; ……展平后叶片呈椭 圆形或卵状披针形 , 10页) (9 。
的药材来源与鉴定项 内容进行 了认真考察和研究 , 发现其中存 在 17 穿 山 甲 . 错误 、 漏缺和不妥之处 , 现分项予 以补正 , 药品标准制订 人员 和 17 1 原 文 鳞 甲 ( 8 供 .. 19页 ) 。 使 用人员参考 。 17 2 补 正 干 燥 鳞 甲 。 .. 1 药 材 来 源 部 分 17 3 注 解 补 上 “ .. 干燥 ” , 后 既符合 实 际 , 与《 又 药典 》 全书 格 《 中国药典》 凡例第( 条记 载 : 药材原 植 ( ) 的科名 、 式 相统 一 。 五) “ 动 物 植( ) 动 物名 、 拉丁学 名 、 用部位 ( 药 矿物药 注 明类 、 、 石名或 2 药 材 鉴 定 项 族 矿 岩石名 、 主要 成 分 ) 采 收 季 节 和 产 地 加 工 等 , 属 各 该 药 材 的 及 均 本条 专对《 药典》 载 的药 材性状 、 别项 中 的药 材鉴定 内 所 鉴 来源范畴 。 以上药 材来源部分 , ” 除拉 丁学名 另文补正外 , 均在本 容进行补正 。以下 先列 出《 药典 》 的中文 药材 名 , 接着列 出错 漏 条予 以补正 。以下先列 出《 典》的中文药 材名 , 中文植物 名 的原文 , 在原文末 尾加 注《 药 如 并 药典》 的页码 。然后 列 出补正 的 内 与 中 文药 材 名 不 同 , 用 括 号 在 中 文 药 材 名 之 后 加 注 中 文 植 物 容和 注解 。 则
中国药典2005版一部 WORD

阿魏化痞膏
拼音名:Awei Huapi Gao
英文名:
书页号:2005年版一部-483
【处方】香附20g厚朴20g三棱20g莪术20g当归20g
生草乌20g生川乌20g大蒜20g使君子20g白芷20g
穿山甲20g木鳖子20g蜣螂20g胡黄连20g大黄20g
蓖麻子20g乳香3g没药3g芦荟3g血竭3g
本品含总氮(N)量不得少于13.0%。
【炮制】阿胶捣成碎块。
阿胶珠取阿胶,烘软,切成丁,照烫法(附录ⅡD)用蛤粉烫至成珠,
内无溏心。
【性味与归经】甘,平。归肺、肝、肾经。
【功能与主治】补血滋阴,润燥,止血。用于血虚萎黄,眩晕心悸,肌
痿无力,心烦不眠,虚风内动,肺燥咳嗽,劳嗽咯血,吐血尿血,便血崩漏,
度,离心,如法清洗3次,倾去上清液,离心管在105℃加热2小时,取出,置干
燥器中冷却30分钟,精密称定,计算,即得。
本品水不溶物不得过2.0%。
挥发性碱性物质取本品约5g,精密称定,置100ml量瓶中,加水使溶解并稀
释至刻度,摇匀,精密量取5ml,置凯氏烧瓶中,立刻加1%氧化镁混悬溶液
5ml,迅速密塞,通入水蒸气进行蒸馏,以2%硼酸溶液5ml为接收液,加甲基红
色谱法(附录ⅥB)试验,吸取上述两种溶液各10μl,分别点于同一硅胶G薄层
板上,以正丁醇-醋酸-水(2:1:1)的上层溶液为展开剂,展开,取出,晾干,
形,壁一面菲薄。
(2)取本品9g,剪碎,加乙醚20ml,超声处理20分钟,滤过,滤液挥干,残
渣加正己烷0.5ml使溶解,作为供试品溶液。另取α-香附酮对照品,加正己烷
制成每1ml含2μl的溶液,作为对照品溶液。照薄层色谱法(附录ⅥB)试验,
八角茴香(中国药典2005版)

《中国药典》2005年版p4【性状】结论:(标准规定:本品为聚合果,多由8个蓇葖果组成,放射状排列于中轴上,蓇葖果长1-2cm,宽0.3-0.5cm,高0.6-1cm;外表面红棕色,有不规则皱纹,顶端呈鸟喙状,上侧多开裂;内表面淡棕色,平滑,有光泽;质硬而脆。
果梗长3-4cm,连于果实基部中央,弯曲,常脱落。
每个蓇葖果含种子1粒,扁卵圆形,长约6mm,红棕色或黄棕色,光亮,尖端有种脐;胚乳白色,富油性。
气芳香,味辛、甜。
)【鉴别】(1)本品粉末显微鉴别显微镜:奥林巴斯C011装片方法:本品粉末图结论:。
(标准规定:本品粉末红棕色。
内果皮栅状细胞长柱形,长200-546um,壁稍厚,纹孔口十字状或人字状。
种皮石细胞黄色,表面观类多角形,壁极厚,波状弯曲,胞腔分枝状,内含棕黑色物;断面观长方形,壁不均匀增厚。
果皮石细胞类长方形、长圆形或分枝状,壁厚。
纤维长,单个散在或成束,直径29-60um,壁木化,有纹孔。
中果皮细胞红棕色,散有油细胞。
内胚乳细胞多角形,含脂肪油滴和糊粉粒。
)(2)取本品粉末1g,加石油醚(60-90℃)--乙醚(1:1)混合液15ml,密塞,振摇15分钟,滤过,滤液于水浴上挥干,残渣加无水乙醇2ml使溶解,作为供试品溶液。
吸取供试品溶液2ul,点于以羧甲基纤维素钠为黏合剂的硅胶G薄层板上,挥干,再点加间苯三酚盐酸试液约2ul,即显色。
结论:。
(规定应显粉红色至紫红色的圆环。
)(3)精密吸取【鉴别】(2)项下的供试品溶液10ul,置10ml量瓶中,加无水乙醇至刻度,摇匀,照紫外-可见分光光度法(附录V A)测定。
测定结果见第页记录。
结论:本品在nm处,有最大吸收。
(规定在259nm波长处有最大吸收)。
(4)取八角茴香对照药材1g,照【鉴别】(2)项下的供试品溶液制备方法,制成对照药材溶液。
另取茴香醛对照品20ul,加无水乙醇2ml 使溶解,作为对照品溶液。
照薄层色谱法(附录ⅥB)试验,吸取[鉴别](2) 项下的供试品溶液及上述两种溶液各10μl,分别点于同一以羧甲基纤维素钠为黏合剂的硅胶G薄层板上,以石油醚(30~60℃)-丙酮-醋酸乙酯(19:1:1)为展开剂,展开,取出,晾干,喷以间苯三酚盐酸试液。
2005药典一部薄层、含量测定检索表
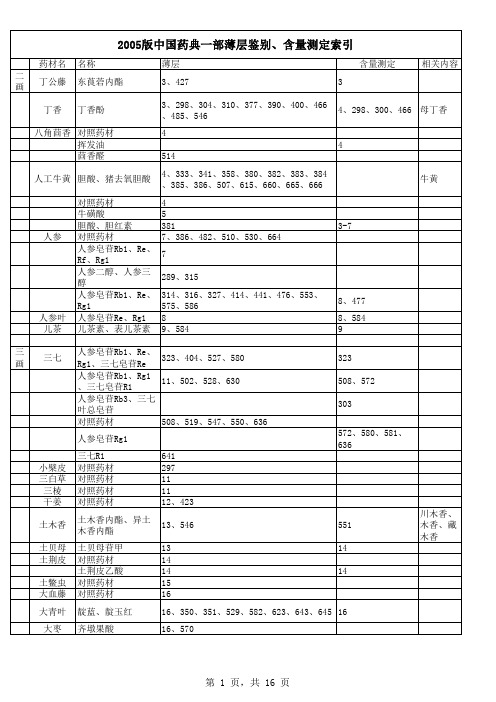
59、294、312、321、322、335、352、354 、355、356、357、358、368、420、423、 435、436、455、479、493、510、519、 564、565、570、585、589、590、601、 608、618、654 59、557、570 60 60 305、349、370、374、388、399、428、 518、520、618、621、625 306、311、399、 480 423、468、479、 520、570 305 60 61 62、452 64 296、297 64、416 65、416 川贝母、湖北贝母 65 、伊贝母、土贝母 66、418、486 418 67 67 67 第 4 页,共 15 页
牛蒡子
对照药材 牛蒡苷
第 3 页,共 15 页
牛膝 毛诃子 升麻 片姜黄 化橘红 丹参
齐墩果酸 对照药材 没食子酸 阿魏酸 异阿魏酸 对照药材 柚皮苷 对照药材 丹参酮ⅡA 原儿茶酸 原儿茶醛 丹酚酸B 丹参素
49、325、361、491、505、570 471 50 50 50 51 51 52、375、452、500、550、584 52、375、394、398、426、481、527、575 、655 361 290、640 52 372、404、501、528、624、653 53 53 54 54 54 55
54
55 55 56 57 56 57 58 58
五画
玉竹 功劳木
58 58
甘草
对照药材
甘草酸铵 甘草苷 甘草次酸
甘草酸 甘青青蓝 对照药材 甘遂 对照药材 石韦 绿原酸 石菖蒲 对照药材 石膏 含水硫酸钙 节裂角茴香 对照药材 龙胆 龙胆苦苷 平贝母 北豆根 仙茅 白及 平贝碱甲 对照药材 蝙蝠葛碱 仙茅苷 对照药材
2005版中国药典西洋参质量标准的修订_王旭

用 HPLC 法测定西洋参中人参皂苷 Rb1 的含量,只能测定其中 1 个成分,此次修订采用梯度洗脱的方法,同时测定西洋参中人
人参及水分的检查同 2000 版药典未作修订,增订总灰 分、酸不溶性灰分浸出物项。
参皂苷 Rg1、Re 与 Rb1 的总量,更全面地控制了质量。 1 仪器、样品与试剂
2.2.1 总灰分[1] 照 2000 版药典灰分测定法测定不同产地 的 10 批西洋参的总灰分,其中最高值为 4 Rg1 and Ginsenoside Re showed the good linear relationships at the range of 4.088~ 20.440 μg (r=0.9999), 0.21~2.13 μg (r=0.9999), 2.5792~12.8960 μg (r=0.9999), respectively. Their average recoveries:Ginsenoside Rb1 was 96.94% (RSD=0.59%), Ginsenoside Rg1 was 99.67% (RSD=1.14%) and Ginsenoside Re was 100.52% (RSD=1.81%), respectively. Conclusion These
·40·
Chinese Journal of Information on TCM
·质量标准研究·
Apr.2005 Vol.12 No.4
2005 版《中国药典》西洋参质量标准的修订
王 旭,王萌萌,邬国庆,张小茜
(北京市药品检验所,北京 100035)
摘要:目的 修订西洋参质量标准,更全面地控制药品质量。方法 采用 HPLC 法,C18 为固定相,用水饱和的正 丁醇回流提取,以流动相 A(乙腈)与流动相 B[水-磷酸(100∶0.1)]进行梯度洗脱;柱温:40 ℃;检测波长:203 nm, 分别控制人参皂苷 Rb1、Re 和 Rgl 的含量。结果 人参皂苷 Rbl 在 4.088~20.440 μg(r =0.9999)、人参皂苷 Rg1 在 0.21~2.13 μg(r =0.9999)、人参皂苷 Re 在 2.5792~12.8960 μg(r =0.9999)范围内呈良好线性关系。加样 回 收 率 人 参 皂 苷 Rb1 为 96.942(RSD=0.59%), 人 参 皂 苷 Rgl 为 99.67%(RSD=1.14%), 人 参 皂 苷 Re 为 100.52%(RSD=1.81%)。结论 此次修订增加了总灰分、酸不溶性灰分、浸出物的检查,并修订了含量测定项。修订 后的标准能更全面地控制药品质量,含量测定方法简便准确,重复性好。
中国药典2005版

江苏省药品检验所 周帼雄
前言
一、概述 《中华人民共和国药典》2005年 版按照第八届药典委员会确定的设计方 案和要求,在全体委员和常设机构工作 人员的努力及各有关部门与单位的支持 下,经过两年多的时间编制完成、成为 建国以来的第八版药典。
本版药典分一部,二部和三部。
舌下片照“崩解时限检查法”(附录Ⅹ A) 检查,除另有规定外,应在5分钟内全部溶化。
口腔贴片 系指粘贴于口腔,经黏膜吸收 后起局部或全身作用的片剂。
口腔贴片应进行溶出度或释放度检查。 咀嚼片 系指于口腔中咀嚼或吮服使片 剂溶化后吞服,在胃肠道中发挥作用或经胃 肠道吸收发挥全身作用的片剂。 咀嚼片口感、外观均应良好,一般应 选择甘露醇、山梨醇、蔗糖等水溶性辅料作 填充剂和黏合剂。咀嚼片的硬度应适宜。 分散片 系指在水中能迅速崩解并均匀 分散的片剂。 分散片中的药物应是难溶性的。分散 片可加水分散后口服,也可将分散片含于口 中吮服或吞服。
【发泡量】阴道泡腾片照下述方法 检查,应符合规定。
检查法 取25ml具塞刻度试管(内 径1.5cm)10支,各精密加水2ml,置 37℃±1℃水浴中5分钟后,各管中分别 投入供试品1片,密塞,20分钟内观察最 大发泡量的体积,平均发泡体积应不少 于6ml,且少于3ml的不得超过2片。
【分散均匀性】分散片照下述方法 检查,应符合规定。
三、 配制注射剂时,可根据药物的性质加入适
宜的附加剂。如渗透压调节剂、pH值调节剂、增溶剂、 助溶剂、抗氧剂、抑菌剂、乳化剂、助悬剂等。所用 附加剂应不影响药物疗效,避免对检验产生干扰,使 用浓度不得引起毒性或过度的刺激。常用的抗氧剂有 亚硫酸钠、亚硫酸氢钠、焦亚硫酸钠,一般浓度为 0.1%~0.2%;常用抑菌剂为0.5%苯酚、0.3%甲酚、 0.5%三氯叔丁醇等。多剂量包装的注射液可加适宜的 抑菌剂,抑菌剂的用量应能抑制注射液中微生物的生 长,加有抑菌剂的注射液,仍应用适宜的方法灭菌, 静脉输液与脑池内、硬膜外、椎管内用的注射液均不 得加抑菌剂。除另有规定外,一次注射量超过15ml的 注射液,不得加抑菌剂。
《中华人民共和国药典》2005年版一部勘误表

《中华人民共和国药典》2005年版一部勘误表页数品名误正11三七【含量测定】测定法“…三者的总量不得少于5.0%。
”【含量测定】测定法“…的总量不得少于5.0%。
”20山豆根【含量测定】第8行“…吸取供试品溶液2~6μl,”【含量测定】第8行“…精密吸取供试品溶液2~6μl,”27川乌【鉴别】(3)“加硫酸液(0.25mol/L)20ml,”【鉴别】(3)“加0.25mol/L 硫酸液20ml,”41云芝【含量测定】单糖精密量取总糖项下的滤液40ml,按总糖项下方法,自“加酚酞指示液l~2滴……”起,同法操作。
【含量测定】单糖精密量取总糖项下的滤液40ml,加酚酞指示液l~2滴,用氢氧化钠试液调节pH值至中性,按总糖项下方法,自“精密加入碘滴定液(0.1mol/L)25m1……”起,同法操作。
52丹参【检查】重金属及有害元素照铅、镉、砷、汞、铜测定法(附录ⅨB原子吸收分光光度法或附录ⅪD电感耦合等离子体质谱法)测定,【检查】重金属及有害元素照铅、镉、砷、汞、铜测定法(附录ⅨB原子吸收分光光度法或电感耦合等离子体质谱法)测定,56水飞蓟【含量测定】第4行“理论板数按水飞蓟宾或异水飞蓟宾峰计算应不低于5000。
”【含量测定】第4行“理论板数按水飞蓟宾峰计算应不低于5000。
”第6行“精密称取水飞蓟宾和异水飞蓟宾混合对照品12mg,”第6行“精密称取水飞蓟宾对照品12mg,”第15行“以水飞蓟宾和异水飞蓟宾峰面积之和计算,”第15行“以水飞蓟宾两个峰面积之和计算,”57水蛭【含量测定】公式中“W”[删去]第16行“W—取样量。
”59甘草【检查】重金属及有害元素照铅、镉、砷、汞、铜测定法(附录ⅨB原子吸收分光光度法或附录ⅪD电感耦合等离子体质谱法)测定,【检查】重金属及有害元素照铅、镉、砷、汞、铜测定法(附录ⅨB原子吸收分光光度法或电感耦合等离子体质谱法)测定,60甘草【含量测定】甘草苷第9行“再称重”【含量测定】甘草苷第9行“再称定重量”第14行“含甘草甘( C21H22O9) 不得少于1.0%”第14行“甘草苷( C21H22O9) 不得少于1.0%”63石膏【检查】砷盐“依法检查(附录ⅨF),”【检查】砷盐“依法检查(附录ⅨF第二法),”68白术【鉴别】(2)“…在与对照品色谱相应的位置上,”【鉴别】(2)“…在与对照药材色谱相应的位置上,”69白芍【检查】重金属及有害元素照铅、镉、砷、汞、铜测定法(附录ⅨB原子吸收分光光度法或附录ⅪD电感耦合等离子体质谱法)测定,【检查】重金属及有害元素照铅、镉、砷、汞、铜测定法(附录ⅨB原子吸收分光光度法或电感耦合等离子体质谱法)测定,81地龙【检查】重金属“…依法检查(附录ⅨE),”【检查】重金属“…依法检查(附录ⅨE第二法),”86西瓜霜【检查】重金属“含量金属不得过百万分之十。
中国药典2005版(中药一部)讲

7
a
(二)一部制剂通则增修订内容介绍 2.1.增修订概况及具体通则(剂型)情况
①概况
概况 通则总数
增订数 修订数 未修订数
05Ch.P 26
3 21 4
8
20Ch.P 26 5 8 13
a
②通则(剂型)具体收载情况
05
1丸剂、2散剂、3颗粒剂、4片剂、5锭剂、6煎
版 共 有 剂 型
膏剂、7胶剂、8糖浆剂、9贴膏剂(巴布膏剂、 橡胶膏剂)、10合剂、11滴丸剂、12胶囊剂、 13酒剂、14酊剂、15流浸膏剂 浸膏剂、16膏 药、17软膏剂、18凝胶剂、19露剂、20茶剂、 21注射剂、22搽剂 洗剂 涂膜剂、23栓剂、24 鼻用制剂(滴鼻剂)、25眼用制剂(滴眼剂)、26
四.每片长度与宽度按中线部位测量,不 得小于标识量 p6
三 . 维 持 含 蔗 糖 量 不 高 于 20 % 的 规 定 (作矫味剂) p8
一.含糖量不低于60%改为不低于45%
(定义,含药材提取物的浓蔗糖水溶液)
单糖浆85﹪g/ml,64.7﹪g/g
药用糖浆65﹪ ,1860﹪g/ml以上
a
注射剂 p12
搽剂 p14
鼻用 制剂 眼用 制剂
四.关于附加剂,可加入渗透压、pH调节 剂、增溶、抗氧、抑菌、乳化、助悬剂 其中抑菌剂:原“注射量超过5ml审慎 选择”改为“一次注射量超过15ml的 注射液不得加”。
四.因溶剂改变的一般质量要求:“以乙 醇为溶剂的检查乙醇量(原相对密度); 油剂除折光率增加相对密度。
我国中药材农药残留污染研究现状

我国中药材农药残留污染研究现状中药是我国宝贵的文化遗产,是中华民族防治疾病、康复保健、繁衍后代的一大法宝。
近年米,世界各国对天然药物的需求日益扩大,尤其对绿色药品尤为关注。
现在,世界每年中药贸易额已达400亿美元,而且每年以10%的速度增长,日前我国中药出口额约6亿美元,仅占世界植物药材市场的3%~5%,这与我国中药大国的地位极不相称。
中药材有效成分的不确定与农药、重金属等有害物质的残留量超标,是中药跻身世界的两大障碍。
我国出口的中草药在欧美等国市场上多次因农药残留超标等原因被查扣,农药残留污染已成为巾药材走向世界的障碍,成为当前中药材生产中急待解决的重要问题。
1中药材中化学农药污染现状和特点1.1中药材中化学农药污染现状目前我国农药市场存在假冒伪劣农药,使之延误病虫害的防治,增加了防治病虫害的难度,使用者无法准确地控制施药剂量和浓度等。
我国制定与中药材生产质量相关的规范、标准起步晚,步伐慢,使得中药材生产行业无法可依,直接导致了现在中药材中安全质量问题积恶难返。
中药材在种植过程中滥用农药,使得大量药剂直接附着在植株上。
由于中药材的种植者缺乏基本的植保常识.未重视农药残留问题,滥用农药的现象十分严重。
有些药材的种植者对农药的选择标准是高效、便宜,很少顾及农药毒性及高残效对中药材质量的影响。
为保产增收,种植者施药次数频繁,且经常是几种高毒农药混配使用,特别是在虫害严重时期,使用农药的浓度加倍,不仪杀死了大量病虫害的天敌,而且使得病虫抗药性增加、防治成本和防治难度越来越大,同时造成了中药材中农药残留超标,加重了农药在药材产区周边环境中的残留,给药材生产带来了持续性的危害,形成了,难以逆转的恶性循环,使药材质量越来越筹。
中药材从环境中被动吸收一些高残留农药。
在中药材种植和生产过程中,忽视种植环境对药材的污染,如:药材生长周同环境土壤、水源、大气的污染等。
虽然高残留的有机氯农药六六六、滴滴涕早在70年代就被禁用,但这些农药半衰期长达60年,容易通过食物链在植物体内形成生物累积。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
香豆素
HPLC
灵芝
灵芝多糖(以无水葡萄糖计,0.5)
多糖
紫外-可见分光光度法(附录V A)
阿胶
总氮13.0
蛋白质
氮测定法(附录IX L第一法)
阿魏
挥发油10.0ml/g
挥发油
挥发油测定法
(附录X D)
陈皮
橙皮苷3.5
黄酮
HPLC
忍冬藤
绿愿酸0.1
有机酸
HPLC
青风藤
青藤碱0.5
生物碱
生物碱
薄层色谱扫描法
黄柏
小檗碱(以盐酸小檗碱计)3.0
生物碱
HPLC
黄精
多糖(以无水葡萄糖计)7.0
多糖类
紫外-可见分光光度法ຫໍສະໝຸດ 黄藤盐酸巴马汀2.0
生物碱
HPLC
菝葜
薯蓣皂苷元0.04
皂苷元
HPLC
菊花
绿原酸0.2
有机酸
HPLC
野菊花
蒙花苷0.8
皂苷
HPLC
蛇床子
蛇床子素1.0
香豆素
HPLC
银杏叶
总黄酮醇苷(槲皮素、山萘酚、异鼠李素)0.40;萜类内酯(银杏内酯A、银杏内酯B、银杏内酯C、白果内酯)0.25
盐酸水苏碱0.5
生物碱
薄层色谱扫描法
益智
挥发油1.0
挥发油
挥发油测定法(附录ⅩD)
浙贝母
贝母素甲+贝母素乙0.08
生物碱
HPLC
莎罗子
七叶皂苷A(0.70)
皂苷
HPLC
桑叶
无水芦丁0.1
黄酮
HPLC
黄芩
黄芩苷9.0
皂苷
HPLC
黄芪
黄芪甲苷0.04
皂苷
HPLC
炙黄芪
黄芪甲苷0.03
皂苷
HPLC
黄连
小檗碱(以盐酸小檗碱计)3.6
黄酮,
萜类内酯
HPLC
猪胆粉
猪去氧胆酸16.0
胆汁酸
薄层色谱扫描法
麻黄
盐酸麻黄碱1.0
生物碱
HPLC
鹿角胶
总氮10.0
蛋白质
氮测定法(附录ⅨL第一法)
淫羊藿
总黄酮(以淫羊藿苷计)5.0,淫羊藿苷0.5
黄酮
紫外-可见分光光度法,HPLC
密蒙花
蒙花苷0.5
皂苷
HPLC
续断
川续断皂苷Ⅵ2.0
皂苷
HPLC
斑蝥
虎杖
大黄素0.6,虎杖苷0.15
蒽醌,
二苯乙烯苷类
HPLC,HPLC(避光操作)
昆布
碘,海带0.35;昆布0.20
无机元素
滴定法
罗布麻叶
槲皮素0.6
黄酮
HPLC
知母
菝葜皂苷元1.0
甾体皂苷元
HPLC
侧柏叶
槲皮苷0.1
黄酮
HPLC
金果榄
盐酸巴马汀0.03
生物碱
薄层色谱扫描法
金钱草
槲皮素+山柰素0.1
黄酮
二苯乙烯苷类
HPLC
龟甲胶
总氮9.0
蛋白质
氮测定法(附录I L第一法)
辛夷
挥发油1.0(ml/g),
木兰脂素0.4
挥发油
挥发油测定法(附录X D)
HPLC
羌活
挥发油2.8ml/g
挥发油
挥发油测定法(附录X D)
沙棘
总黄酮(以无水芦丁计1.5),异鼠李素0.1
黄酮
紫外-可见分光光度法,HPLC
补骨脂
补骨脂素+
紫外可见分光光度法,薄层色谱扫描法
厚朴
厚朴酚+和厚朴酚2.0
酚类
HPLC
厚朴花
厚朴酚+和厚朴酚0.2
酚类
HPLC
砂仁
挥发油,阳春砂、绿壳砂不得少于3.0;海南砂1.0
挥发油
挥发油测定法
(附录X D)
牵牛子
咖啡酸+咖啡酸乙酯1.2
有机酸
HPLC
骨碎补
柚皮苷0.5
黄酮
HPLC
钟乳石
碳酸钙95.0
无机物
滴定法
(附录X D),HPLC
前胡
白花前胡甲素0.9
香豆素
HPLC
洋金花
东莨菪碱0.15
莨菪烷型生物碱
HPLC
穿山龙
薯蓣皂苷元1.1
甾体皂苷元
HPLC
穿心莲
穿心莲内酯+脱水穿心莲内酯0.8
二萜内酯
HPLC
秦艽
龙胆苦苷2.0
苷类
HPLC
秦皮
秦皮甲素+秦皮乙素1.0
生物碱
HPLC
莲子心
莲心碱0.2
生物碱
HPLC
生物碱
HPLC
牡丹皮
丹皮酚1.2
挥发油(酚类)
HPLC
体外培育牛黄
胆酸6.0
胆红素35.0
胆汁酸类
胆汁色素类
薄层色谱扫描法,紫外-可见分光光度法
何首乌
2,3,5,4′-四羟基二苯乙烯-2-O-β-D-葡萄糖苷(1.0)
二苯乙烯苷类
HPLC
制何首乌
2,3,5,4′-四羟基二苯乙烯-2-O-β-D-葡萄糖苷(0.7)
莪术
挥发油1.5
挥发油
挥发油测定法
(附录X D)
荷叶
荷叶碱0.1
生物碱
HPLC
桔梗
总皂苷6.0
皂苷
重量法
夏天无
原阿片碱0.3
生物碱
HPLC
夏枯草
熊果酸0.12
有机酸
HPLC
射干
次野鸢尾黄素0.1
黄酮
HPLC
徐长卿
丹皮酚1.3
酚类
HPLC
高良姜
桉油精0.15
挥发油
HPLC
粉葛
葛根素0.3
黄酮
HPLC
益母草
香加皮
4-甲氧基水杨醛0.2
萜类
HPLC
香薷
挥发油0.6,麝香草酚+香荆芥酚0.16
挥发油,单萜
挥发油测定法
(附录X D),GC
重楼
重楼皂苷I+重楼皂苷II(0.80)
甾体皂苷
HPLC
独一味
木犀草素0.15
黄酮
HPLC
独活
蛇床子素0.5
香豆素
HPLC
姜黄
挥发油7.0,姜黄素1.0
挥发油,酚类
挥发油测定法
红景天苷0.5
苷类
HPLC
远志
远志酸0.7
皂苷元
HPLC
赤芍
芍药苷1.8
苷类(酚苷)
HPLC
花椒
挥发油1.5ml/g
挥发油
挥发油测定法(附录X D)
芥子
芥子碱(以芥子碱硫氰酸盐)0.5
生物碱
HPLC
芦荟
芦荟苷:库拉索芦荟18.0;好望角芦荟6.0
羟基蒽醌苷类
HPLC
苏合香
肉桂酸5.0
有机酸
HPLC
杜仲
松脂醇二葡萄糖苷0.1
木脂素苷
HPLC
杜仲叶
绿原酸0.08
有机酸
HPLC
豆蔻
挥发油4.0(ml/g),桉油精3.0
挥发油,单萜
挥发油测定法(附录X D),GC
两头尖
竹节香附素0.2
三萜皂苷
HPLC
两面针
两面针碱0.25
生物碱
薄层色谱扫描法
连翘
连翘苷0.15
木脂素
HPLC
吴茱萸
吴茱萸碱+
吴茱萸次碱0.15
(附录X D)
胡芦巴
胡芦巴碱0.45
生物碱
HPLC
胡黄连
胡黄连苷I+胡黄连苷II(9.0)
生物碱
HPLC
胡椒
胡椒碱3.0
酰胺
HPLC
南五味子
五味子酯甲0.12
木脂素
HPLC
枳壳
柚皮苷4.0
黄酮
HPLC
枳实
辛弗林0.3
生物碱
HPLC
栀子
栀子苷1.8
环烯醚萜苷
HPLC
枸杞子
多糖1.8,甜菜碱0.3
多糖,生物碱
玉竹多糖(以葡萄糖计6.0)
多糖
紫外-可见分光光度法
功劳木
盐酸小檗碱0.2
生物碱
HPLC
甘松
挥发油2.0
挥发油
挥发油测定法(附录XD)
甘草
甘草酸2.0,甘草苷1.0
五环三萜皂苷,黄酮
HPLC
石韦
绿原酸0.3
有机酸
HPLC
石菖蒲
挥发油1.0
挥发油
挥发油测定法(附录X D)
石榴皮
鞣质
鞣质
鞣质含量测定法(附录ⅡD)
石膏
含水硫酸钙CaSO4·2H2O(95%)
无机物
滴定法
煅石膏
硫酸钙(CaSO4)
无机物
滴定法
龙胆
龙胆苦苷1.0
环烯醚萜苷
HPLC
仙茅
仙茅苷0.1
苷类
HPLC
白芍
芍药苷1.6
苷类(酚苷)
HPLC
白芷
欧前胡素0.08
香豆素
HPLC
白矾
含水硫酸铝钾KAl(SO4)2·12H2O(99.0)
无机物
滴定法
白鲜皮
HPLC
金银花
绿原酸1.5,木犀草苷0.1
黄酮