MDR技术文件清单
欧盟医疗器械法规MDR技术文件清单与要求
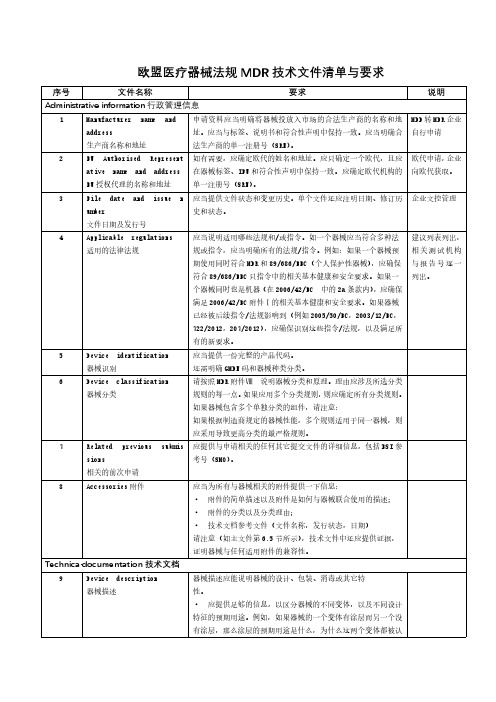
欧盟医疗器械法规MDR技术文件清单与要求序号文件名称要求说明Administrative information行政管理信息1Manufacturer name and address生产商名称和地址申请资料应当明确将器械投放入市场的合法生产商的名称和地址。
应当与标签、说明书和符合性声明中保持一致。
应当明确合法生产商的单一注册号(SRN)。
MDD转MDR企业自行申请2EU Authorised Represent ative name and addressEU授权代理的名称和地址如有需要,应确定欧代的姓名和地址。
应只确定一个欧代,且应在器械标签、IFU和符合性声明中保持一致。
应确定欧代机构的单一注册号(SRN)。
欧代申请,企业向欧代获取。
3File date and issue n umber文件日期及发行号应当提供文件状态和变更历史。
单个文件还应注明日期、修订历史和状态。
企业文控管理4Applicable regulations 适用的法律法规应当说明适用哪些法规和/或指令。
如一个器械应当符合多种法规或指令,应当明确所有的法规/指令。
例如:如果一个器械预期使用同时符合MDR和89/686/EEC(个人保护性器械),应确保符合89/686/EEC只指令中的相关基本健康和安全要求。
如果一个器械同时也是机器(在2006/42/EC中的2a条款内),应确保满足2006/42/EC附件Ⅰ的相关基本健康和安全要求。
如果器械已经被后续指令/法规影响到(例如2005/50/EC,2003/12/EC,722/2012,207/2012),应确保识别这些指令/法规,以及满足所有的新要求。
建议列表列出,相关测试机构与报告号逐一列出。
5Device identification 器械识别应当提供一份完整的产品代码。
还需明确GMDN码和器械种类分类。
6Device classification 器械分类请按照MDR附件Ⅷ说明器械分类和原理。
欧盟新法规MDR培训

CER: Clinical evaluation report临床评价报告 CIE:
方向:重
Clinical Investigations and Evaluation临床调查与评价 PMS:post-market surveillance上市后监管(PMSP/PMSR)
过程(几 份计划!)
PSUR:periodic safety update report定期安全性更新报告
一个组织的每个参与角色都需要一个SRN代码。
即使一个制造商有多个生产地点,作为制造职能,多个点也仍然只需要一 个SRN代码,除非这些地点在不同法定制造商的标签(及证书、IFU等)上 面被标出来。
欧代AR,虽然是同时作为n家制造商的欧代,但其职能是一样的,又是一 个组织,所以可以申请一个SRN。
MDR的主要变化(2)
范围扩充、部分调整 保存期限,EUDAMED,UDI
合规负责人
MDR的主要变化
NO. Item
·可持续 ·公开透明 ·可预测
1
法规本身的扩容
经济运营商,数据库导致
2
执行过程 独立的微生物审核,风管Review
3
强化制造商责任
4
加强产品批准前监管
5
适用范围增大
6
加强可追溯性和透明度
7
市场后监管力度和警戒
书的器械材将不得 投入市场
MDR certificates医疗器械法规 (MDR)认证
NB designation under MDR 医疗器械法规 (MDR) 下的公告机构的授权
过渡期要求
MDR的发布之路
• 指令(Directive)升级为法规(Regulation)
当前取得MDR资质的公告机构
欧盟医疗器械2017∕745 法规(MDR)(附录 XI 基于产品合规性验证的符合性评估 )

附录XI基于产品合规性验证的符合性评估1.基于产品合规性验证的符合性评估的目的是确保器械符合已发布EC 型式检验证书中所说明的形式,并满足本法规中的适用规定要求。
2.如已根据附录X 颁发EC 型式检验证书,制造商可申请表第A 部分所述流程(生产质量保证),或本附录第B 部分所述流程(产品验证)。
3.通过豁免上述第1 节和第2 节,本附录所述程序加上附录II 和III 所述技术文件也可适用于IIa 类器械制造商。
第A 部分:生产质量保证4.制造商应确保实施批准的相关器械生产质量管理体系,并按照第 6 节的规定进行最终验证,且接受第7 节所述的监管。
5.制造商应履行第4 节所规定的义务,并根据第19 条和附录IV 起草并保存符合性评估流程所涵盖器械型号的EU 符合性声明。
通过发布EC 符合性声明,制造商应确定并声明有关器械是否符合EC 型式检验证书中所说明的型式,以及是否满足本法规中的适用规定要求。
6.质量管理体系6.1制造商应向公告机构提出申请表,评估自己的质量管理体系。
申请表应当包括:- 附录IX 第2.1 节所列的所有要素;- 附录II 和III 所述批准型式的技术文件;- 附录X 第4 节所述EC 型式检验证书副本;若提出申请表后,EC 型式检验证书由同一公告机构颁发,则技术文件及其更新信息和所颁发证书的参考资料应包含在申请中。
6.2质量管理体系的实施应确保器械是否符合EC 型式检验证书中所说明的型式,以及是否满足本法规中的适用于各阶段器械的规定要求。
制造商为其质量管理体系而采用的所有要素、要求和规定,必须以系统和有序的方式记录在质量手册、书面政策和程序之中,例如质量程序、质量计划和质量记录。
该文件尤其应包括对附录IX 第2.2 节(a)、(b)、(d)和(e)所列所有要素的适当说明。
6.3附录IX 第2.3 节第一和第二段的规定适用。
若质量管理体系可确保器械符合EC 型式检验证书中所说明的形式,并满足本法规中的适用规定要求,则欧盟公告机构应出具欧盟质量保证证书。
MDR技术文件清单

具体内容
具体内容描述
a) 产品名称或商品名、基本描述、预期用途、预期使用者
b)制造商给申请器械分配的基本UDI-DI信息
c)预期病人群体、诊断治疗或监测医疗情况,适应症、禁忌症、警示信
息情况
d)器械原理、配件清单、型号规格清单
e)器械的风险分类等级、根据MDR附录VIII适用的分类规则
器械描述与规 范,包括变动和
c)协调标准、通用规范或其他适用的解决方案
d)文件的精确识别以提供符合性证据
收益/风险分
a)MDR附录I中的条款1到条款8涉及的收益风险分析
5 析和风险管
-
b)MDR附录I中的条款3涉及的采取措施和风险管理结果
理
通常包括风险管理计划和风内容描述
a)各类证明产品符合通用性能和安全要求的测试方案、数据、报告和结
a设计过程介绍设计和生产b生产流程及过程确认情况连续监测和成品检验要求相关数据要包3信息含在技术文件中c设计生产过程涉及的所有场地信息包括外包方mdr附录i中23条要求全部列出相互适用的符合性信息判断是否适用满足并描述相关检测报告风险报告验证和确认报告标签说明书样稿临床相关报告等并包括用于这些要求的措施的判断确认和验证4基本安全和基本安全和性a适用与器械的基本安全和性能要求以及不适用条款的解释性能要求能要求检查表b用于证明与适用的基本安全和性能要求符合的方法c协调标准通用规范或其他适用的解决方案d文件的精确识别以提供符合性证据收益风险分amdr附录i中的条款1到条款8涉及的收益风险分析5析和风险管bmdr附录i中的条款3涉及的采取措施和风险管理结果理通常包括风险管理计划和风险管理报告序号主题具体内容具体内容描述a各类证明产品符合通用性能和安全要求的测试方案数据报告和结论特别是
MDR技术文件清单
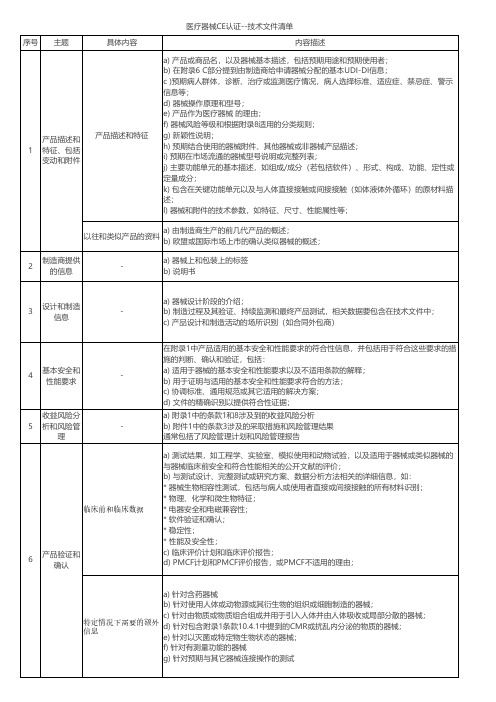
序号主题具体内容内容描述产品描述和特征a) 产品或商品名,以及器械基本描述,包括预期用途和预期使用者;b) 在附录6 C部分提到由制造商给申请器械分配的基本UDI-DI信息;c )预期病人群体,诊断、治疗或监测医疗情况,病人选择标准、适应症、禁忌症、警示信息等;d) 器械操作原理和型号;e) 产品作为医疗器械 的理由;f) 器械风险等级和根据附录8适用的分类规则;g) 新颖性说明;h) 预期结合使用的器械附件、其他器械或非器械产品描述;i) 预期在市场流通的器械型号说明或完整列表;j) 主要功能单元的基本描述,如组成/成分(若包括软件)、形式、构成、功能、定性或定量成分;k) 包含在关键功能单元以及与人体直接接触或间接接触(如体液体外循环)的原材料描述;l) 器械和附件的技术参数,如特征、尺寸、性能属性等;以往和类似产品的资料a) 由制造商生产的前几代产品的概述;b) 欧盟或国际市场上市的确认类似器械的概述;2制造商提供的信息-a) 器械上和包装上的标签b) 说明书3设计和制造信息-a) 器械设计阶段的介绍;b) 制造过程及其验证、持续监测和最终产品测试,相关数据要包含在技术文件中;c) 产品设计和制造活动的场所识别(如合同外包商)4基本安全和性能要求-在附录1中产品适用的基本安全和性能要求的符合性信息,并包括用于符合这些要求的措施的判断、确认和验证,包括:a) 适用于器械的基本安全和性能要求以及不适用条款的解释;b) 用于证明与适用的基本安全和性能要求符合的方法;c) 协调标准、通用规范或其它适用的解决方案;d) 文件的精确识别以提供符合性证据;5收益风险分析和风险管理-a) 附录1中的条款1和8涉及到的收益风险分析b) 附件1中的条款3涉及的采取措施和风险管理结果通常包括了风险管理计划和风险管理报告临床前和临床数据a) 测试结果,如工程学、实验室、模拟使用和动物试验,以及适用于器械或类似器械的与器械临床前安全和符合性能相关的公开文献的评价;b) 与测试设计、完整测试或研究方案、数据分析方法相关的详细信息,如:* 器械生物相容性测试,包括与病人或使用者直接或间接接触的所有材料识别;* 物理、化学和微生物特征;* 电器安全和电磁兼容性;* 软件验证和确认;* 稳定性;* 性能及安全性;c) 临床评价计划和临床评价报告;d) PMCF计划和PMCF评价报告,或PMCF不适用的理由;特定情况下需要的额外信息a) 针对含药器械b) 针对使用人体或动物源或其衍生物的组织或细胞制造的器械;c) 针对由物质或物质组合组成并用于引入人体并由人体吸收或局部分散的器械;d) 针对包含附录1条款10.4.1中提到的CMR或扰乱内分泌的物质的器械;e) 针对以灭菌或特定物生物状态的器械;f) 针对有测量功能的器械g) 针对预期与其它器械连接操作的测试1产品描述和特征、包括变动和附件6产品验证和确认医疗器械CE认证--技术文件清单。
最新MDR一类医疗器械全套技术文件模板(中英文对照)

最新MDR一类医疗器械全套技术文档模板(中英文对照)CE TECHNICALDOCUMENTATION Based on REGULATION (EU) 2017/745Product Name: {填写申报产品名称}Model:{填写申报产品的具体型号}Document No.: {填写本文档编号}Edition: {填写本文档版本号}{填写申请者的企业名称}ContentRationale for the qualification as a medical device and the risk class attributedProduct Name:{填写申报产品名称}Model:{填写申报产品的具体型号}Document No.: {填写本文档编号}Edition: {填写本文档版本号}Drafted by: Date: {填写本文档编写日期}Checked by: Date: {填写本文档审核日期}Approved by: Date: {填写本文档批准日期}{填写申请者的企业名称}Revision records:1.Rationale for the qualification as a medical device{填写申报产品可作为器械的合理的理由}【可从产品的预期用途,适应症方面考虑】(参考示例:According to the definition of medical device in REGULATION (EU) 2017/745 as below and the application of{填写申报产品名称}(参考示例:CRP mask), we consider that{填写申报产品名称}(参考示例:CRP mask)is a medical device.Define of medical device:“medical device” means any instrument, apparatus, appliance, software, implant, reagent, material or other article intended by the manufacturer to be used, alone or in combination, for human beings for one or more of the following specific medical purposes:—diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of disease,—diagnosis, monitoring, treatment, alleviation of, or compensation for, an injury or disability,—investigation, replacement or modification of the anatomy or of a physiological or pathological process or state,—providing information by means of in vitro examination of specimens derived from the human body, including organ, blood and tissue donations,and which does notachieve its principal intended action by pharmacological, immunological or metabolic means, in or on the human body, but which may be assisted in its function by such means.The following products shall also be deemed to be medical devices:—devices for the control or support of conception;—products specifically intended for the cleaning, disinfection or sterilisation of devices as referred to in Article 1(4) and of those referred to in the first paragraph of this point.)2.Rationale for the risk class attributed{填写申报产品风险分类的基本原理}(参考示例:The risk class is attributed according to Annex VIII CLASSIFICATION RULES in REGULATION (EU) 2017/745.{填写申报产品名称}(参考示例:CPR mask)is a{填写申报产品的类别}【根据产品实际情况填写申报产品所属类别如是属于非侵入、侵入还是有源】(参考示例:non-invasive)device. The rules of classification for{填写申报产品的类别}【根据产品实际情况填写申报产品所属类别如是属于非侵入、侵入还是有源】(参考示例:non-invasive)devices is in{填写申报产品分类所适用的分类法规}(参考示例:Rule1-Rule4), the details are as below.{填写申报产品分类所适用的分类法规原文}(4. NON-INVASIVE DEVICES4.1. Rule 1All non-invasive devices are classified as class I, unless one ofthe rules set out hereinafter applies.4.2. Rule 2All non-invasive devices intended for channelling or storingblood, body liquids, cells or tissues, liquids or gases for thepurpose of eventual infusion, administration or introduction intothe body are classified as class IIa:—if they may be connected to a class IIa, class IIb or class IIIactive device; or—if they are intended for use for channelling or storing blood or other body liquids or for storing organs, parts of organs or body cells and tissues, except for blood bags; blood bags are classified as class IIb.In all other cases, such devices are classified as class I.4.3. Rule 3All non-invasive devices intended for modifying the biological or chemical composition of human tissues or cells, blood, otherbody liquids or other liquids intended for implantation or administration into the body are classified as class IIb, unless thetreatment for which the device is used consists of filtration,centrifugation or exchanges of gas, heat, in which case they areclassified as class IIa.All non-invasive devices consisting of a substance or a mixtureof substances intended to be used in vitro in direct contact withhuman cells, tissues or organs taken from the human body orused in vitro with human embryos before their implantation oradministration into the body are classified as class III.4.4. Rule 4All non-invasive devices which come into contact with injuredskin or mucous membrane are classified as:—class I if they are intended to be used as a mechanical barrier,for compression or for absorption of exudates;—class IIb if they are intended to be used principally for injuriesto skin which have breached the dermis or mucous membraneand can only heal by secondary intent; —class IIa if they are principally intended to manage themicro-environment of injured skin or mucous membrane; and—class IIa in all other cases.This rule applies also to the invasive devices that come intocontact with injuredmucous membrane.)According to above rules, {填写申报产品名称}(参考示例:CPR mask)is classified as Class I according to{填写产品适用的分类规则}(参考示例:rule 1).)Description and specificationProduct Name:{填写申报产品名称}Model:{填写申报产品的具体型号}Document No.: {填写本文档编号}Edition: {填写本文档版本号}Drafted by: Date: {填写本文档编写日期}Checked by: Date: {填写本文档审核日期}Approved by: Date: {填写本文档批准日期}{填写申请者的企业名称}Revision records:1 product or trade name and a general description of the device including its intended purpose and intended users;1.1 Name of the DeviceProduct name: {填写申报产品名称}1.2 Product description{对产品进行简要描述}【可从产品的组成,是否含附件以及所属的类别,是否需要灭菌等方面,进行简要描述】1.3 Intended Purpose{填写申报产品的预期用途}1.4 Intended User{填写申报产品的目标使用者}(参考示例:hospital staff)2 The intended patient population and medical conditions to be diagnosed, treated and/or monitored and other considerations such as patient selection criteria, indications, contra-indications, warnings;2.1 Intended patient population{填写产品适用的患者人群}(参考示例:adult)2.2 Medical conditions{填写产品适用的疾病阶段或程度}(参考示例:The patient requires ventilation)2.3 Other considerations2.3.1 Patient selection criteria{填写申报产品适用的病人的选择标准}(参考示例:the years old of patient over18)2.3.2 Indications{填写申报产品的适应症}2.3.3 Contra-indications,{填写申报产品的禁忌症}2.3.4 Warnings{填写申报产品的警告}3 Principles of operation of the device and its mode of action, scientifically demonstrated if necessary3.1 Principles of operation{填写申报产品的操作原理}3.2 Mode of action{填写申报产品的作用方式}4 The rationale for the qualification of the product as a devicePlease refer to Document 1 “Rationale for the qualification as a medical device and the risk class attributed”.5 The risk class of the device and the justification for the classification rule(s) applied in accordance with Annex VIII;Please refer to Document 1 “Rationale for the qualification as a medical device and the risk class attributed”.6 An explanation of any novel features;{填写申报产品是否使用新技术的描述} (参考示例:This is a traditional technology. It has been used all over the world for many years. There are no novel features.)7 A description of the accessories for a device, other devices and other products that are not devices, which are intended to be used in combination with it;{对申报器械是否有附件,以及预期与其他非器械联合使用的简要描述} (参考示例:The actual device do not contain accessories. It is not used in combination with other devices.)8 A description or complete list of the various configurations/variants of the device that are intended to be made available on the market;{详细列出申报产品的所有的型号,并描述各型号之间的差异 }9 A general description of the key functional elements9.1 Its parts/components (including software if appropriate)9.1.1 Parts/ComponentsThe parts of {填写申报产品名称} are as below.{列出申报产品组件 }9.1.2 Software{此处对申报产品的软件进行描述}.9.2 FormulationIt is consisted of components as section 9.1.1. It does not contain special formulation.9.3 Its compositionPlease refer to section 10.1.1.9.4 Functionality{对关键原件的功能进行描述}9.5 qualitative and quantitative composition.{对关键元件的定性和定量进行描述}9.6 Labelled pictorial representations(e.g. diagrams, photographs, and drawings), clearly indicating key parts/components, including sufficient explanation to underst and the drawings and diagrams;The picture of key parts/components for actual device are as below:{填写申报产品的关键部件的图片或图示}【提供带标记的图形指示关键部件/组件,如图表、照片、图纸等】10 A description of the raw materials incorporated into key functional elements and those making either direct contact with the human body or indirect contact with the body, e.g., during extracorporeal circulation of body fluids;{填写预期与人体间接接触和直接接触的材料说明}(示例见下文,包括不限于下列表格中的内容)11 Technical specifications{对申报产品的技术规格进行描述}【包括但不限于特征、尺寸、性能属性,通常出现在用户阅读的产品规范中,例如宣传册、目录、相似出版物等】Technical SpecificationsProduct Name:{填写申报产品名称}Model:{填写申报产品的具体型号} Document No.: {填写本文档编号}Edition: {填写本文档版本号}Drafted by: Date: {填写本文档编写日期}Checked by: Date: {填写本文档审核日期}Approved by: Date: {填写本文档批准日期}{填写申请者的企业名称}Revision records:1.Raw materials{填写原材料信息}2.Drawings of components{请详细列出各种组件的图片或图纸}【若有多个组件图片,请依次列出】3. Quality control procedures{对申报产品的质量控制进行简要描述}(参考示例:Our company has established EN ISO 13485 quality system. The manufacture process is continuously monitored.Firstly, the materials are monitored. All incoming suppliers shall be selected by the company in accordance with the requirements of the Supplier management control procedures. When incoming materials of the supplier are delivered to the factory, the quality department shall formulate incoming inspection specifications and conduct incoming inspection in accordance with the inspection requirements. The defective incoming materials shall be handled in accordance with the company's procedure document control procedure for nonconforming products.Secondly, final product is monitored. Before product delivery, the final inspection should be conducted. Meanwhile, deal with the unqualified products according to thecontrol procedure of unqualified products.Thirdly, ex-factory process is monitored.According to the inspection specification, the inspector shall carry out factory inspection on the products, make inspection records and mark the quality status as required, and deal with the unqualified products according to the control procedure of unqualified products.)Information to be supplied by the manufacturerProduct Name:{填写申报产品名称}Model:{填写申报产品的具体型号}Document No.: {填写本文档编号}Edition: {填写本文档版本号}Drafted by: Date: {填写本文档编写日期}Checked by: Date: {填写本文档审核日期}Approved by: Date: {填写本文档批准日期}{填写申请者的企业名称}Revision records:er Manual and Label{放入申报产品的使用手册和标签}(参考示例:Please refer to***User Manual and ***Label.){请放入申报产品的说明书}【对申报产品的说明书要求请参考MDR 法规第附录I第III章中对说明书,以及EN ISO 15223-1:2016标准中对说明书的要求】{请放入申报产品的标签}【对申报产品的标签的要求请参考MDR 法规第附录I第III章中对标签,以及EN ISO 15223-1:2016标准中对标签的要求】Reference to previous generations of the device andto similar devicesProduct Name:{填写申报产品名称}Model:{填写申报产品的具体型号}Document No.: {填写本文档编号}Edition: {填写本文档版本号}Drafted by: Date: {填写本文档编写日期}Checked by: Date: {填写本文档审核日期}Approved by: Date: {填写本文档批准日期}{填写申请者的企业名称}Revision records:1.An overview of the previous generation or generations of the device produced by the manufacturer, where such devices exist{若申报产品有前代产品的话,请对前代产品进行简要概述}【参考示例:The actual device is the First-generation products, we consider there is no previous generation 】2. An overview of identified similar devices available on the Union or international markets, where such devices exist.{对欧盟以及国际市场上的类似产品进行简要概述}Design and manufacturing informationProduct Name:{填写申报产品名称}Model:{填写申报产品的具体型号}Document No.: {填写本文档编号}Edition: {填写本文档版本号}Drafted by: Date: {填写本文档编写日期}Checked by: Date: {填写本文档审核日期}Approved by: Date: {填写本文档批准日期}{填写申请者的企业名称}Revision records:rmation that allows the understanding of thedesign and manufacturing ofa device1.1Information that allows the understanding of thedesign of a device{填写申报产品的设计开发程序}(参考示例:The actual device are designed according to the Design and development Process as below.)1.2Information that allows the understanding of themanufacturing of a device1.2.1manufacturing processes{列出申报产品的生产程序}1.2.2Process validation{填写生产过程中的各种验证}.【应放入完整的测试数据或链接】(参考示例:The process validation are please refer to * * * Final product test report,* * * Packaging seal validation report)1.2.3 Continuous monitoring{对申报产品生产过程中的连续监测进行描述}(参考示例:Our company has established EN ISO 13485 quality system. The manufacture process is continuously monitored.Firstly, the materials are monitored. All incoming suppliers shall be selected by the company in accordance with the requirements of the Supplier management control procedures. When incoming materials of the supplier are delivered to the factory, the quality department shall formulate incoming inspection specifications and conduct incoming inspection in accordance with the inspection requirements. The defective incoming materials shall be handled in accordance with the company's procedure document control procedure for nonconforming products. Secondly, final product is monitored. Before product delivery, the final inspection should be conducted. Meanwhile, deal with the unqualified products according to the control procedure of unqualified products. Thirdly, ex-factory process is monitored.According to the inspection specification, the inspector shall carry out factory inspection on the products, make inspection records and mark the quality status as required, and deal with the unqualified products according to the control procedure of unqualified products.)1.2.4 Final product testing{列出最终产品的所有验证测试}2 Design calculations relevant to the intended use of the product{填写实现申报产品预期用途的设计原理}.3 Technology{对申报产品的当前技术生产工艺进行简要描述}.(参考示例:From the information of Section 2 of Document 5 Reference to previous generations of the device and to similar devices, the technology of actual device is mature. The design is safe and have been established for a number of years. Actual device have been performing as intended during that time such information is likely tobe sufficient to cover this requirement.)4 Identification of all sites4.1 Company addressThe company registration address:{填写公司注册地址} the manufacturing address:{填写生产地址}.4.2 Supplier addressThe supplier addresses of critical materials are as below:4.3 Sub-contractor address{填写分包商的地址}General safety and performance requirementsProduct Name: {填写申报产品名称}Model:{填写申报产品的具体型号}Document No.: {填写本文档编号}Edition: {填写本文档版本号}Drafted by: Date: {填写本文档编写日期}Checked by: Date: {填写本文档审核日期}Approved by: Date: {填写本文档批准日期}{填写申请者的企业名称}Revision records:1.General safety and performance requirements{放入一般安全和性能要求的符合性证明}(参考示例:Please refer to **** Checklist for General safety and performance requirements Compliance analysis)requirements Compliance analysis REGULATION (EU) 2017/745Conformity Assessment Procedure according to the following Annex of the Directive (pls. tick): Declare the conformity of their products by issuing the EU declaration of conformity referred to in Article 19(MDR regulation EU 2017/745) after drawing up the technical documentation set out in Annexes II and III.requirements Compliance analysisPage 3 of 104Page 4 of 104Page 5 of 104Page 6 of 104Page 7 of 104Page 8 of 104Page 10 of 104Page 11 of 104Page 12 of 104Page 14 of 104。
MDR申请提交文档清单

1. Executive summary(option)摘要(选项)2. Declaration of conformity符合性声明3. Devicedescri tion of器械描述4. Technical specification技术规范5. Previous and similar generation以前的或类似器械的引用6. Labels and IFU (Instruction for use)标签和使用说明书7.GSPR(General Safety &Performance Requirements) checklist通用安全和性能要求清单8. List of standards 标准/法规清单9. Benefit-risk analysis and riskmanagement收益-风险分析和风险管理10. Description of the design设计说明11. Production V&V*生产1)Electrical safety电气安全2)Electromagnetic compatibility电磁兼容3)Mechanical safety机械安全 4)Reliability and lifetime 可靠性和寿命 5)Diagnostic device with measuring function 具有诊断和测试功能的器械 6)Combination with other device与其连接的器械 7)Human factor and usability人的因素和可用性(非专业人员使用) 8)Software verification & validation 软件验证&确认 9)Other pre-clinical test; device specific bench test 其他临床前测试;设备特定货架试验 10)Bioburden and sterilization微生物污染水平和灭菌 11)Packaging qualification and shelf life 包装质量和保质期 12)Packaging testing for handing and transport 搬运和运输的包装试验 13)Hygienic reprocessing of device设备的卫生后处理 14)Biological evaluation 生物学评价 15)Other pre- clinical test; in-vitro/animal studies 其他临床前测试;体外和动物实验 16)Clinical Evaluation 临床评价 17)SSCP (Summary of Safety and Clinical Performance )安全性和临床性能总结 18)Implant card植入物卡 19)Medicinal product 含有药物产品 20)Tissue or cells of animal or human ongin 动物或人类来源的组织或细胞 21)Absorbed or locally dispersed substances 可吸收或局部扩散的物质 22)CMR or endocrine disruptive substances 含有CMR (致癌,致突变或生物毒性)或内分泌物质的器械 23)Disposal consideration 12. Description of manufacturing process and their validation and QC 制造过程的描述及其验证和质量控制13. Post-market surveillance(PMS)上市后监督14. Installation, repair&service, and maintenance 安装,维修&服务,维护15. Design change records(option)设计更改记录(选项)MDR申请提交文档清单。
mdr-世卫指南-文档资料36页

Xpert MTB/RIF 结果
Xpert +
Xpert –
合计
33
0
33
52
2
54
31
12
43
116
14
130
灵敏度
100% 96,3%
97,7%
72,1%
89,2%
Marlowe, J Clin Microbiology 2019
快速药敏试验的使用
WHO 2019
2019 世卫指南 建议2
MDR-TB患者治疗期间,建议使用痰涂片和培养进 行治疗监测,而不是单独应用痰涂片。
- 最好的实施:每月监测 - 培养:比单独应用痰涂片更早发现治疗失败 - 治疗初期痰涂片阴性:每月进行涂片监测(与培养比较)导致治 疗失败发现延迟的风险在统计学上显著高于痰涂片阳性患者
均数: 中位数
均数: 中位数
涂片 (503)
液体培养 (946)
固体培养 (539)
Migliori, ERS-online course, 2019
GeneXpert – 检测利福平耐药的时间
均数: 中位数
均数: 中位数
均数: 中位数
分子生物学 DST(19)
传统DST (110)
Migliori, ERS-online course, 2019
在治疗MDR-TB患者时, 应使用乙硫异烟胺(或丙 硫异烟胺)
2019 世卫指南 建议6
在治疗MDR-TB患者时,强化期(治疗的初始阶段, 即应用注射剂阶段)应包括四种可能有效的二线抗 结核药品(包括二线注射用药卡那霉素,阿米卡星 或者卷曲霉素中的一种)以及吡嗪酰胺
欧盟MDR技术文档怎么写

欧盟MDR技术文档怎么写
撰写欧盟MDR技术文档需要注意以下几个方面:
1. 目录结构:技术文档应具有清晰的目录结构,方便读者查找信息。
一般可以包括导言、产品说明、技术规格、测试过程、性能评估等
章节。
2. 引言部分:在引言部分介绍产品的背景、目的和主要功能。
还可
以包括MDR规定的相关规范和标准的说明。
3. 产品说明:详细描述产品的特性、结构、组成和功能。
包括使用
方法、操作步骤和注意事项等信息。
4. 技术规格:列出产品的技术规格和性能指标。
例如尺寸、重量、
功耗、工作温度范围、存储容量等。
5. 测试过程:描述产品的测试过程和方法。
包括原型测试、认证测试、性能测试等内容。
应包括测试方法、测试环境、测试数据和结果。
6. 性能评估:对产品的性能进行评估和分析。
包括产品的可靠性、
安全性、持久性等方面。
7. 安全性评估:对产品的安全性进行评估和分析。
包括风险评估、安全措施、风险管理等方面。
8. 符合性声明:在文档最后可以添加产品的符合性声明,说明产品符合MDR要求的相关规定和标准。
在撰写过程中需要确保信息准确、完整,遵循欧盟MDR相关要求和规定。
同时,应使用清晰的语言和易于理解的表达方式,以方便读者理解和应用该技术文档。
MDR所需资料清单

要有驻厂检验员和生产商的签名。 在产品发货之前,ITP 放行单必须送审至驻厂检验员并征求同意。
3、 章 B—材质证书和材料试验报告
此章节包括所有的材质证书以及主要钢结构工程的材质测试报告。 根据材质种类(板材、角钢、钢管等)必须建立一些分章节,每个分 章节要有 “材料登记表” , 登记表中所有交付的材料必须列出,并 且每个部件要有它唯一的名称,并包括以下各项:产品描述、材料规 格和等级、证书号和交货数量。 在该章中包含了对材料进行追踪的材料追踪表; 每张图纸都要建立并根据图号分类; 编号要和零件表中所给的号码相一致,并要标出以下一些相关内容: 材料描述、与材料登记表中相对应的唯一的材料编号 所有的材料和部件必须要有材料证书,在终检之前必须递交材料证 书; 当碰到没有所需要的材料尺寸时,这时需用材料拼接而成。 例如:有一块编号为部件 2 的板材,要求长度 7380MM,但是板材的 最大长度只有 3000MM,因此要用 3000+3000+1380 进行拼接,使总 长达到 7380: 在零件表中所列的是板材 2 的长度,而在材料追踪表中,板材 2 要分 成 2A、2B、2C 并标出相应的长度和唯一的编号; 在这种情况下需要一份草图, 草图中要标出工程号、 图号、 材料编号、
10、章 I—不符合报告和特许权
在制造和预装配过程中,技术问题可能会导致部件的缺失; 在这种情况下要求提供此技术问题表格(TQ) ; 此种情况下必须使用单独的“TQ”表格或者“OSD”报告并附有单 独的报告编号; 所有情况必须记录在“TQ 登记表”上,这个登记表要时刻更新并同 “TQ”一起发送
11、章 J—机械和性能试验报告 12、章 K—电器证书和报告 13、章 L—设备、仪器证书和报告 14、章 M—图纸 15、章 N—其他各种证书、报告和记录 16、章 O—行车、起重机等起重设备的法定认可证书
MDR认证技术文档

MDRCE认证技术文档的要求
CE技术文件是欧医疗器械指令中很車要的一个事项,它的目的是要求企业准备充份的技术资料和证明,供主管机关抽查,或发生诉讼纠纷时使用。
医疗器械MDR法规要求"技术档案“可能包含:企业的质量手册和程序文件,企业简介及欧洲代表名称、联系方式,CE符台性声明(或称自我保证明),若该产品是和其它设备联合运用,则应有整体符合基本要求的证明材料,主要内容如下:
1、产品名称、分类
2、产品概述(包括类型和预期用途〕
a产品的历史沿革
b产品的预期用途
c产品配合使用的附件、配合件和其它设备满单
d技术性能参数
e 产品的图示与样品(照片、图纸和原理图)
f产品所用原材料及供应商
3、使用该产品的协调标准/或其它标准
4、风险分析评估结论和预防惜施
5、生产质量控制
a产品资料和控制文档(包括产品生产工艺流程图〕
b过程确认和验证
--关键工序/特殊过程的验证
--产品的灭菌方法和确认的描述
c产品质量控制措施
d产品稳定性和效期的描述
6、包装和标识
a包装材料说明
b使用说明书
c标签
7、技术评价
a产品检验报告及相关文献
b技术概要及观点
8、风险管理
a潜在风险报告及相关文献
b潜在风险的概要及观点
9、临床评价
a产品临床测试报告及相关文献
b临床使用概述及观点
附1、产品出厂检验报告
附2、产品型式检验报告
附3、基本要求检杳表。
MDR申请提交文档清单

MDR申请提交文档清单
MDR(医疗器械监管条例)的申请提交文档清单是指在进行医疗器械注册、变更、延续等申请过程中需要提交的文件的清单。
根据MDR的相关规定,以下是一个大致的清单,其中包含了一些可能需要提交的文件:
1.申请表格:包括医疗器械注册、变更、延续等申请表格。
2.产品说明书:详细说明医疗器械的适用范围、结构、材料、性能指标、使用方法、注意事项等信息。
3.生产工艺:描述医疗器械的生产工艺流程、工艺控制点、原材料选择和使用等信息。
4.质量管理体系:提供医疗器械制造商的质量管理体系文件,包括质量手册、程序文件、记录等。
5.临床试验报告:如果医疗器械需要进行临床试验,需要提交相应的临床试验报告,包括试验设计、试验结果、安全性和有效性评价等信息。
7.技术文件:包括医疗器械的设计文件、技术文件、测试报告、验证结果等技术资料。
8.包装和运输文件:提供医疗器械的包装规范、包装测试报告、运输条件和要求等信息。
9. GMP认证:如果医疗器械需要符合GMP(Good Manufacturing Practice)要求,需要提供GMP认证证书。
10.产品样品:提交医疗器械的样品供审核,以便审查机构进行产品的实物检验和性能测试。
11.经销授权文件:如适用,需要提供医疗器械的经销授权文件,证明供应商与制造商之间的合作关系。
12.收费证明:提供缴纳相关费用的收据或证明。
请注意,上述清单并非绝对,具体的申请提交文档清单可能会因不同的医疗器械类型、申请类型和国家的法规要求而有所差异。
因此,在实际申请过程中,应根据相关法规和规定进行详细查询,并按照规定提交相应的文件。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
a)含药物成份的器械,应提供药物成分来源、质量、安全性方面信息
b)含人源和动物源成份的器械,应提供报告证明符合GSPR第13条要求
c)含可吸收成份的器械,应提供成分人体吸收、分布、代谢、排泄、毒
特定器械的特 殊要求
性研究资料 d)含有致癌、致突变、有毒物质的器械,应提供必需使用这些物质理由 e)无菌医疗器械,应提供包装、灭菌、无菌屏障相关确认报告,应报告
c)协调标准、通用规范或其他适用的解决方案
d)文件的精确识别以提供符合性证据
收益/风险分
a)MDR附录I中的条款1到条款8涉及的收益风险分析
5 析和风险管
-
b)MDR附录I中的条款3涉及的采取措施和风险管理结果
理
通常包括风险管理计划和风险管理报告
序号 主题
具体内容
具体内容描述
a)各类证明产品符合通用性能和安全要求的测试方案、数据、报告和结
序号 主题
具体内容
具体内容描述
a) 产品名称或商品名、基本描述、预期用途、预期使用者
b)制造商给申请器械分配的基本UDI-DI信息
c)预期病人群体、诊断治疗或监测医疗情况,适应症、禁忌症、警示信
息情况
d)器械原理、配件清单、型号规格清单
e)器械的风险分类等级、根据MDR附录VIII适用的分类规则
器械描述与规 范,包括变动和
有材料的识别
c)设计/生产过程涉及的所有场地信息(包括外包方)
MDR附录I中23条要求全部列出相互适用的符合性信息,判断是否适用、
Байду номын сангаас
满足并描述相关检测报告、风险报告、验证和确认报告、标签说明书样
稿、临床相关报告等,并包括用于这些要求的措施的判断、确认和验证
4
基本安全和 基本安全和性 a)适用与器械的基本安全和性能要求,以及不适用条款的解释 性能要求 能要求检查表 b)用于证明与适用的基本安全和性能要求符合的方法
以往和类似产 a)制造商同类旧款产品介绍、 品资料 b)欧盟或国际市场主流类似产品情况介绍
序号 主题
具体内容
具体内容描述
2
制造商提供 的信息
-
a)最小包装、销售包装、运输包装标签
b)产品说明书。
a)设计过程介绍
3
设计和生产 信息
-
b)生产流程及过程确认情况、连续监测和成品检验要求,相关数据要包 含在技术文件中
f)器械作为医疗器械的理由
1
器械描述和 规范
附件
g)新颖性说明 h)预期组合使用的器械附件、其他器械或非医疗器械产品描述
i)产品关键功能模块介绍,如组成/定性或定量成份(包括软件)、形式 、结构、功能等
j)关键和接触人体的原材料、技术规范等
k)关键功能模块介绍、关键和接触人体的原材料
l)器械和附件的关键技术参数,如特征、尺寸、性能属性等技术规范
论,特别是: * 生物相容性,包括与病人或使用者直接或间接接触的所有材料的识别
* 物理/化学/微生物特征
* 电气安全和电磁兼容性
临床前研究报 * 软件确认 告和临床数据 * 产品稳定性(寿命)
* 产品性能和安全性方面
b) 临床评估计划和报告、上市后临床跟踪计划及报告(或不进行上市
产品验证和
6
确认
后临床跟踪的理由)。 c)适用时,实验室符合GLP的证明。如果某些测试未进行,需要有合理 的理由。
微生物测试、热原测试、灭菌剂残留测试等信息
f)有测量功能的器械,应说明如何确保产品的准确度
特定器械的特 殊要求
序号 7
主题
具体内容
上市后监督
上市后监督
PMCF计划 上市后监督
PMCF评价报告
g)预期与其他器械联用的设备,应描述如何确保连接其他器械时的产品 安全、性能符合要求
具体内容描述 第一次CE认证审核要有上市后监督计划,取得CE证书后监督审核就要求 有“定期安全更新报告” (PSUR,IIa/IIb/III类器械) 上市后监督报告(I类器械)或定期安全更新报告(PSUR,IIa/IIb/III 类器械)