色谱定性定量分析
色谱定性定量方法

既充分利用红外光谱、质谱等适于分析分子结构、官能 团或物质的摩尔质量等特点,克服了它们不易检定复杂 物质的困难,又充分利用了色谱的高效分离能力
2.与其它方法结合定性
①与化学方法结合进行定性 将试样经过一些特殊试剂处理,发生物理 变化或化学反应后,其色谱峰将会提前、 移后或完全消失 比较处理前后色谱图的差异,以及在柱后 用化学试剂鉴定流出物,就可初步定性鉴 别试样中含有哪些官能团
2.与其它方法结合定性
②与红外光谱、质谱及核磁共振谱联合定性
用相对校正因子
把混合物中的不同组份的峰面积校正成相当
于某一标准物质的峰面积,用于计算各组份的 含量
1.定量依据和校正因子
相对校正因子fis是指某组份i的绝对校正因子与标准物质s的绝对 校正因子之比值,通常简称为校正因子,即
标准物质:
f is
fi fs
苯(用于热导检测器)
正庚烷(用于氢火焰离子化检测器) 质量校正因子、摩尔校正因子和体积校正因子
常用标准物:苯、正丁烷、对二甲苯、环己烷、2,3,4-三甲基戊烷
对于组份比较简单的已知范围的混合物试样,可采用此法进行定 性。也可利用文献上的r2,1值或色谱手册中的r2,1值对照定性。
1.利用色谱保留参数定性
②加入已知物增加峰高法
首先用被测试样作色谱图,然后将已知纯物质加到试 样中去,在相同的条件下作色谱图,对比这两个色谱 图
100 %
Ai为任一组份的峰面积,fi为任一组份的质量校正因子 归一法的优点是无需标样,结果准确,操作简便,操作条件(如 进样量、流速等)变化对测定结果影响较小,宜于分析多组份试 样中各组份的含量。
第三章_定性和定量分析

6
优点:测得Ix值与文献值对照就可定性。 缺点:1. 要有正构烷烃纯样。 2.柱子与柱温要与文献规定相同 3. 对于多官能团的化合物和结构比 较复杂的天然产物,无法用保留 指数定性。
4
3. 峰高加入法 如果未知样品较复杂, 如果未知样品较复杂 , 可在未知混合物中加 入纯样,看未知物中哪个峰高增加, 入纯样 , 看未知物中哪个峰高增加 , 来确定未知 物中可能的成分。 物中可能的成分。
进样量“ 进样量“低”
5
4.保留指数定性法 保留指数又称Kovasts指数,是一种重视性较 其他保留数据都好的定性参数,可根据所用固定 相和柱温直接与文献值对照,而不需标准样品。
30
校正因子测定方法 准确称量被测组分和标准物质,混合后,在 实验条件下进样分析(注意进样量应在线性范围 之内),分别测量相应的峰面积,然后通过公式 计算校正因子,如果数次测量数值接近,可取其 平均值。
31
3.常用的定量计算方法
(l)归一化法 归一化法是气相色谱中常用的一种定量方 法。应用这种方法的前提条件是试样中各组分必 须全部流出色谱柱,并在色谱图上都出现色谱峰。
7
为乙酸正丁酯在阿皮松L柱上的流出 [例]图19-15为乙酸正丁酯在阿皮松 柱上的流出 为乙酸正丁酯在阿皮松 曲线(柱温100℃)。由图中测得调整保留距离为: 由图中测得调整保留距离为: 曲线(柱温 ℃)。由图中测得调整保留距离为 乙酸正丁酯310.0mm,正庆烷 乙酸正丁酯 ,正庆烷174.0mm,正辛烷 , 373.4mm,求乙酸正丁酯的保留指数。 ,求乙酸正丁酯的保留指数。
色谱定性和定量分析

一.保留值定性1.利用纯物质对照定性优点: 简单缺点: 要有纯样 , 适用于已知物,操作条件要稳定
优点: 比绝对法重现性好缺点: 也需要纯样 , 比绝对法麻烦3.加入已知物增加峰高法在未知样品加入纯样看哪个峰高增加的 组分即可能为这种已知物。
二.定量校正因子f 为什么要用f? ∵不同 组分有不同的响应值例如用TCD , N2作载气测O2 , H2 的百分含量若H2 、O2峰面积相同, 百分含量相同就不对 。 不能用下式计算:
∵H2 的热导系数大,TCD响应大,但 实际含量小∴必须用校正因子.
C50%50%
f0.1 1
A50050
3. 内标法(外加标准法)不能全出峰或只需测某几个组分时采用方法 :准确称取样品,加入一定量内标物,根据重量及 峰面积求出某组分的含量Ws 内标物重量As 内标物峰面积A i 被测物峰面积fs 内标物重量校正因子fi 被测物重量校正因子W 样品重 一般选内标物为基准 , fs= 1
准确定量分析时,应该用自己测定的校正因子,而不用 值∵ 校正因子随检测器类别,使用载气的不同而不
1.重量校正因子 w -- 重量A -- 峰面积
2.摩尔校正因子
文献 同
3. 相对校正因子的测定方法f ’值可引用文献值 , 也可以自己测定。标准物质 , TCD是苯,FID是正庚烷。准确称量被测组分wi和标准组分ws 的重量在线线范围内进样测 Ai,As求f ’ w或f ’ M多次测定 , 求平均值。
烦
总结 :归一法与 内标法: 是相对法,与操作条件无关 , 误差小外标法 : 简单 , 是绝对法,与操作条件有关作业: P147 17 18
色谱定性定量分析方法

(1)绝对校正因子 某组分i通过检测器的量与检测器对该组分的响应信号之比
测定方法:将已知量的被测标准物质注入色谱仪,根据进样 量及色谱图上的峰面积或峰高计算出绝对校正因子
(2)相对校正因子 组分i与基准物(标准物)s的绝对校正因子之比
检测器不同,所选用的基准物不同 热导检测器——苯 氢火焰离子化检测器——正庚烷
(3)内标法
若试样中所有组分不能全部出峰,或仅需测定试样中某个或 某几个组分的含量时,可以采用内标法 将一定的标准物(内标物s)加入到一定量的试样中,混合均 匀后进样,从色谱图上分别测出组分i和内标物s的峰面积 (或峰高)
或:
内标法中常以内标物为基准,即fs=1.0,则:
■ 内标法最关键是选择合适的内标物,对内标物的 要求:
1.定量校正因子
■ 色谱定量分析是基于被测物质的量与其峰面积的 正比关系。但由于同一检测器对不同的物质具有 不同的响应值,所以两个相等量的物质出的峰面 积往往不相等,或者说,相同的峰面积并不意味 着相等物质的量。这样就不能用峰面积来直接计 算物质的量。
■ 因此,在计算组分的量时需将面积乘上一个换算 系数,使组分的面积转换成相应物质的量。即必 须将峰面积A乘上一个换算系数进行“校正”。
例:苯、甲苯、乙苯的相对校正因子的测定:分别称取一定 量的三种物质,在25 mL容量瓶中定容。取一定量注入色谱 仪,获得色谱图,测量其峰面积,以苯为基准物,计算各组 分相对校正因子。
组分 质量/g 1
峰面积/mm2
2
3
平均
相对校 正因子
苯(标 准物)
2.22
442
Hale Waihona Puke 440438440
甲苯 2.22 429
例:试样混合物中仅含有甲醇、乙醇和正丁醇,测得峰高分
chapter5 色谱定性、定量分析方法

2012-5-20
一、色谱定性鉴定方法
色谱分析对象除少数外,大多数物质在分析前都需要预处理 例如, 色谱分析对象除少数外,大多数物质在分析前都需要预处理。例如, 预处理。 样品中含有大量的水,乙醇或被强烈吸附的物质,可导致色谱柱性能变坏。 样品中含有大量的水,乙醇或被强烈吸附的物质,可导致色谱柱性能变坏。 一些非挥发性物质进入色谱柱,本身还会逐渐降解,造成严重噪声。 一些非挥发性物质进入色谱柱,本身还会逐渐降解,造成严重噪声。还有 些物质,如有机酸,极性很强,挥发性很低,热稳定性差( 些物质,如有机酸,极性很强,挥发性很低,热稳定性差(液相色谱不考 必须先进行化学处理,才能进行色谱分析。 虑)。必须先进行化学处理,才能进行色谱分析。 色谱法是一种高效、快速的分离分析技术,它可以在很短时间内分离 色谱法是一种高效、快速的分离分析技术, 几十种甚至上百种组分的混合物,这是其他方法无法比拟的。但是, 几十种甚至上百种组分的混合物,这是其他方法无法比拟的。但是,由于 色谱法定性分析主要依据是保留值 所以需要标准样品等。 定性分析主要依据是保留值, 色谱法定性分析主要依据是保留值,所以需要标准样品等。而且单靠色谱 法对每个组分进行鉴定,往往不能令人满意——分离强 定性差。 分离强、 法对每个组分进行鉴定,往往不能令人满意 分离强、定性差。 近年来,色谱与质谱、光谱等联用 既充分利用色谱的高效分离能力。 联用, 近年来,色谱与质谱、光谱等联用,既充分利用色谱的高效分离能力。 又利用了质谱、光谱的高鉴别能力, 又利用了质谱、光谱的高鉴别能力,加上运用计算机对数据的快速处理和 检索,为未知物的定性分析开辟了一个广阔的前景。 检索,为未知物的定性分析开辟了一个广阔的前景。
色谱定性与定量

仪器分析中各分析定量定性的依据定量分析是依据统计数据,建立数学模型,并用数学模型计算出分析对象的各项指标及其数值的一种方法。
定性分析则是主要凭分析者的直觉、经验,凭分析对象过去和现在的延续状况及最新的信息资料,对分析对象的性质、特点、发展变化规律作出判断的一种方法。
1、气相色谱:色谱峰保留值和面积,这样气相色谱可根据这两个数据进行定性定量分析。
色谱峰保留值是定性分析的依据,而色谱峰面积则是定量分析的依据。
2、紫外光谱:最大吸收波长λ、摩尔吸收系数ε及吸收曲线的形状不同是进行物质定性分析的依据。
进行定量分析依据朗伯-比耳定律:A=εbc3、核磁:定量分析以结构分析为基础,在进行定量分析之前,首先对化合物的分子结构进行鉴定,再利用分子特定基团的质子数与相应峰谱的峰面积之间的关系进行定量测定。
定量分析的根据:吸收能量的大小取决于核的多少。
以磁场强度为横坐标提供定性分析所依据的参数,以吸收能量为纵坐标,纵坐标对应于不同H0的出峰面积就是定量分析参数。
4、质谱:利用电磁学原理,对物质气相离子依其质荷比(m/e)进行分离和分析的方法。
被分析的样品首先离子化,然后利用离子在电场或磁场中的运动性质,将离子按质荷比(m/e)分开并按质荷比大小排列成谱图形式,根据质谱图可确定样品成分、结构和相对分子质量。
5、原子吸收:原子吸收光谱法进行定量分析的依据是:试样中待测元素的浓度与待测元素吸收辐射的原子总数成正比,即A=k'C 。
定量分析方法有标准曲线法和标准加入法两种。
6、红外:红外光谱的定性主要根据图谱中的:基团的特征吸收频率红外光谱的定量是根据图谱中的:特征峰的强度7、离子:利用离子交换的原理,连续对多种阴离子进行定性和定量的分析。
保留时间定性,峰高或峰面积定量。
8、荧光:物质吸收的光,称为激发光;物质受激后所发射的光,称为发射光或荧光。
根据荧光的光谱和荧光强度,对物质进行定性或定量测定9、差热:定性分析:定性表征和鉴别物质依据:峰温、形状和峰数目方法:将实测样品DTA曲线与各种化合物的标准(参考)DTA曲线对照。
气相色谱仪的定性、定量分析

常用峰面积定量被测组分经
校正过的峰面积(或峰高)占样品中各组分 经校正过的峰面积(或峰高)的总和的比例
来表示样品中各组分含量的定量方法。 当试样中所有组分均能流出色谱柱,且
完全分离,并在检测器上都能产生信号时, 可用归一化法计算组分含量。
4、标准曲线法 标准曲线法也称外标法或直接比较法, 是一种简便、快速的定量方法,具体方法与 分光光度分析中的标准曲线法相似。 优点:绘制好标准工作曲线后测定工作 就变得相当简单,可直接从标准曲线上读出
含量,因此特别适合于大批样品分析。缺点: 每次样品色谱分析的色谱操作条件(检测器 的响应性能、柱温、流动相流量及组成、进 样量、柱效等)很难完全相同,因此容易出 现圈套误差。
这个结论并不准确可靠。
(2)双柱法定性。若要得到更为准确可靠 的结论,可再用另一根极性完全不同的色谱 柱,做同样的对照比较。如果结论同上,那 么最终的定性结果相对更为可靠。
(3)色谱操作条件不稳定时的定性。这时 可以采用相对保留值定性或用已知标准物增
加峰高法定性。 ① 相对保留值定性; ② 用已知标准物增加峰高法定性。 2、利用保留指数定性 在利用已知标准物直接对照定性时,已
缺点是必须在所有样品中加入内标物, 选择合适的内标物比较困难,内标物的称量 要准确,操作较复杂。
3、标准加入法 标准加入法是一种将欲测组分的纯物质 加入到待测样品中,然后在相同的色谱条件 下,分别测定加入欲测组分纯物质前后欲测 组分的峰面积(或峰高),从而计算欲测组 分在样品中的含量的方法。
优点:不需要别处的标准物质作内标物, 只需要欲则组分的纯物质,进样量不必十分 准确,操作简单,是色谱分析中较常用的定 量分析方法。缺点:要求加入欲测组分前后 两次色谱测定的色谱操作条件完全相同,否 则将引起分析测定的误差。
6--第二章色谱的定性与定量

二、定量校正因子
为何引入定量校正因子? 色谱定量分析是基于被测物质的量与其峰面积 的正比关系。但由于同一检测器对不同的物质具有 不同的响应值,所以两个相等量的物质出的峰面积 往往不相等,这样就不能用峰面积来直接计算物质
的量。这就需要引入“定量校正因子”来进行校正。
绝对校正因子 一定操作条件下,进样量(mi)与响应信号(峰面积Ai)成 正比:
若各组分的f值相近或相同,例如同系物中沸点接近的 各组分,则上式可简化为:
ω i= Ai A1+A2+…+An
×100%
对于狭窄的色谱峰,也有用峰高代替峰面积来进行定量 测定。当各种条件保持不变时,在一定的进样量范围内,峰 的半宽度是不变的,因为峰高就直接代表某一组分的量。 ω i= hi fi´´ f1 ´´ h1+ f2 ´´ h2+…+ fn ´´ hn
第六节 色谱定性方法
一、根据色谱保留值进行定性分析 1.为什么色谱保留值可以作为定性分析的依据?
因为在确定的色谱分析条件下,各物质都有确定不变的保留值。 保留值是最常用的色谱定性方法。
2.根据保留值定性的优劣
方法简便,但应用局限性大。
3.定性方法 (1)简单情况:对于较简单的多组分混合物,其所有待测
式中Mi,Ms分别为被测物和标准物质相对分子量。
3、体积校正因子fV 如果以体积计量(气体试样)则体积校正因子就是 摩尔校正因子,因为1mol任何气体在标准状态下其 体积都是22.4L。 mi /Mi *22.4 Ai fi ’(V) = fM fV = = fS ’ ms/Ms *22.4 (V) As
.
.fi ×100%
此法是通过测量内标物和待测组分的峰面积的相对值来进行计算的,因此,由于 操作条件变化所引起的误差可以得到抵消,结果比较准确。
色谱定性定量分析方法

⑥稳定性(stability):
意义: 考察分析样品与试剂在一定时间内稳定性。 内容:
根据样品与试剂测定时实际可能所处的环 境进行考察。
⑦耐用性( robustness ):
意义: 考察测定条件发生小变动时测定结果的变化。
内容:
流动相的组成和pH、商品柱的品牌尺寸、 柱温等
广泛用于药物中的杂质、体内外代谢产物的结构鉴定
重现性: 不同实验室,不同人测定的精密度 1、色谱信号的测量:
意义: 待测物浓度与响应值成线性关系的浓度范围;
相对保留值 α, (t-t0)/(tr -t0)
2、选择合适的离子源,利用LC-MS获得杂质的准分量不同浓度的对照品,比较测定值和加入值确定。
ELSD响应的自然对数与样品的浓度或质量呈线 性关系;
质谱(MS-ESI)检测器高浓度时的响应与样品 的质量可能呈二次或更复杂的方式。
四、色谱分析方法验证
目的:
证明所采用的色谱分析方法适合于相应的检验 要求,判断能否用于药品分析。
效能指标:评价分析方法的尺度
效能指标包括: 精密度、准确度、专属性、检测限、定量限、
tr
内容: LC-ESI-MS的
要求,判断能否用于药品分析。 内容: 药物制剂含量测定时的专属性考察内容:
重复性 广药泛品用 质于量药标物准中分的析杂方质法:、验体证内外代同谢产一物的实结构验鉴定室,同一人多次测定的精密度
中间精密度 2药、品选质择量合标适准的分离析子方源法,验利证用LC:-MS同获得一杂质实的准验分子室离子,峰。不同人,不同仪器测定的精密度
线性与范围、耐用性、稳定性、系统适用性等
不同分析测定方法的要求
药品质量标准分析方法验证 药物制剂人体生物利用度和生物等效性试验
第十一章 色谱分析法——定性定量分析
气相色谱法的定性分析
1、知道气相色谱流出曲线及常用的基本术语。 2、知道气相色谱的定性和定量方法
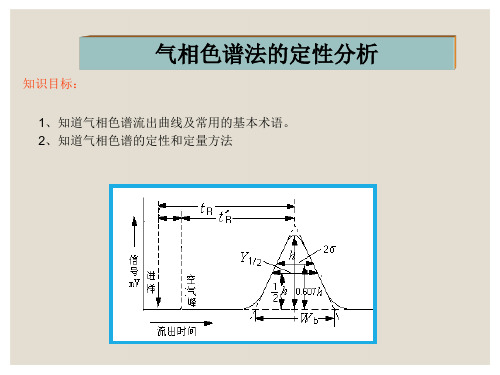
一、色谱流出曲线
色谱流出曲线:以组分电信号为纵坐标,流出时间为横坐标所得的曲线称为色谱流 出曲线或色谱图。该曲线反映了试样在色谱柱分离的效果,是组分定性和定量的依 据,同时也是研究色谱动力学和热力学的依据。
空气峰有时有,有时没有。
tM
②保留时间(tR):组分从进样到柱后出现浓度极大值时所需的时间。
③调整保留时间(t R ’): (1) t′R = tR-tM (2)反映组分在固定相中停 留的时间
(3)在实验条件一定时, t′R 决定于组分的性质,是定性 的基本参数。
(2) 相对保留值r21 组分2与组分1调整保留值之比:
内标法 当组分不能全部流出色谱柱,或检测器对样品中某些组分不产生信号,或只测
定样品中某一组分,采用内标法可获得准确结果。
1、测定步骤 (1)称取样品m样(其中:样品中待测组分i的质量用mi表示) (2)选定内标物。称取内标物ms。 (3)将内标物加入到已准确称量的样品中去。 (4)进样,测定待测组分的峰面积Ai和内标物的峰面积As。
气相色谱的定量分析 一、定量依据
样品中组分的质量与组分色谱峰的面积或峰高成正比。
m i = f i ·A i 或 m i = f i ·h i
组
绝色
分
对谱
的
校峰
质
正面
量
因积
子
文献查出
①准确测定Ai和hi ②准确求出f i ③计算mi
峰 高
峰面积A 1、定义:色谱峰与峰底基线所围成区域的面积叫峰面积。
c.将所测组分的相对保留值ris与手册数据对比作出定性判断。
气相色谱常用定量和定性方法ppt课件

定量注意事项
• 一般定量以峰面积为基准 • 所有参加计算的峰形正常(谱峰不前伸、不拖尾、不过载)的情
况下,也可以以峰高为基准进行计算 • 分子量相差不大或分子量较大的同系物校正因子相差不大,可直
接用峰面积(或峰高)定量
谢 谢!
准物S的调整保留时间ti’和ts ’ : ai,s = ti’/ ts ’
(2)计算ai,s并与文献相应值比较定性。 2.3.1.3特点 可消除实验条件不一致带来的误差。
2.3.2保留指数(I)定性法
2.3.2.1依据
保留指数I只与柱温和固定相的性质和被测物质的性质有关,与色谱柱 的尺寸、固定相的液膜厚度、载气流量、流速无关。
校正因子与待测物/标准物的性质和检测器的类型有关,可查文献, 也可测定
3.2.1定量校正因子的分类
• 质量校正因子
• 摩尔校正因子
• 体积校正因子
• fM ′ =fV ′
fm
f' m(i)
f' m(s)
m(i) A(s) m(s) A(i)
fM
f' M (i)
f' M (s)
m(i) A(s)M (s) m(s) A(i)M (i)
• 绝对校正因子:用已知准确浓度的标准 样品
3.3常用的定量计算方法
3.3.1 归一化法 3.3.2 外标法 3.3.3 单点校正法 3.3.4 内标法 3.3.5 标准加入法 3.3.6 加内标的标准加入法
3.3.1 归一化法
3.3.1.1 方法
当样品中各组分都能出峰时,将各组分的含量之和
按100%计算的定量方法。
2024/1/26
1
主要内容
1.什么是色谱定性和定量分析 2.常用的色谱定性分析方法 3.常用的色谱定量分析方法
色谱的定性和定量分析

第四章色谱的定性和定量分析色谱分析分三个阶段:仪器调试;色谱操作条件选择;定性定量分析。
气相色谱法是用载气将试样带入分离柱。
各成分在柱中分离后用检测器测定,通常是未知试样与标准試样的保留时间及峰面积比较,进行定性定量分析。
色谱法分离较容易,往往是定性较困难。
用t R定性时,因t R与分子结构有关,但两者间相关规律远未阐明.因为色谱信息少,响应信号缺乏典型的分子结构特征,因此不能鉴定未知的新的化合物,只能鉴定已知的化合物。
第一节定性分析色谱定性分析就是要确定各色谱峰所代表的化合物。
由于各种物质在一定的色谱条件下均有确定的保留值,因此保留值可作为一种定性指标。
目前各种色谱定性方法都是基于保留值的。
但是不同物质在同一色谱条件下,可能具有相似或相同的保留值,即保留值并非专属的。
因此仅根据保留值对一个完全未知的样品定性是困难的。
因为许多化合物可能在同一时间流出色谱柱,因此仅仅依靠气相色谱本身是不能对一个完全未知的化合物进行定性的。
然而当样品限定时,如果在了解样品的来源、性质、分析目的的基础上,对样品组成作初步的判断,再结合下列的方法则可确定色谱峰所代表的化合物。
气相色谱将变成一个强有力的工具。
也可以通过比较气相色谱图来确定样品是否相同,例如油轮里的原油样品可以和海上浮油比较以确定油轮是否应对原油的泄漏负责,GC对于排除可疑性是很有用的,如果您从先前的实验中知道异辛烷在1.9 分钟出峰,那么一个在1.5分钟出的峰就不会是异辛烷,那么它是什么呢?幸运的是您不必要考虑所有的有机化合物的样品信息,如果限定化合物范围。
例如您不会期望在烷烃中找到苯系物,当一个未知的峰被初步确定后,还必须在别的不同性质的色谱柱上重现以得到确认,如果一个化合物在基于沸点分离的柱甲基硅氧烷和聚乙二醇极性柱上有正确的保留时间,此定性很可能就是正确的。
GC在处理已知样品组分并且要求定量时是特别有用的。
一、保留值定性(一)利用纯物质对照定性1.利用保留时间t R对照定性色谱分析的的基本依据是保留时间。
色谱定性和定量分析方法

Identification
2019/9/22
二、 色谱定量分析方法 1. 峰面积的测量
(1)峰高(h)乘半峰宽(Y 1/2)法:近似将色谱峰当作等腰三角形。此法算 出的面积是实际峰面积的0.94倍:
A = 1.064 h·Y1/2 (2)峰高乘平均峰宽法:当峰形不对称时,可在峰高0.15和0.85处分别测定峰 宽,由下式计算峰面积:
fi' Ai
f
' s
AS
ci
%
mi W
100
ms
fi' Ai
f
' s
AS
W
100
ms W
fi' Ai
f
' s
AS
100
2019/9/22
内标法特点
(1) 内标法的准确性较高,操作条件和进样量的稍许变动对定量结果的影响 不大。
(2) 每个试样的分析,都要进行两次称量,不适合大批量试样的快速分析。 (3)若将内标法中的试样取样量和内标物加入量固定,则:
Ai Ai
)
100
i 1
特点及要求: 归一化法简便、准确; 进样量的准确性和操作条件的变动对测定结果影响不大; 仅适用于试样中所有组分全出峰的情况。
2019/9/22
(2)外标法
外标法也称为标准曲线法。 特点及要求: 外标法不使用校正因子,准确性较高, 操作条件变化对结果准确性影响较大。 对进样量的准确性控制要求较高,适用于大批量试样的快速分析。
1.0 DEG/MI N
HEWLET PTACKAR
5972A
D
Mass Selectiv eDetecto r
6.色谱分析中的定性与定量方法

色谱定量分析
色谱定量分析
• 绝对校正因子:单位峰面积对应的物质量:fi = mi/Ai • 定量校正因子与检测器响应值成倒数关系: • fi=1/Si • 相对校正因子fi :即组分的绝对校正因子与标准物质 的绝对校正因子之比。
fi mi / Ai mi As fi f s ms / As ms Ai
• 化学反应定性:利用化学反应,使样品中某些 化合物与特征试剂反应,生成相应的衍生物。 • 柱前反应:被分离混合物进入色谱柱前与某些 特征性试剂反应,观察色谱图上某些色谱峰发 生消失、提前或滞后而断定有无此类化合物。 • 柱 上: 如装有5A分子筛的前置柱,可吸附C3C11的正构烷烃,KOH处理的石英粉可将羧酸 和酚除去(吸附)等。 • 柱 后: 柱后流出物收集后,加入特征试剂与 其反应,可对未知物定性。
色谱定量分析
• 叠加内标法:以样品中已有的组分做内标,比 较该组分加入前后面积的改变,计算被测组分 含量
A1 mS ci % 100 A2 A1 W
• 特点:要求两次进样量完全相同。
色谱定量分析
• 叠加内标法:两次进样量不同时的处理
'
• 当 m i 、 m S 为质量单位时,为质量相对校正因子;当 m i 、 mS用摩尔单位时相应于摩尔校正因子。
色谱定量分析
• 归一化法:
mi ci % 100 m1 m2 mn
f i ' Ai
' ( f i Ai ) i 1 n
100
归一化法的特点: • 对同系物可认为校正因子一样,通过峰面积直接测定; • 进样量的准确性和操作条件的变动对测定结果影响不 大; • 试样中所有组分必须全出峰并且无分解反应发生。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
A Ax
Cx Cs
③内标法
用一定量的纯物质作内标物,加入到准确称量的试样中,根
据被测试样和内标物的质量比及相应的色谱峰面积之比,计
算被测组分的含量
mi fi Ai ms fs As
mi fi Ai ms f s As
mi
f is
Ai ms As
i
mi m试样
100 %
f is
4影响定量分析结果准确度的因素
样品制备
前处理方法,转化率,溶剂,供试液储存,
进样技术
与外标法关系密切,手动进样,阀进样
色谱条件
流动相流速,流动相组成,温度,柱效,色谱峰的对称性
检测器性能
线性关系,浓度型检测器,质量型监测器,灵敏度
分离度——大则影响小
5分析方法的评价——方法学考察
mi=fiAi
这就是色谱定量分析的依据
绝对校正因子主要由仪器的灵敏度所决定,并与分 析的操作条件有密切关系
2.峰面积的测量
①准确测量法
用计算机测量 通常高级色谱仪配有计算机,能自动计算峰面积
用自动积分仪测量 它能自动测出曲线所包围的面积,测量精度达
0.2~2%
上述两种方法分析速度快,线性范围广,对小峰 或不规则的峰法结合定性
①与化学方法结合进行定性 将试样经过一些特殊试剂处理,发生物理
变化或化学反应后,其色谱峰将会提前、移 后或完全消失
比较处理前后色谱图的差异,以及在柱后 用化学试剂鉴定流出物,就可初步定性鉴别 试样中含有哪些官能团
2.与其它方法结合定性
②与光谱、质谱联合定性
线性关系
测绘标准曲线,指标:相关系数r>0.9999, 获得线性范围
精密度
平行测定多次,指标:变异系数(RSD%) 应从样品制备做起
准确度
加样回收实验,指标:回收率%——接近100% 应从样品制备做起
也可用内标校正曲线法进行定量测定
④ 标准加入法
是一种特殊的内标法。设样品中待测组分含量为Wi
首先在一定色谱条件下测定样品的色谱图,得待测组分的峰面积Ai
然后在该样品中加入已知量为△Wi的待测组分,并在相同色谱条件 下测得加入后的待测组分峰面积A’i。
计算公式:wi fi Ai
wi wi fi Ai'
Ai ms As m试样
100 %
ms和mi分别为内标物和被测组份的质量
As和Ai分别为内标物和被测组份的峰面积
fis为组份i相对于内标物s的相对校正因子
选择内标物有四个要求:
(a)内标物应是该试样中不存在的纯物质; (b)它必须完全溶于试样中,并与试样中各组份的色谱峰能完全分离; (c)加入内标物的量应接近于被测组份; (d)色谱峰的位置应与被测组份的色谱峰的位置相近,或在几个被测 组份色谱峰中间。
②外标法
A标准曲线法 用标样配成一系列不同浓度的标准溶液,在一定操作条件 下,分别定量进样测量,绘制峰面积对标液浓度的标准曲 线。并在相同条件下,注入相同体积的被测试样测出峰面 积,在标准曲线上求出被测组份的含量 B外标一点法(单点校正) C外标两点法(两点校正)
外标法简便,不需用校正因子,但需要标样并且进样量要 求十分准确,操作条件也需严格控制
色谱仪与光谱或质谱仪等联用,实际上是光谱仪和质谱仪等起着 检测器的作用,将复杂组份的混合物经色谱柱分离为单组份,再利 用红外光谱、质谱或核磁共振谱等进行定性分析
既充分利用光谱、质谱等适于分析分子结构、官能团或物质的摩 尔质量等特点,克服了它们不易检定复杂物质的困难,又充分利用 了色谱的高效分离能力
色谱定性定量方法
一、色谱定性方法
定性依据 ★主要依据是每个组份的保留值
★一般需要标准样品,离开已知纯物质的对照, 就无法识别各色谱峰代表何种组份
★对某一未知试样,单独用色谱法定性十分困 难,因此常需与化学分析及其它仪器分析方法 相结合
1.利用色谱保留参数定性
①保留值定性
在一定的固定相和恒定的操作条件(如柱温、流动相流速、固 定或液VR含)量,、一柱般长不和受柱其径它等组)份下的,影每响种,物表质现都为有每一一定组的份保的留特值征(值tR
2.峰面积的测量
②近似测量法 对称峰可用峰高乘半高峰宽法:
A 1.065 hWh / 2
在相对计算时可省略系数,狭窄峰还可将半高峰 宽视为常数以峰高计算
1 不对称峰可用峰高乘平均峰宽法: A 2 h(W0.15 W0.85 )
平均峰宽是指在峰高0.15和0.85处分别测量的峰 宽的平均值
wi wi Ai'
wi
Ai
wi
wi Ai' 1
Ai
标准加入法
优点:不需要另外的标准物质为内标,只需要待测组分的 纯物质,进样量不必十分准确,若在阳平前处理之前就加 入已知准确量的的测组分,则完全可以补偿待测组分在前 处理过程中的损失。对样品中的其他组分可作为内标。
要求:待测组分加入前后的色谱条件要完全一致。
内标法的优点:
测定的结果较为准确,由于通过测量内标物及被测组份的峰面积的相对 值来进行计算,因而在一定程度上消除了操作条件等的变化所引起的误 差, 在样品前处理前加入内标,再进行处理,可部分补偿待测组分在样 品前处理中的损失。
内标法的缺点:
A.操作程序较为麻烦,每次分析时内标物和试样都要准确称量 B.有时寻找合适的内标物也有困难
1.利用色谱保留参数定性
②加入已知物增加峰高法
首先用被测试样作色谱图,然后将已知纯物质加 到试样中去,在相同的条件下作色谱图,对比这两 个色谱图
若后一色谱图中某一色谱峰相对增高时,则该色谱 峰的组份原则上与加入已知纯物质是同一种化合物
当试样组份比较复杂,峰间距离太近,或操作条 件不易控制稳定,很难准确地测定其保留值时,可 采用此法进行定性
联用方法是解决复杂未知物定性问题最有效的方法之一,其中特 别是色谱-质谱联用分析,是当今分离和鉴定未知物最好的手段
二、色谱定量方法
1.定量依据和校正因子
在一定的操作条件下,被测组份的质量mi与检测器 产生的响应信号(色谱图上表现为峰面积)Ai成正比, 比例系数称为峰面积绝对校正因子fi,即
因此可利用已知物的保留值和未知物的保留值对照进行定性
利用绝对保留值定性时,要求严格控制色谱操作条件,否则重 现性较差
某若些采操用作相条对件保差留异值所r带2,1来作的定影性响分析,它仅与柱温有关,则可消除
常用标准物:苯、正丁烷、对二甲苯、环己烷、2,3,4-三甲基戊 烷
对于组份比较简单的已知范围的混合物试样,可采用此法进行 定性。也可利用文献上的r2,1值或色谱手册中的r2,1值对照定性。
3.常用定量方法
①归一法
若试样中各组份都能流出色谱柱并在所用的检测器上都能产 生信号显示出色谱峰,则可测量所有组份色谱峰的峰面积, 由下列公式计算各组份的含量
i
A1 f1
Ai A2 f 2
fi
An
fn
100 %
Ai为任一组份的峰面积,fi为任一组份的质量校正因子 归一法的优点是无需标样,结果准确,操作简便,操作条件 (如进样量、流速等)变化对测定结果影响较小,宜于分析 多组份试样中各组份的含量。
1.利用色谱保留参数定性
③双柱法定性
将试样和标准物的混合物分别在两根极性相差较 大的色谱柱上进行色谱分离
观察标准物和未知物色谱峰在这两根柱子上是否 始终重合,如两色谱峰始终重合,可判断为同一组 份,否则不是同一组份。
可以避免不同组份由于保留值的偶然一致性,可 能发生的定性错误。