实验用色谱纯甲醇及乙腈验收记录表-甲醇
保健食品中总皂苷的测定

附件:一、保健食品中大蒜素的测定1 范围本方法适用于以大蒜及其制品为原料的保健食品中大蒜素(三硫二丙烯,C6H10S3)的含量测定。
本方法最低检出浓度为0.0430mg/mL;取100mg试样,提取定容5.0mL时,试样中大蒜素最低检出质量浓度为0.20g/100g。
本方法最佳线性范围: 0.320mg/mL~3.00 mg/mL。
2 原理:根据大蒜素为挥发性油成分,经有机溶剂提取,用气相色谱仪分析,采用外标法定量。
3 试剂除特殊说明,所用试剂均为分析纯。
3.1 无水乙醇3.2 正己烷3.3标准溶液:称取0.1000g标准品,置于10mL容量瓶中,用正己烷定容至刻度,该溶液中含大蒜素浓度为10.0mg/mL。
此溶液可在冰箱中保存七天。
取该溶液1.0mL,置于10mL容量瓶中,用正己烷定容至刻度,此溶液含大蒜素浓度为1.0mg/mL。
4 仪器4.1气相色谱仪:附氢火焰(FID)检测器4.2数据处理机或积分仪4.3分析天平:万分之一4.4超声清洗机4.5离心机:3000r/min5分析方法5.1试样提取5.1.1固体试样:称取试样适量(相当于含大蒜素5mg,精确至0.001g),加无水乙醇适量,密塞,超声70min,取出冷却,加正己烷定容(调节大蒜素含量约为1mg/mL),振摇,静置分层后,取上层溶液进样。
5.1.2液体试样吸取试样适量(相当于含大蒜素5mg,精确至0.001g),置于分液漏斗中,加5mL 正己烷振摇提取1min ,静置(或离心)分层后,取上层溶液进样。
5.2 气相色谱参考条件5.2.1 色谱柱:HP-5(30m×0.25mm)5.2.2 柱箱温度:100℃(3min )min)10(℃200℃150℃/min 20℃/min 10−−−→−−−−→−5.2.3 进样口温度:220℃ 5.2.4 检测器温度:250℃ 5.2.5 载气:氮气(1mL/min )5.2.6 氢气:40mL/min ;空气:400mL/min 5.2.7 进样量:1μL5.3 定性分析:在参考操作条件下,以对照品与试样比较保留时间定性。
QuEChERS_结合液相色谱质谱联用技术测定动物源性食品中N-_二甲基亚硝胺

QuEChERS结合液相色谱质谱联用技术测定 动物源性食品中N-二甲基亚硝胺赵广西1,刘志梅2(1.秦皇岛市食品药品检验中心,河北秦皇岛 066000;2.秦皇岛市市场监督管理局,河北秦皇岛 066000)摘 要:目的:建立高效液相色谱质谱联用技术定量检测动物源性食品中的N-二甲基亚硝胺。
方法:样品前处理粉碎后经乙腈提取,低温离心除去油脂,QuEChERS净化,采用多反应监测模式测定。
结果:N-二甲基亚硝胺校准曲线在2.0~200.0 ng·mL-1线性关系良好,相关系数为0.998 5,方法检出限和定量限分别为0.5 μg·kg-1和3.0 μg·kg-1,回收率为86.8%~90.8%,相对标准偏差在2.2%~3.1%。
结论:该方法取样量少,试剂消耗量少,提取和净化流程简便,定性和定量准确度好,可用于检测动源性食品中N-二甲基亚硝胺。
关键词:高效液相色谱-质谱联用技术;QuEChERS;N-二甲基亚硝胺;多重反应监测Determination of N-Dimethylnitrosamine in Food of Animal Origin by QuEChERS Combined with LC-MS/MSZHAO Guangxi1, LIU Zhimei2(1.Qinhuangdao Municipal Food and Drug Inspection Center, Qinhuangdao 066000, China; 2.QinhuangdaoAdministration of Market Regulation, Qinhuangdao 066000, China)Abstract: Objective: To establish a high performance liquid chromatography-mass spectrometry method for the quantitative determination of N-dimethylnitrosamine in animal derived food. Method: The sample was extracted by acetonitrile after pretreatment and grinding, and the oil was removed by low temperature centrifugation, purified by QuEChERS, and determined by multi-reaction monitoring mode. Result: The calibration curve of N-dimethylnitrosamine had a good linear relationship in the range of 2.0~200.0 ng·mL-1, and the correlation coefficient was 0.998 5. The detection limit and quantitative limit of the method were 0.5 μg·kg-1 and 3.0 μg·kg-1, respectively. The recovery rate was 86.8%~90.8%, and the relative standard deviation was 2.2%~3.1%. Conclusion: This method has the advantages of less sampling, less reagent consumption, simple extraction and purification process, good qualitative and quantitative accuracy, and can be used to detect N-dimethylnitrosamine in animal-derived food.Keywords: high performance liquid chromatography-mass spectrometry; QuEChERS; N-dimethylnitrosamine; multiple reaction monitoring肉、鱼等食品中富含蛋白质和脂肪,在煎、炸、烤等热处理过程中会产生较多的胺类化合物,易与亚硝酸盐反应生成亚硝胺类化合物,其中主要是N-亚硝基二甲胺。
HG-JC-52.D0 农药产品中隐性成分检测方法细则

农药产品中隐性成分检测方法细则1. 概述本检测方法细则是根据本实验室仪器的实际配置情况及现有的检测能力进行编写,适用于本实验室使用气相色谱质谱联用仪对农药产品中的隐性成分的定性检测。
2. 适用范围本检测方法细则适用于本实验室农药产品中隐性成分的定性检测。
3. 依据标准农药管理条例4. 试验方法4.1 试剂和溶液乙腈:色谱纯标准品:溴甲烷(100ug/mL甲醇)、氟乙酰胺(100ug/mL乙醇)、1,2-溴乙烷(99.0%)、二溴氯丙烷(98.5%)、灭多威(99.0%)、甲胺磷(99.2%)、灭线磷(100ug/mL丙酮)、杀虫脒(99.5%)、硫线磷(100ug/mL丙酮)、氧乐果(100ug/mL丙酮)、治螟磷(100ug/mL丙酮)、甲拌磷(98.6%)、α-六六六(100ug/mL正己烷)、内吸磷(100ug/mL丙酮)、久效磷(100ug/mL丙酮)、克百威(98.6%)、特丁硫磷(100ug/mL丙酮)、地虫硫磷(100ug/mL丙酮)、γ-六六六(100ug/mL正己烷)、氯唑磷(100ug/mL甲醇)、磷胺(100ug/mL丙酮)、β-六六六(100ug/mL正己烷)、δ-六六六(100ug/mL正己烷)、甲基对硫磷(99.0%)、三氯杀螨醇(98.1%)、对硫磷(100ug/mL丙酮)、水胺硫磷(99.0%)、甲基异柳磷(95.9%)、甲基硫环磷(97.2%)、硫环磷(100ug/mL丙酮)、克线磷(100ug/mL乙酸乙酯)、氟虫腈(99.0%)、O,P’-滴滴涕(100ug/mL正己烷)、除草醚(1000ug/mL异辛烷)、硫丹(98.0%)、P,P’-滴滴涕(100ug/mL正己烷)、蝇毒磷(100ug/mL丙酮)、毒死蜱(99.0%)、戊唑醇(99.0%)、螺螨酯(99.0%)、多效唑(99.0%)、虫螨腈(99.0%)、联苯菊酯(99.0%)、啶虫脒(99.0%)。
4.2 仪器气相色谱质谱联用仪:Agilent7890A-5975C色谱柱:DB-1301(60m×0.25mm×0.25μm),色谱数据处理机(或工作站)4.3 试验条件程序升温:初始温度45℃,保持8min,以30℃/ min程序升温至120℃,保持5 min,再以30℃/ min程序升温至200℃,保持5min, 再以60℃/ min程序升温至270℃,保持18min总运行时间42.3min;载气:氦气(纯度不低于99.999%);进样口温度:250℃;恒流模式,流速为1.5mL/min;进样量:1μL;分流比:20:1;全扫描模式,扫描范围:10-500amu;电子轰击源(EI);电离能70eV;离子源温度:230℃;GCMS接口温度:270℃;四级杆温度:150℃。
甲醇分析原始记录表

工作曲线
标液来源: 标液编号:
备 注
序号
组分
回归方程
检出限
r
1
2
3
4
5
6
7
8
9
10
分析/日期:校核/日期:复核/日期:
甲醇分析原始记录表
第 共 页
项目编号:分析项目:样品性质:收样日期:分析日期:环境温度:℃相对湿度:%RH
甲醇分析原始记录表
第 页 共 页
项目编号:分析项目:样品性质:收样日期:分析日期:环境温度:℃相对湿度:%RH
分析方法:仪器型号规格:仪器管理编号:检测地点:.
测试条件
仪器型号:
色谱柱类型:□填充柱 □毛细管柱
□热解析□直接进样□溶剂解吸□自动进样
仪器编号:
色谱柱型号:
N2: MPa H2: ml/min 分流: ml/min
样品编号
定容体积( )
测定值 ()
标况体积(L)
空白值()
结果值()
计算公式
分析/日期:校核/日期:复核/日期:
甲醇分析原始记录表(续表)
第 页 共 页
项目编号:分析项目:样品性质:收样日期:分析日期:环境温度:℃相对湿度:%RH
样品编号
定容体积( )
测定值 ()
标况体积(L)
空白值()
结果值()
计算公式
分析/日期:校核/日期:复核/日期:
高效液相色谱法测定室内空气中16种醛、酮类化合物

高效液相色谱法测定室内空气中16种醛、酮类化合物发布时间:2022-07-13T01:18:42.147Z 来源:《中国科技信息》2022年5期3月作者:杨卉杨云王赛[导读] 利用高效液相色谱对2,4-二硝基苯肼衍生化生成的16种醛酮-DNPH化合物的方法进行研究。
杨卉杨云王赛天津市环科检测技术有限公司摘要:利用高效液相色谱对2,4-二硝基苯肼衍生化生成的16种醛酮-DNPH化合物的方法进行研究。
考察了流动相、仪器条件等对分离效果的影响。
以纯水-乙腈-甲醇为流动相进行梯度洗脱,对16种醛酮-DNPH进行定量分析。
试验结果表明,在C18色谱柱上,流速为1.0mL/min,柱温为30度时,16种醛酮-DNPH化合物具有良好的线性范围,每个化合物的相关系数在0.999以上,最低检出限为0.001mg/m3~0.002mg/m3,平行样相对偏差在0.7%~9.3%,样品加标回收率为78.1%~100%。
关键词:醛酮类化合物,高效液相色谱,室内空气1.引言醛和酮都是含有羰基官能团的化合物。
空气中醛酮类化合物分低分子醛酮化合物和高分子醛酮化合物。
环境中最关注的是低分子醛酮化合物,一方面低分子醛酮化合物的毒性大,另一方面它是光化学烟雾中的主要成分。
近年来,由于室内装修的兴起,装修使用的建筑、装饰材料中的脲醛树脂、胶合板油漆、染料以及新家具等产生的气体中也有醛类物质,所造成的室内环境污染问题也越来越引起人们的重视。
资料提示,室内空气污染物浓度多为室外的2~5倍,甚至可高达100多倍[1,2],醛、酮类化合物会刺激皮肤与粘膜及毒害中枢神经系统。
因此,室内空气检测过程的可靠性及分析结果的准确度对了解室内空气质量状况尤为关键。
2 实验部分2.1方法原理采用饱和 2,4-二硝基苯肼(DNPH)盐酸溶液吸收室内空气中的醛、酮类化合物,在酸性介质中醛、酮类化合物与吸收液中的 2,4-二硝基苯肼(DNPH)反应,生成稳定的 2,4-二硝基苯腙类衍生物,用二氯甲烷溶剂萃取、浓缩后,用高效液相色谱分离,紫外检测器检测。
基于离子液体萃取分离甲醇-乙腈共沸物机理的综合实验教学设计

ISSN 1002-4956 CN11-2034/T实验技术与管理Experimental Technology and Management第38卷第3期202丨年3月Vol.38No.3Mar.2021D O I: 10.16791 /j .cnki.sjg.2021.03.012基于离子液体萃取分离甲醇-乙腈共沸物机理的综合实验教学设计朱久娟,范寒寒,梁树平,孙兵(中国地质大学(北京)数理学院,北京100083 )摘要:结合大地质类物理化学和仪器分析的教学要求及学科前沿方向,该文设计了离子液体笮取分离甲醇-乙腈共沸物机理的综合实验s通过动态汽液相平衡釜-气相色谱联用实验方案,以丨-r基-3-甲基咪唑=氟甲磺酸盐([BM IM]OTF)为萃取剂、甲醇-乙腈共沸物系为研究对象,通过研究离子液体对甲醇-乙腈物系等压相平衡的影响,分析离子液体萃取分离共沸物系的机理.该实验结合物理化学、仪器分析、大学化学等专业课程内容,将学科基础知识与大型仪器创新研究有机融合,夯实学生理论知识,激发学生科研兴趣,提高学生创新能力=关键词:萃取分离;共沸物分离;实验教学中图分类号:06-3;G642.423 文献标识码:A 文章编号:1002-4956(2021)03-0057-04Comprehensive experiment teaching design based on separation of methanol-acetonitrile azeotrope with ionic liquidZHU Jiujuan,FAN Hanhan,LIANG Shuping,SUN Bing(School of Science, China University of Geosciences (Beijing), Beijing 100083, China)Abstract: Combined with the teaching requirements and frontier direction of physical chemistry and instrumental analysis in geology, a comprehensive experiment on the extraction and separation mechanism of methanol acetonitrile azeotrope by ionic liquid is designed. The effect of ionic liquid on isobaric phase equilibrium of methanol acetonitrile system is studied, and the mechanism of extraction separation of azeotropic system by ionic liquid is analyzed by using l-butyl-3-methylimidazolium trifluoromethanesulfonate ([BMIM] OTF) as extractant and methanol acetonitrile azeotrope system as the research object. This experiment combines physical chemistry, instrumental analysis, college chemistry and other professional courses, organically integrates the basic knowledge of the discipline with the innovative research of large-scale instruments, tamps students' theoretical knowledge, stimulates their interest in scientific research, and improves their innovative ability.Key words: extraction; azeotrope separation; experiment teaching萃取分离技术是目前分离共沸物应用最为广泛的 分离技术[1_2],离子液体作为一种新型无污染的绿色萃 取剂,具有可设计性强、稳定性好、难挥发性和良好 的导电性等特点,不仅可以解决传统萃取剂(有机溶剂、无机盐、复合溶剂)分离过程中的能耗问题,同时还可以减轻无机盐对设备的腐蚀,降低有机溶剂对 环境的污染[3_41。
中药饮片—— 甲醇量原始记录

甲醇量检验原始记录
编号:
品名
规格
温度
编码(批号)
请验日期
湿度
色谱条件与系统适用性试验
采用(6℃)氰丙基苯基-(94℃)二甲基聚硅氧烷为固定液的毛细管柱;起始温度为40℃,维持2分钟,以每分钟3℃的速率升温至65℃,再以每分钟25℃的速率升温至200℃,维持10分钟;进样口温度为200℃;检测器(FID)温度220℃;分流进样,分流比为1:1;顶空进样平衡温度为85℃,平衡时间为20分钟。理论板数按甲醇峰计算应不低于10000,甲醇峰与其色谱峰的分离度应大于1.5。
供试品溶液与对照品溶液的制备
取供试液作为供试品溶液。精密量取甲醇ml,置100ml量瓶中,加水稀释至刻度,摇匀,精密量取ml,置100ml量瓶中,加水稀释至刻度,摇匀,作为对照品溶液,浓度为,批号()。
测定方法
分别精密量取对照品溶液与供试品溶液各3ml,置10ml顶空进样瓶中,密封,顶空进样。按外标法以峰面积计算,即得。
含量计算
供试品溶液平均峰面积
对照品溶液平均峰面积
甲醇量=——————————————×对照浓度×100%
计算结果:
RSD=
实测结果
供试液含甲醇量为(ml/ml)
标准规定
供试液含甲醇量不得过0.05%(ml/ml)。
结论
□(均)符合规定□(均)不符合规定
梯度洗脱反相高效液相色谱法测定恩替卡韦的有关物质

梯度洗脱反相高效液相色谱法测定恩替卡韦的有关物质蒋银妹1,罗秀琴2,徐燕2,毕开顺13(1.沈阳药科大学药学院,沈阳110016;2.长沙市华美医药研究所,长沙410007)摘要 目的:建立测定恩替卡韦有关物质的梯度洗脱反相高效液相色谱法。
方法:采用W aters C18色谱柱(5μm,416mm×250 mm),以乙腈-水(3∶97)为流动相A,乙腈为流动相B;梯度洗脱条件:0~10m in,A从100%→90%;10~15m in,A为90%;15~25m in,A从90%→60%;25~28m in,A从60%→100%;以110mL・m in-1的流速进行梯度洗脱,检测波长为254n m。
结果:在上述色谱条件下,恩替卡韦与各中间体杂质及降解杂质均能有效分离,分离度大于210;检测限为014ng,精密度良好(RS D为018%)。
结论:本方法操作简便,专属性强,灵敏度高,可用于恩替卡韦有关物质的测定。
关键词:恩替卡韦;有关物质;反相高效液相色谱法;梯度洗脱中图分类号:R917文献标识码:A文章编号:0254-1793(2009)04-0676-04HP LC deter m i n ati on of rel ated subst ances i n entecavi rJ I A NG Yin-mei1,LUO Xiu-qin2,XU Yan2,B I Kai-shun13(1.Shenyang Phar maceutical University,Shenyang110016,China;2.I nstitute of HameiMedicine of Changsha City,Changsha410007,China.) Abstract O bjecti ve:T o establish an RP-HP LC method f or related substances deter m inati on of ent ecavir.M eth2 od:The W aters C18colu mn(5μm,416mm×250mm)was used.Mobile phase A:acet onitrile-water(3∶97);Mo2 bile phase B:acet onitrile.Conditi on of the gradient eluti on:0-10m in,A:100→90%;10-15m in,A:90%;15-25m in,A:90→60%;25-28m in,A:60%→100%;The gradient eluti on was purf or med with a fl ow rate of110 mL・m in-1,the detecti on wavelength was254nm.Results:Good chr omat ographic separati on of entecavir and its inter mediate p r oducts and by-p r oducts were obtained.The detecti on li m it is014ng and the p recisi on was good with RSD of018%.Conclusi on:The method is convenient,highly s pecific,sensitive and suitable for deter m inati on of the related substances of entecavir.Key words:entecavir;related substance;RP-HP LC;gradient eluti on 恩替卡韦化学名为2-氨基-9-[(1S,3R, 4S)-4-羟基-3-羟甲基-2-亚甲基环戊基]-1,9-二氢-6H-嘌呤-6-酮一水合物,是由百时美施贵宝公司自主研发的一种核苷类似物,已于2005年3月30日在美国获得美国食品药品监督管理局(F DA)的上市批准,剂型有片剂(规格:015, 110mg・片-1)和口服液(规格:0105mg・mL-1),适用于病毒复制活跃、血清转氨酶(ALT)持续升高或有肝脏组织学显示活动性病变的成人慢性乙型肝炎患者。
高效液相色谱法同时测定化妆品中9种4-羟基苯甲酸酯类防腐剂

化学分析计量CHEMICAL ANAL Y SIS AND METERAGE第30卷,第5期2021年5月V ol. 30,No. 5May 202146doi :10.3969/j.issn.1008–6145.2021.05.011高效液相色谱法同时测定化妆品中9种4-羟基苯甲酸酯类防腐剂邢海艳,孙丽,吴旭,鲁素雅,金鹏,金华(淮安市食品药品检验所,江苏淮安 223005)摘要 建立同时测定化妆品中9种4-羟基苯甲酸酯类防腐剂的高效液相色谱法。
样品经甲醇溶解,超声提取,使用Waters Symmtery C 18(2.1 mm ×250 mm ,5 μm )色谱柱,用0.1%磷酸溶液–乙腈为流动相进行梯度洗脱,采用二极管阵列检测器进行检测,检测波长为254 nm ,外标法定量。
9种防腐剂的质量浓度在0.1~100 μg /mL 范围内与对应的色谱峰面积线性关系良好,相关系数为0.999 3~0.999 7,检出限为0.65~2.22 μg /g ,定量限为2.18~7.39 μg /g 。
样品在25、50、250 μg /g 3个加标水平时的平均回收率为95.73~104.06%,测定结果的相对标准偏差为0.58~5.28%(n =6)。
该方法简单、准确、灵敏,可用于化妆品中9种4-羟基苯甲酸酯类防腐剂的测定。
关键词 高效液相色谱法;4-羟基苯甲酸酯类;化妆品;防腐剂中图分类号:O657.7 文献标识码:A 文章编号:1008–6145(2021)05–0046–05Simultaneous determination of nine kinds of 4-hydroxybenzoate preservatives in cosmeticsby high performance liquid chromatographyXing Haiyan ,Sun Li ,Wu Xu ,Lu Suya ,Jin Peng ,Jin Hua(Huai ’an Institute for Food and Drug Control, Huai ’an 223005, China)Abstract A method for simultaneous determining of nine kinds of 4-hydroxybenzoate preservatives in cosmetics by high performance liquid chromatography was established. The sample was dissolved in methanol and extracted by ultrasound. Waters Symmtery C 18 (2.1 mm ×250 mm ,5 μm) column was used for gradient elution with 0.1% phosphoric acid solution–acetonitrile as mobile phase. Diode array detector was used for detection, the detection wavelength was 254 nm and the external standard method was used for quantitative analysis. The mass concentration of nine preservatives had a good linear relationship with the chromatographic peak area in the range of 0.1–100 μg /mL, the correlation coef fi cient was 0.999 3–0.999 7, the detection limits were 0.65–2.22 μg /g and the quantification limits were 2.18–7.39 μg /g. The average recoveries of standard addition in three different levels of 25, 50 and 250 μg /g were 95.73–104.06%, and the relative standard deviations of the determination results were 0.58–5.28%(n =6). The method is simple, accurate and sensitive, which can be used for the determination of nine kinds of 4-hydroxybenzoate preservatives in cosmetics.Keywords high performance liquid chromatography; 4-hydroxybenzoates; cosmetics; preservative通信作者 鲁素雅,硕士,主管药师,从事化妆品、药品等研究,****************收稿日期 2021–03–03引用格式 邢海艳,孙丽,吴旭,等.高效液相色谱法同时测定化妆品中9种4-羟基苯甲酸酯类防腐剂[J ].化学分析计量,2021,30(5): 46.Xing Haiyan, Sun Li, Wu Xu, et al. Simultaneous determination of nine kinds of 4-hydroxybenzoate preservatives in cosmetics by high performance liquid chromatography [J ]. Chemical Analysis and Meterage ,2021,30(5): 46.防腐剂是指天然或合成的化学成分,用于加入食品、药品、化妆品等以延迟微生物生长或化学变化引起的产品腐败,从而能够长久保持产品的安全性和稳定性[1–3]。
高效液相色谱法检测防腐橡胶木中的碘代丙炔基丁基氨基甲酸酯

高效液相色谱法检测防腐橡胶木中的碘代丙炔基丁基氨基甲酸酯作者:李晓文韩利平李彤彤张景朋蒋明亮来源:《热带作物学报》2021年第11期摘要:橡胶木中淀粉含量远高于其他树种,易发霉、蓝变。
碘代丙炔基丁基氨基甲酸酯(3-iodo-2-propynyl butyl carbamate,IPBC)绿色环保,对霉菌、蓝变菌有特效,尤其适用于橡胶木保护。
目前橡胶木中IPBC分析方法的研究报道较少,其防腐处理材的质量监督尚缺乏有效手段。
为了准确分析防腐橡胶木中的IPBC,建立了一种高效液相色谱(high-performance liquid chromatography,HPLC)方法,检验了该方法的检出限、加标回收率等性能,并比较了不同提取方法、提取溶剂、木粉粒径等因素对防腐橡胶木中IPBC提取效果的影响。
建立的HPLC方法使用C18反相色谱柱,乙腈-水(55︰45,V/V)为流动相,流速1.0 mL/min,检测波长204 nm,IPBC保留时间约6.45 min。
在2~80 mg/L浓度范围内,线性关系良好,相关系数0.9998;检出限0.2 mg/L,平均加标回收率99.61%。
超声法提取的效果显著优于索氏抽提器法;以甲醇或甲醇-水(9︰1, V/V)混合液为溶剂超声提取的效果较好,乙酸乙酯和石油醚不适于提取木材中的IPBC。
以甲醇-水(9︰1, V/V)混合液为溶剂制备样品可改善色谱峰形,有助于提高分析方法的准确性。
木粉粒径差异对IPBC超声提取的影响较小。
高温条件会导致IPBC降解,严重影响分析结果准确性,因此在整个分析过程中应避免高温条件。
本方法检出限低、重现性好,准确性高,适于分析防腐木材中微量的IPBC。
关键词:橡胶木;高效液相色谱;碘代丙炔基丁基氨基甲酸酯;提取方法;防腐木材中图分类号:S782.33 文献标识码:AAbstract: Rubber wood is known to be very susceptible to mold and sap stain fungi attacks because of its extraordinary high starch content. 3-iodo-2-propynyl butyl carbamate (IPBC) is suitable for rubber wood preservation as it is highly effective in mold and sap stain fungi, and environmentally friendly. However, few studies on the quantitation methods of IPBC in treated rubber wood have been reported. To determine IPBC in treated rubber wood accurately, a high-per¬formance liquid chromatography (HPLC) method was developed, and the optimization of extracting IPBC from rubber wood was conducted by comparing the effects of different extraction methods, solvents, and particle sizes of wood powder. The HPLC conditions were as follows:C18 reversed-phase column, acetonitrile-water (55︰45, V/V) as mobile phase, flow rate 1.0 mL/min, detection wavelength 204 nm, the retention time of IPBC about 6.45 min. There was a good linear correlation between the mass concentration of IPBC and the peak area with a correlation coefficient of 0.9998 in the range of 2~80 mg/L. The limit of detection (LOD) was 0.2 mg/L,and the average recovery of spiked samples was 99.61%. The comparison of extraction methods showed that ultrasonic extraction was significantly better thanthe Soxhlet extraction. Methanol or methanol-water (9︰1, V/V) mixed solution was the best solvent to extract IPBCfrom wood, but ethyl acetate and petroleum ether were not suitable for extracting. Using methanol-water (9︰1, V/V) asthe solvent could eliminate the leading peak, which was helpful to improve the accuracy of the analytical method. Thedifference in wood powder particle size had little effect on the analysis results of IPBC extracted by ultrasonic. The enormous degradation of IPBC due to high temperature greatly affected the analysis results, therefore, high-temperature conditions should be avoided during the analysis process. With the advantages of low detective limit, good reproducibility, and high accuracy, the developed method may have great potential for the analysis of IPBC in preservative woods.Keywords: rubber wood; high-performance liquid chromatography (HPLC); 3-iodo-2-propynyl butyl carbamate (IPBC); extraction method; treated woodDOI: 10.3969/j.issn.1000-2561.2021.11.031橡胶树(Hevea brasiliensis)是重要的热带经济林木,广泛种植于东南亚、非洲等60多个国家和地区。
HPLC外内标法测定含量

测定
(1)安装C18。反相柱,按仪器操作说明依次打开色谱仪各单元的电源。 (2)色谱条件:C18一ODS色谱柱(5um,4.6 mm×250 mm);流动相 比例:乙腈一0.1%磷酸水(20:80,V/V);流动相流速:1.0 mL/ min;检测波长:273 nm。 (3)换上流动相,待基线稳定后,采用进样器依次吸取浓度分别为 0.016、0.08、0.16、0.4、0.8 g/L扑热息痛和0.02、0.10、 0.20、0.50、1.0 g/L阿司匹林、内标物咖啡因浓度为0.2 g/L的 5种混合对照品溶液各10uL进样,待组分色谱峰出完后,按“停止”键 停止分析。记录各色谱峰面积(保留时间、峰面积),以对照品峰面积与 内标物峰面积之比为纵坐标(y),以浓度为横坐标(x),绘制标准曲线,进 行回归分析,求扑热息痛和阿司匹林的线性方程。 (4)采用进样器吸取供试品溶液10uL,进样,通过保留时间确定扑热息 痛、阿司匹林、咖啡因色谱峰的位置,记录扑热息痛、阿司匹林、咖啡 因色谱峰面积(保留时间、峰面积),并计算待测样品溶液中扑热息痛、 阿司匹林的含量。
测定
(1)安装C18反相柱,按仪器操作说明依次打开色谱仪各单元的电源。 (2)色谱条件。C18一ODS色谱柱(53um,4.6 mm×250 mm);流动相 比例:甲醇-0.5%冰乙酸水(1:1);流动相流速:1.0 mL/min;检 测波长:238 nm。 (3)换卜流动相,待基线稳定后,采用进样器依次吸取浓度分别为 0.004,0.02,O.04,0.1,0.2,0.4 g/L的栀子苷对照品溶 液各10仙L进样,待组分色谱峰出完后,按“停止”键停止分析。记录 栀子苷色谱峰面积(保留时间、峰面积),以栀子苷进样浓度(g/L)为横 坐标,色谱峰面积A为纵坐标,绘制标准曲线,进行回归分析。 (4)采用进样器吸取栀子样品溶液10¨L,进样,通过保留时间确定栀子 苷色谱峰的位置,记录栀子苷色谱峰面积(保留时间、峰面积),并计算 栀子药材中栀子苷的含量。
饲料中维生素D3的测定 高效液相色谱法-最新国标

1范饲料中维生素D3的测定高效液相色谱法围本文件描述了饲料中维生素D3的高效液相色谱测定方法。
本文件中“第一法皂化提取法”适用于配合饲料、精料补充料、浓缩饲料、复合预混合饲料、维生素预混合饲料中维生素D3的测定,定量限为12.5μg(500IU)/kg。
本文件中“第二法直接提取法”适用于维生素预混合饲料中维生素D3的测定,定量限为25mg (100万IU)/kg。
2规范性引用文件下列文件中的内容通过文中的规范性引用而构成本文件必不可少的条款。
其中,注日期的引用文件,仅该日期对应的版本适用于本文件;不注日期的引用文件,其最新版本(包括所有的修改单)适用于本文件。
GB/T6682分析实验室用水规格和试验方法GB/T20195动物饲料试样的制备3术语和定义本文件没有需要界定的术语和定义。
4第一法皂化提取法注意:使用的器皿不含有氧化性物质;分液漏斗活塞玻璃表面不涂油,处理过程在避光下操作;提取过程在通风柜中操作。
4.1原理试样用碱溶液皂化后,高含量的预混合饲料皂化液经液液萃取或固相萃取净化、浓缩后,直接上反相高效液相色谱仪分离、测定,外标法测定;而低含量试样的皂化液则需在液液萃取或离线或在线固相萃取净化后,经一维色谱柱进一步分离净化,然后切换至二维反相色谱柱分离、测定,外标法定量。
4.2试剂或材料除非另有规定,仅使用分析纯试剂。
4.2.1水:GB/T6682,一级。
4.2.2无水乙醇:色谱纯。
4.2.3无水乙醇。
4.2.4石油醚(沸程30℃-60℃)。
4.2.5甲醇:色谱纯。
4.2.6乙腈:色谱纯。
4.2.7甲酸。
4.2.8异丙醇:色谱纯。
4.2.9L-抗坏血酸。
4.2.10二丁基羟基甲苯(BHT)。
4.2.11无水硫酸钠。
4.2.24.2.12氢氧化钾溶液(500g/L ):称取500g 氢氧化钾,加水溶解,冷却后用水定容至1L ,混匀。
4.2.13乙醇溶液I(70%,体积分数):量取无水乙醇70mL,用水稀释,定容至100mL ,混匀。
饲料中胆汁酸的测定-最新国标

饲料中胆汁酸的测定警示——使用本文件的人员应有正规实验室工作的实践经验。
本文件并未指出所有可能的安全问题。
使用者有责任采取适当的安全和健康措施,并保证符合国家有关规定的条件。
1范围本文件描述了饲料中胆汁酸(胆酸、猪去氧胆酸、鹅去氧胆酸)的液相色谱-串联质谱和高效液相色谱测定方法。
本文件适用于配合饲料、浓缩饲料、精料补充料和添加剂预混合饲料中胆汁酸(胆酸、猪去氧胆酸、鹅去氧胆酸)的测定。
本文件液相色谱-串联质谱法的检出限为0.03mg/kg,定量限为0.10mg/kg;高效液相色谱法的检出限为3mg/kg,定量限为10mg/kg。
2规范性引用文件下列文件中的内容通过文中的规范性引用而构成本文件必不可少的条款。
其中,注日期的引用文件,仅该日期对应的版本适用于本文件;不注日期的引用文件,其最新版本(包括所有的修改单)适用于本文件。
GB/T6682分析实验室用水规格和试验方法GB/T20195动物饲料试样的制备3术语与定义本文件没有需要界定的术语和定义。
4液相色谱-串联质谱法4.1原理试样中的胆汁酸(胆酸、猪去氧胆酸、鹅去氧胆酸)用0.1%乙酸甲醇提取,经HLB固相萃取柱净化,液相色谱-串联质谱仪检测,外标法定量。
4.2试剂和材料除另有说明外,所用试剂均为分析纯。
4.2.1水:GB/T6682,一级。
4.2.2甲醇:色谱纯。
4.2.3正己烷:色谱纯。
4.2.4乙酸。
4.2.5乙腈:色谱纯。
4.2.60.1%乙酸甲醇溶液:移取1.0mL乙酸(4.2.4),用甲醇(4.2.2)定容至1000mL,混匀。
4.2.750%乙腈水溶液:移取50mL乙腈(4.2.5),用水定容至100mL,混匀。
4.2.8胆酸标准储备溶液(1g/L):称取胆酸标准品(CAS号:81-25-4)10mg(精确至0.01mg)于10mL容量瓶中,用甲醇溶解并定容。
4.2.9猪去氧胆酸标准储备溶液(1g/L):称取猪去氧胆酸标准品(CAS号:83-49-8)10mg(精确至0.01mg)于10mL容量瓶中,用甲醇溶解并定容。
甲醇检验原始记录1
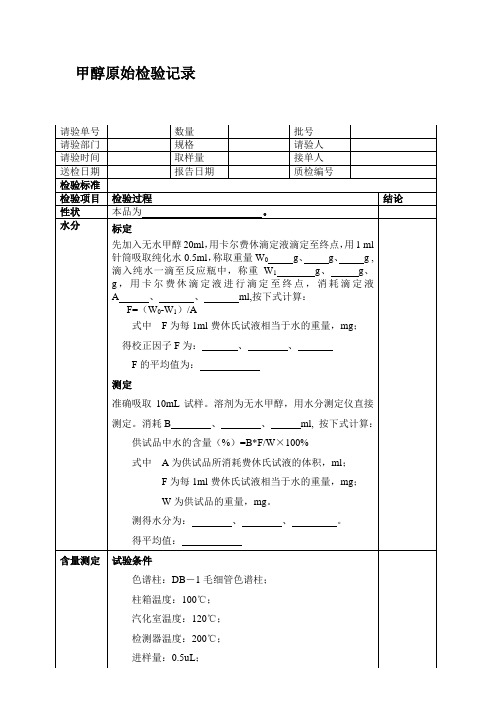
甲醇原始检验记录
请验单号数量批号
请验部门规格请验人
请验时间取样量接单人
送检日期报告日期质检编号
检验标准
检验项目检验过程结论性状本品为。
水分标定
先加入无水甲醇20ml,用卡尔费休滴定液滴定至终点,用1 ml
针筒吸取纯化水0.5ml,称取重量W0g、g、g ,
滴入纯水一滴至反应瓶中,称重W1 g、g、
g,用卡尔费休滴定液进行滴定至终点,消耗滴定液
A 、、ml,按下式计算:
F=(W0-W1)/A
式中F为每1ml费休氏试液相当于水的重量,mg;
得校正因子F为:、、
F的平均值为:
测定
准确吸取10mL试样。
溶剂为无水甲醇,用水分测定仪直接
测定。
消耗B 、、ml, 按下式计算:
供试品中水的含量(%)=B*F/W×100%
式中A为供试品所消耗费休氏试液的体积,ml;
F为每1ml费休氏试液相当于水的重量,mg;
W为供试品的重量,mg。
测得水分为:、、。
得平均值:
含量测定试验条件
色谱柱:DB-1毛细管色谱柱;
柱箱温度:100℃;
汽化室温度:120℃;
检测器温度:200℃;
进样量:0.5uL;
氮气流速:30mL/min;
空气流速:300mL/min;
氢气流速:30mL/min;计算方法
面积归一化法。
由气相色谱仪型号:
图谱编号:
得含量为:。
甲氨基阿维菌素苯甲酸盐含量的两种检测方法

第52卷第9期 辽 宁 化 工 Vol.52,No. 9 2023年9月 Liaoning Chemical Industry September,2023甲氨基阿维菌素苯甲酸盐含量的两种检测方法段元红,张晓婉,袁敏,李向阳(山东省农药检定所,山东 济南 250100)摘 要:采用两种不同的高效液相色谱法对8种不同农药产品中的甲氨基阿维菌素苯甲酸盐含量进行了分析测定,并对结果进行了比较分析,确定了各方法的优缺点。
两种方法互为补充,可满足实验室中绝大多数农药产品中甲氨基阿维菌素苯甲酸盐的含量测定。
关 键 词:甲氨基阿维菌素苯甲酸盐;高效液相色谱法;含量测定中图分类号:TQ450.7 文献标识码: A 文章编号: 1004-0935(2023)09-1402-03甲氨基阿维菌素苯甲酸盐(Emamectin benzoate,以下简称甲维盐)是基于阿维菌素衍生的一种新型半合成抗生素杀虫剂,具有低毒、高效、广谱等特点,广泛应用于蔬果、粮食、经济作物等害虫的防治[1]。
随着人类环保意识的增强,低毒、低残留的农药越来越受到市场的青睐。
甲维盐以其绿色环保的特点在市场的占有率越来越高。
关于甲维盐的配方研究也是层出不穷[2-4]。
目前,绝大多数国内农药生产厂家都有剂型不同、含量不同、配方不同的甲维盐系列产品在生产。
为了能更好的监督市场上的甲维盐产品质量,国家在2006年就制定了甲维盐原药及乳油的检测标准[5-6],并在2019年、2020年又制定了新标准[7-8]。
国内外也有许多对不同剂型的甲维盐单剂、复配制剂检测方法的研究报道[9-14]。
但每一种检测方法都有其局限性,并不能适用于所有甲维盐产品。
通过多年来实验室对甲维盐产品的分析检测经验,本文选取介绍了2种甲维盐高效液相色谱分析法,对8种农药产品中的甲维盐含量进行了测定,经过对检测结果的分析比较,发现此2种方法互为补充,可适用于绝大多数甲维盐产品。
1 试验部分1.1 主要试剂甲醇;色谱纯;乙腈:色谱纯;水:高纯水;氨水:分析纯;三乙胺:分析纯;甲氨基阿维菌素苯甲酸盐标样≥98.0%;5.7%甲氨基阿维菌素苯甲酸盐悬浮剂(样品a);12%甲维·虫螨腈悬浮剂:甲维盐含量2.0%(样品b)。
UPLC-MS

第34卷第3期2023年9月广西科技大学学报JOURNAL OF GUANGXI UNIVERSITY OF SCIENCE AND TECHNOLOGY Vol.34No.3 Sept.2023UPLC-MS/MS法同时测定发酵面制品中7种甜味剂蒋叶灵1,侯绪和*2,吕保樱2(1.桂林市食品药品检验所,广西桂林541200;2.广西科技大学医学部,广西柳州545005)摘要:应用超高效液相色谱-串联质谱法(ultra-high performance liquid chromatography-tandem mass spectrom‐etry,UPLC-MS/MS)实现发酵面制品的7种甜味剂(糖精钠、甜蜜素、安赛蜜、阿斯巴甜、阿力甜、纽甜、三氯蔗糖)同时检测。
样品经机械震荡、超声萃取,亚铁氰化钾溶液和乙酸锌沉淀蛋白,经0.22μm滤膜过滤预处理,以Agilent Eclipse Plus C18柱(2.1mm×50.0mm,1.8μm)和乙腈-水为流动相梯度洗脱;电喷雾负离子模式(ESI-)下采用多重反应监测(multiple reaction monitoring,MRM)模式进行分段采集。
实验结果:7种甜味剂在0.01~0.50μg/mL浓度范围内线性关系较好,相关系数r2均大于0.9991,检出限为3.0×10-6~3.0×10-4g/kg,定量限为1.0×10-5~1.0×10-3g/kg,样品平均加标回收率为84.5%~96.5%,相对标准偏差为1.3%~8.1%(n=6)。
实验结果表明:该方法快速、准确、重现性好,在各种发酵面制品检测中有广泛的应用前景。
关键词:甜味剂;发酵面制品;超高效液相色谱-串联质谱法(UPLC-MS/MS)中图分类号:TS213.2DOI:10.16375/45-1395/t.2023.03.0180引言发酵面制品是指以面粉为主要成分,经由和面-发酵-熟制而成的一类食品,包括馒头、包子、花卷、蒸糕等,是我国北方居民的主要粮食,占其主食的70%以上[1],其中馒头是60%以上消费者的主食[2]。
化妆品中利多卡因等7种物质的检测方法-中国食品药品检定研究院
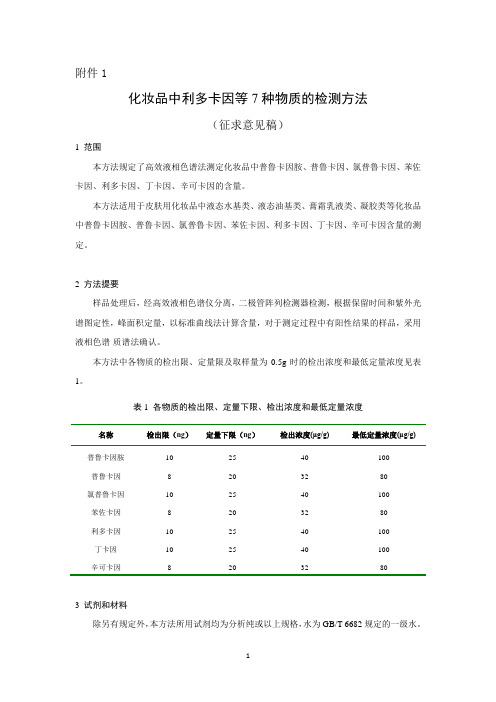
附件1化妆品中利多卡因等7种物质的检测方法(征求意见稿)1 范围本方法规定了高效液相色谱法测定化妆品中普鲁卡因胺、普鲁卡因、氯普鲁卡因、苯佐卡因、利多卡因、丁卡因、辛可卡因的含量。
本方法适用于皮肤用化妆品中液态水基类、液态油基类、膏霜乳液类、凝胶类等化妆品中普鲁卡因胺、普鲁卡因、氯普鲁卡因、苯佐卡因、利多卡因、丁卡因、辛可卡因含量的测定。
2 方法提要样品处理后,经高效液相色谱仪分离,二极管阵列检测器检测,根据保留时间和紫外光谱图定性,峰面积定量,以标准曲线法计算含量,对于测定过程中有阳性结果的样品,采用液相色谱-质谱法确认。
本方法中各物质的检出限、定量限及取样量为0.5g时的检出浓度和最低定量浓度见表1。
表1 各物质的检出限、定量下限、检出浓度和最低定量浓度名称检出限(ng)定量下限(ng)检出浓度(μg/g)最低定量浓度(μg/g)普鲁卡因胺10 25 40 100普鲁卡因8 20 32 80氯普鲁卡因10 25 40 100苯佐卡因8 20 32 80利多卡因10 25 40 100丁卡因10 25 40 100辛可卡因8 20 32 803 试剂和材料除另有规定外,本方法所用试剂均为分析纯或以上规格,水为GB/T 6682规定的一级水。
3.1 甲醇,色谱纯。
3.2 乙腈,色谱纯。
3.3 正己烷,色谱纯。
3.4 三氯乙酸,优级纯。
3.5 甲酸,优级纯。
3.6 磷酸,相对密度=1.685,w(H3PO4)=85%,优级纯。
3.7 磷酸氢二钠。
3.8 氨水,优级纯,含量(NH3)= 25%~28%。
3.9 混合标准储备溶液:称取普鲁卡因胺、氯普鲁卡因、丁卡因、利多卡因标准品各0.05g (精确到0. 00001g),普鲁卡因、苯佐卡因、辛可卡因标准品各0.04g(精确到0. 00001g)置于同一50 mL容量瓶中,加甲醇(3.1)使溶解并定容至刻度,摇匀。
3.10 流动相的配制:流动相A:0.01 moL/L Na2HPO4 水溶液,H3PO4调pH值至7.0流动相B:甲醇4 仪器和设备4.1 高效液相色谱仪,二极管阵列检测器。