吡嗪盐酸盐的合成实验报告
一锅法制备1-(2,3-二氯苯基)哌嗪盐酸盐

第20卷第4期2008年12月江苏工业学院学报JOU RN AL OF JIAN GSU P OLY T ECHN IC U N IVERSIT YVo l120No14Dec12008文章编号:1673-9620(2008)04-0032-04一锅法制备1-(2,3-二氯苯基)哌嗪盐酸盐*朱晓斌,徐崇福,陈苗(江苏工业学院化学化工学院,江苏常州213164)摘要:1-(2,3-二氯苯基)哌嗪盐酸盐是重要的医药中间体。
以二乙醇胺为起始原料,无溶剂条件下经氯代反应制得B,B c -二氯代二乙基胺盐酸盐,不经分离直接滴加2,3-二氯苯胺和相转移催化剂,经环合反应合成目标化合物1-(2,3-二氯苯基)哌嗪盐酸盐。
产率4813%,纯度9517%。
讨论了各步反应的影响因素。
产物经红外光谱、核磁共振氢谱分析验证与目标化合物的结构一致。
该方法经济环保,省时,易操作。
关键词:1-(2,3-二氯苯基)哌嗪盐酸盐;一锅法反应;相转移催化剂中图分类号:O6261415文献标识码:AO ne Pot Synthesis of1-(2,3-Dichlorophenyl)Piperazinium C hlorideZH U Xiao-bin,XU Chong-fu,CH EN Miao(School of Chemistry and Chemical Eng ineering,Jiangsu Polytechnic University,Changzhou213164,China)Abstract:1-(2,3-Dichloro pheny l)piperazinium chloride is an im por tant pharmaceutical interm ed-i ate.Diethanolam ine w as chlor inated w ithout so lvent to g ener ate B,B c-dichlo rodiethylammo nium chlo ride in situ,fo llow ed by addition of2,3-dichlo roaniline and a phase transfer catalyst to cyclolize and y ield the targ eted com po und1-(2,3-dichloro phenyl)piperazinium chloride w ith4813%yield and9517%pur-i ty.T he structure of the pr oduct w as consistent w ith that of the ex pected result by IR and NM R spectros-copy.The process applied w as environment benign,time-saving and operation fr iendly.Key words:1-(2,3-Dichlorophenyl)piper azinium chlo ride;one po t reaction;phase transfer cataly st苯基哌嗪类化合物具有不同程度的5-H T受体[1,2]阻断活性,并且将该部分与其他部分载体相结合则可以使化合物具有中枢神经调节作用或中枢及外周降压活性。
4_羟基吡啶盐酸盐的表征_戎红仁

有限公司提 供 , 并经本 实验室 用 H P LC 法复测 认 量 , 结果见表 1 。
可 。 将 H P 溶于 盐酸 , 分别调 整溶液的 pH 值为 1
表 1 HP 盐酸 盐 (A)和 (B)的分析数据
或 3 .5 后 , 在水 浴上蒸 干后 , 相应 地得到 盐酸盐 :
(A)和(B)。(A)经真空干燥 箱中在 < 50 ℃干 燥
为了确定(A)和(B)是两个不同的化合物 , 扫描 并对比它们的 X-射线粉末衍射图谱 , 见图 1 。谱线 依其相对强度的顺序列出见表 2 。
pH S-2 精密酸度计由上海雷磁仪器厂生产 , 配
用上海精密科学仪器有限公司生产 E-201- C pH 复
合电极 , pH 计经用标准 缓冲溶液(pH = 4 .00 和
其他试剂均为分析纯 。 1 .1 .2 仪器和工作条件
全自动多晶 X-射线粉末衍射仪由日本 理学生 产 , Rigaku D / M AX - 2550 PC 型 Cu K α(λ = 0 .15405 nm)射线 。 电压 40 kV 。 电 流 100 mA 。 2θ角从 5 .0°扫描到 50°。 扫描速 度 :2°/ mi n 。 取 3 次测定的平均值 。
59.6, 61 .8 18.7, 15 .1 15.0, 12 .8 6 .68, 10 .3
11 .46 49 .13 6 .14
49 .08 11 .45 19 .63 14 .50
表 1 中氯含量和水含量都和理论值相符 , 可以 确定两个 H P 的盐酸盐(A)和(B)的组成分别和分 子式 H P · H Cl 和(H P)2 ·H Cl · H 2O 符合 。 2 .2 盐酸盐的 X-射线粉末衍射图
UPLC-MS

基因毒性杂质研究专栏㊀基金项目:中国食品药品检定研究院关键技术研究基金(No.GJJS-2022-4-2)ꎻ#同为第一作者ꎬ∗同为通信作者作者简介:袁松ꎬ男ꎬ硕士ꎬ助理研究员ꎬ研究方向:化学药品质量控制ꎬE-mail:yuansong@nifdc.org.cnꎻ李婕ꎬ女ꎬ硕士ꎬ副主任药师ꎬ研究方向:化学药品质量控制ꎬE-mail:lijie@nifdc.org.cn通信作者:刘阳ꎬ男ꎬ博士ꎬ研究员ꎬ研究方向:药品质量安全ꎬTel:010-53851571ꎬE-mail:yangliu@nifdc.org.cnꎻ张庆生ꎬ男ꎬ硕士ꎬ主任药师ꎬ研究方向:药品质量安全ꎬTel:010-53851375ꎬE-mail:zqs@nifdc.org.cnUPLC-MS/MS法测定磷酸西格列汀中基因毒性杂质NTTP袁松#ꎬ李婕#ꎬ张娜ꎬ张龙浩ꎬ刘阳∗ꎬ张庆生∗(中国食品药品检定研究院ꎬ国家药品监督管理局化学药品质量研究与评价重点实验室ꎬ北京102629)摘要:目的㊀建立磷酸西格列汀原料药及制剂中基因毒性杂质Nitroso-STG-19(NTTP)的超高效液相色谱-串联质谱(UPLC-MS/MS)检测方法ꎮ方法㊀色谱柱为EclipsePlusC18RRHD(3.0mmˑ150mmꎬ1.8μm)ꎬ以含0.1%甲酸的水溶液为流动相Aꎬ0.1%甲酸的乙腈溶液为流动相Bꎬ梯度洗脱ꎬ流速为0.35mL min-1ꎬ柱温为30ħꎬ进样器温度为10ħꎬ进样体积为5μLꎮ采用多反应监测(MRM)模式ꎬ对磷酸西格列汀原料药及制剂中的NTTP进行定量检测ꎮ结果㊀NTTP在0.54~53.93ng mL-1范围内具有良好的线性关系ꎮ检测限和定量限分别为0.02ng mL-1和0.08ng mL-1ꎮ原料药及制剂在低㊁中㊁高3个浓度的平均加样回收率范围(n=3)分别为105.43%~107.21%(RSDɤ3.2%)及115.03%~120.20%(RSDɤ3.6%)ꎮ应用该方法对12批原料药及3批制剂中的NTTP进行检测ꎬ结果显示均有检出ꎮ结论㊀该方法灵敏度高㊁专属性强ꎬ可用于测定磷酸西格列汀原料药及制剂中的NTTPꎬ可为磷酸西格列汀原料药及制剂的质量控制提供参考ꎮ关键词:磷酸西格列汀ꎻ基因毒性杂质ꎻNitroso-STG-19ꎻ含量测定ꎻ超高效液相色谱-串联质谱中图分类号:R927.1㊀文献标志码:A㊀文章编号:2095-5375(2023)12-1000-005doi:10.13506/j.cnki.jpr.2023.12.009DeterminationofgenotoxicimpurityNTTPinSitagliptinPhosphatebyUPLC-MS/MSYUANSong#ꎬLIJie#ꎬZHANGNaꎬZHANGLonghaoꎬLIUYang∗ꎬZHANGQingsheng∗(NMPAKeyLaboratoryforQualityResearchandEvaluationofChemicalDrugsꎬNationalInstitutesforFoodandDrugControlꎬBeijing102629ꎬChina)Abstract:Objective㊀ToestablishanUPLC-MS/MSmethodfordeterminationofgenotoxicimpurityNitroso-STG-19(NTTP)inSitagliptinPhosphate.Methods㊀TheseparationofNTTPwasperformedonaEclipsePlusC18RRHD(3.0mmˑ150mmꎬ1.8μm)withthemobilephaseconsistingof0.1%formicacidaqueoussolution(mobilephaseA)and0.1%formicacidmethanolsolution(mobilephaseB)underagradientelutionataflowrateof0.35mL min-1andacolumntem ̄peratureof30ħ.Multiplereactionmonitoring(MRM)wasperformedonatriplequadrupolemassspectrometerinpositivemode.Results㊀Thecalibrationcurveswasingoodlinearityintherangeof0.54~53.93ng mL-1.Thelimitofdetectionwas0.02ng mL-1ꎬandthelimitofquantificationwas0.08ng mL-1.Therecoveries(n=3)ofactivepharmaceuticalingredients(API)andtabletsatlowꎬmiddleꎬandhighspikedconcentrationswere105.43%~107.21%(RSDɤ3.2%)and115.03%~120.20%(RSDɤ3.6%)respectively.UsingthedevelopedmethodꎬwedetectedNTTPin12batchesofAPIand3batchesofpreparation.TheresultsshowedthatNTTPwasdetectedinallbatches.Conclusion㊀ThemethodwassensitiveandaccurateꎬwhichcanbeappliedforthequantificationsofNTTPinSitagliptinPhosphateꎬprovidingreferenceforqualitycontrolofSita ̄gliptinPhosphate.Keywords:SitagliptinPhosphateꎻGenotoxicimpurityꎻNTTPꎻContentdeterminationꎻUPLC-MS/MS㊀㊀N-亚硝胺类化合物(N-nitrosaminesꎬNAs)是一类结构通式为R1(R2)N-N=O的化合物ꎬ其中R1和R2为烷基或芳烃ꎬ国际癌症研究机构于1987年将NAs列入致癌物清单ꎮ2017年世界卫生组织发布的致癌清单中ꎬ有近16个短脂肪链的N-亚硝胺类化合物被列为2类致癌物质[1]ꎮNAs常常存在于食品㊁饮用水㊁烟草㊁化妆品等物质中[2-5]ꎬ人们长期接触含NAs的这些物质会产生潜在危害ꎮ自2018年欧洲药品管理局(EuropeanMedicinesAgencyꎬEMA)宣布在缬沙坦原料和制剂中检测出N-亚硝基二甲胺(NDMA)后ꎬ各国药品监管机构纷纷加强对药品中NAs的监测ꎬ并对多批含有NDMA或其他亚硝胺类杂质的沙坦类药物进行召回ꎬ并要求对产品中NAs存在进行风险评估及建立合适的控制策略[6-9]ꎮ西格列汀(结构式见图1)是一种二肽基肽酶-4(DPP-4)抑制剂ꎬ可以刺激胰高血糖素的释放并减少胰岛素的分泌ꎬ从而发挥降糖作用ꎬ是一种常见的降糖药ꎬ可与其他药联合用药来治疗包括超重肥胖㊁胰岛素治疗不佳㊁肥胖型2型糖尿病等疾病ꎬ治疗效果良好[10-12]ꎮ2022年报道表明西格列汀合成中间体三氟甲基-5ꎬ6ꎬ7ꎬ8-四氢-1ꎬ2ꎬ4-三唑并-[4ꎬ3-α]-吡嗪盐酸盐(TTP)[13]可能与亚硝酸盐反应生成Nitroso-STG-19(NTTPꎬ见图1)ꎮNTTP是新发现的一种存在于西格列汀中的基因毒性杂质ꎬ可能是以TTP为原料合成西格列汀时产生或是最终产品中残留的TTP与辅料中微量的亚硝酸盐反应生成NTTPꎮEMA和美国食品药品监督管理局(FDA)最初都将其最大摄入量设定为37ng d-1ꎬ但FDA为了避免西格列汀产品的短缺ꎬ将最大摄入量调整为246.7ng d-1ꎬ暂未召回该产品[14-15]ꎮ目前国内外尚无对西格列汀中的NTTP检测方法的报道ꎬ本文建立了一个超高效液相色谱-串联质谱(UPLC-MS/MS)检测西格列汀中NTTP的方法ꎬ为西格列汀相关原料药与制剂的药品质量控制和监管提供参考ꎮ图1㊀西格列汀㊁TTP和NTTP的结构式1㊀材料1.1㊀仪器㊀高效液相色谱-串联三重四极杆质谱仪(美国Waters公司)ꎬ配备电喷雾离子源(ESI)和MassLynxV4.2数据处理系统ꎻXP205DR型电子分析天平(瑞士Mettler公司)ꎻMilli-Q超纯水纯化系统(美国Millipore公司)ꎮ1.2㊀药物与试剂㊀乙腈㊁甲酸㊁甲酸铵(质谱级ꎬ美国FisherScientific)ꎬ水为超纯水ꎬ对照品NTTP来自本实验室合成ꎬ采用高分辨质谱(见图2A)和核磁(见图2B㊁C)对其结构进行了确证ꎮ2㊀方法与结果2.1㊀溶液的制备2.1.1㊀标准曲线溶液的制备㊀取NTTP对照品约10mgꎬ精密称定ꎬ置100mL量瓶中ꎬ加水使溶解并稀释至刻度ꎬ作为对照品储备液ꎻ精密量取对照品储备液适量ꎬ以水为稀释剂逐级定量稀释制成每1mL中含NTTP0.54㊁1.08㊁4.31㊁10.79㊁26.97㊁53.93ng的溶液ꎬ作为系列线性溶液ꎮ2.1.2㊀供试品溶液的制备㊀取磷酸西格列汀原料药约100mgꎬ精密称定ꎬ置10mL量瓶中ꎬ加水适量ꎬ超声使溶解ꎬ用水稀释至刻度ꎬ摇匀ꎬ作为原料药的供试品溶液ꎮ取磷酸西格列汀片10片ꎬ精密称定ꎬ研细ꎬ混匀ꎬ精密称取约相当于磷酸西格列汀100mg的细粉ꎬ置15mL离心管中ꎬ精密加入水10mLꎬ超声并振摇10minꎬ滤过ꎬ取续滤液作为片剂的供试品溶液ꎮ2.2㊀色谱及质谱条件2.2.1㊀色谱条件㊀采用AgilentEclipsePlusC18RRHD(3.0mmˑ150mmꎬ1.8μm)色谱柱ꎬ以含0.1%甲酸的水溶液为流动相Aꎬ0.1%甲酸的乙腈溶液为流动相Bꎬ梯度洗脱:0~8.0minꎬ40%Bң100%Bꎻ8.0~8.1minꎬ100%Bң40%Bꎻ8.1~12.0minꎬ40%Bꎻ流速为0.35mL min-1ꎬ柱温为30ħꎬ进样器温度为10ħꎬ进样体积为5μLꎮ2.2.2㊀质谱条件㊀采用电喷雾离子源(ElectrosprayIonizationSourceꎬESI)ꎬ正离子检测模式ꎬ毛细管电压为4.0kVꎬ脱溶剂气温度为500ħꎬ脱溶剂气流量为1000L h-1ꎬ锥孔气流量为150L h-1ꎬ离子源温度为150ħꎮ监测模式为多反应监测(Multiplere ̄actionmonitorꎬMRM)ꎬ以m/z221.83ң191.95作为定量离子对ꎬ锥孔电压为10Vꎬ碰撞能量为10Vꎬ以离子对m/z221.83ң164.71作为定性离子对ꎬ锥孔电压为10Vꎬ碰撞能量为20Vꎮ采集时间为2.3~8.0minꎮ2.3㊀方法学考察2.3.1㊀专属性试验㊀取水作为空白溶剂和 2.1.1项下约4ng mL-1的对照品溶液分别进样ꎬ记录色谱图ꎬ在所建立的色谱和质谱条件下ꎬNTTP的保留时间为2.62min(见图3A)ꎬ峰型良好ꎬ空白溶剂对检测无干扰ꎮ图2㊀结构确证-高分辨质谱图(AꎬH-ESI+)㊁核磁共振氢谱图(B)和碳谱图(C)2.3.2㊀系统精密度㊀取 2.1.1 项下约4ng mL-1的线性溶液连续进样6次ꎬ得NTTP峰面积的RSD为1.13%ꎬ结果表明系统精密度良好ꎮ2.3.3㊀重复性㊀2.3.3.1㊀原料药重复性㊀取磷酸西格列汀原料药(批号:SGZ1040006)ꎬ按 2.1.2 项下方法平行制备6份原料药供试品溶液ꎬ进样检测ꎬ按标准曲线法计算NTTP含量ꎬ计算得6份原料药供试品溶液中NTTP含量的RSD为7.2%ꎮ结果表明方法对原料药测定具有良好的重复性ꎮ2.3.3.2㊀片剂重复性㊀取磷酸西格列汀片ꎬ按 2.1.2 项下方法平行制备6份片剂供试品溶液ꎬ进样检测ꎬ按标准曲线法计算NTTP含量ꎬ计算得6份片剂供试品溶液中NTTP含量的RSD为3.3%ꎬ结果表明方法对片剂测定具有良好的重复性ꎮ2.3.4㊀线性㊀分别取 2.1.1 项下系列线性溶液ꎬ进样检测ꎬ记录色谱图ꎮ以质量浓度为横坐标(X)ꎬNTTP峰面积为纵坐标(Y)进行线性回归ꎬ在0.54~53.93ng mL-1浓度范围内线性结果为Y=5100.84X+539.79(r=1.0000)ꎮ结果显示NTTP在其线性范围内峰面积与进样浓度之间呈良好的线性关系ꎮ2.3.5㊀回收率试验2.3.5.1㊀原料药回收率试验㊀取磷酸西格列汀原料药(批号:GZ1040006)约100mgꎬ精密称定ꎬ置10mL量瓶中ꎬ分别用 2.1.1 项下25ng mL-1(高浓度)㊁10ng mL-1(中浓度)㊁4ng mL-1(低浓度)的线性溶液溶解并稀释至刻度ꎬ摇匀ꎬ作为原料药回收率溶液ꎬ每个浓度点平行制备3份ꎬ进样检测ꎬ结果见表1ꎬ低㊁中㊁高浓度点的回收率分别为107.21%(RSD2.9%ꎬn=3)㊁105.95%(RSD2.5%ꎬn=3)㊁105.43%(RSD3.2%ꎬn=3)ꎬ结果表明原料药回收率良好ꎮ2.3.5.2㊀片剂回收率试验㊀取磷酸西格列汀片(批号:200515JA)ꎬ精密称定ꎬ研细ꎬ混匀ꎬ精密称取细粉适量(约相当于西格列汀100mg)ꎬ置10mL量瓶中ꎬ分别用 2.1.1 项下25ng mL-1(高浓度)㊁10ng mL-1(中浓度)㊁4ng mL-1(低浓度)的线性溶液溶解并稀释至刻度ꎬ摇匀ꎬ滤过ꎬ取续滤液作为片剂回收率溶液ꎬ每个浓度点平行制备3份ꎬ进样检测ꎬA.空白溶剂ꎻB.对照品溶液ꎻC.片剂供试品溶液图3㊀空白溶剂㊁对照品溶液和片剂供试品溶液提取的离子流色谱图结果见表3ꎬ低㊁中㊁高浓度点的回收率分别为115.03%(RSD2.1%ꎬn=3)㊁115.95%(RSD3.6%ꎬn=3)㊁120.20%(RSD1.4%ꎬn=3)ꎬ结果表明片剂回收率在可接受范围内ꎮ表1㊀原料药加样回收率结果编号加入量/ng检测量/ng本底/ng回收率(%)平均回收率(%)RSD(%)143.1548.722.80106.42243.1547.972.84104.59107.212.9343.1550.632.90110.614107.87117.312.96106.015107.87119.992.85108.59105.952.56107.87114.182.82103.247269.66297.642.84109.328269.66282.852.83103.84105.433.29269.66281.093.01103.122.3.6㊀检测限与定量限㊀取 2.1.1 项下约0.5ng mL-1的线性溶液ꎬ以水为稀释剂逐步稀释ꎬ分别在信噪比为3ʒ1和10ʒ1时作为检测限和定量限ꎬ测得NTTP的检测限和定量限分别为0.02ng mL-1和0.08ng mL-1ꎮ2.3.7㊀提取效率考察㊀取磷酸西格列汀原料药(批号:SGZ1040006)和磷酸西格列汀片(批号:T024447)ꎬ分别按 2.1.2 项平行制备2份溶液ꎬ分别超声并振摇10㊁20minꎬ进样检测ꎬ按标准曲线法计算NTTP含量ꎬ计算得原料药2份供试品溶液中NTTP含量相对标对偏差为3.32%ꎬ片剂2份供试品溶液中NTTP含量相对标对偏差为2.02%ꎬ结果表明超声并振摇10min可提取完全ꎮ表2㊀片剂加样回收率结果编号加入量/ng检测量/ng本底/ng回收率(%)平均回收率(%)RSD(%)143.1550.580117.22243.1549.840115.50115.032.1343.1548.490112.384107.87128.890119.495107.87120.180111.41115.953.66107.87126.150116.957269.66328.640121.878269.66324.350120.28120.201.49269.66319.390118.442.3.8㊀溶液稳定性㊀取 2.1.1 项下约1ng mL-1的对照品溶液ꎬ放置于进样盘ꎬ间隔6h进样检测ꎮ0h和6h时NTTP峰面积的相对偏差为2.77%ꎬ结果表明对照品溶液10ħ时6h内稳定ꎮ2.4㊀样品测定㊀按 2.1 项下制备磷酸西格列汀供试品溶液ꎬ照 2.2 项下条件进样检测ꎬ记录色谱图(典型图谱见图2B㊁C)ꎬ以峰面积按标准曲线法计算供试品溶液中NTTP的含量ꎮ4家原料厂家的12批样品中检出NTTP含量为0.014~0.20μg g-1ꎻ有3批磷酸西格列汀片中检测出NTTPꎬ含量为1.26~1.41μg g-1ꎮ3㊀讨论NTTP在水中易溶ꎬ以水为溶剂配制浓度约为1μg mL-1的对照品溶液ꎬ分别采用ESI离子源和APCI离子源下针泵进样对NTTP的响应及定量/定性离子进行考察ꎬ结果表明NTTP在ESI离子源正离子采集模式下响应更好ꎬ继续对ESI+条件进行优化ꎬ最终确定NTTP定量离子对为m/z221.83ң191.95ꎬ锥孔电压为10Vꎬ碰撞能量为10eVꎬ定性离子对为m/z221.83ң164.71作为ꎬ锥孔电压为10Vꎬ碰撞能量为20eVꎮ以水为溶剂配制含磷酸西格列汀和NTTP浓度均约为100μg mL-1的混合溶液ꎬ采用紫外检测器对NTTP峰与西格列汀峰之间的分离情况进行了考察ꎬ分别试验了甲醇-水㊁乙腈-水系统ꎬ试验证明当流动相为乙腈-水时NTTP和西格列汀出峰较快ꎬ且可完全分离ꎬ水中加入甲酸铵后NTTP的质谱响应降低明显且峰型变差ꎬ加入甲酸可以增强离子化效率提高质谱响应ꎮ最终以含0.1%甲酸的水溶液为流动相Aꎬ0.1%甲酸的乙腈溶液为流动相Bꎬ并进行梯度洗脱ꎮ在最终确定的色谱条件下ꎬ磷酸西格列汀在2.2min即可洗脱完全ꎬ质谱采集时间设定为2.3~8.0minꎬ高浓度的磷酸西格列汀经液相洗脱后直接从废液排出ꎬ以避免对质谱的污染ꎮ磷酸西格列汀片未明确最大单日剂量ꎬ按推荐剂量100mg d-1计ꎻEMA和FDA最初都将NTTP最大摄入量设定为37ng d-1ꎬ但FDA为了避免西格列汀产品的短缺ꎬ将NTTP最大摄入量调整为246.7ng d-1[15]ꎻ如按37ng d-1计算ꎬ则磷酸西格列汀中NTTP的残留限度为0.37μg g-1ꎬ所检原料药中NTTP均未超过此限度ꎬ但片剂中NTTP含量已超限度值ꎻ如按246.7ng d-1计算ꎬ则磷酸西格列汀中NTTP的残留限度为2.47μg g-1ꎬ原料药和片剂中NTTP均未超出此限度ꎮ片剂中NTTP含量显著高于原料药ꎬ可能是由于终产品中的TTP残留继续与极微量的亚硝酸盐继续反应生产NTTPꎬ关于TTP残留量与NTTP残留量之间的相关性还需进行进一步的研究ꎮ4㊀结论本研究建立了磷酸西格列汀原料药及片剂中NTTP的UPLC-MS/MS检测方法ꎬ并进行了相关方法学验证ꎬ结果表明ꎬ所建立的方法具有良好的专属性㊁灵敏度和准确度ꎬ可准确测定磷酸西格列汀原料药及片剂中潜在的NTTP含量ꎬ有助于磷酸西格列汀的市场监管ꎬ保障药品质量安全ꎮ参考文献:[1]㊀袁松ꎬ黄海伟ꎬ于颖洁ꎬ等.UPLC-MS/MS法同时测定氯沙坦钾和缬沙坦中7个亚硝胺类基因毒性杂质[J].药物分析杂志ꎬ2021ꎬ41(7):1218-1225.[2]PARKJEꎬSEOJEꎬLEEJYꎬetal.DistributionofsevenN-NitrosaminesinFood[J].ToxicolResꎬ2015ꎬ31(3):279-298. [3]KRASNERSWꎬMITCHWAꎬMCCURRYDLꎬetal.For ̄mationꎬprecursorsꎬcontrolꎬandoccurrenceofnitrosaminesindrinkingwater:areview[J].WaterResꎬ2013ꎬ47(13):4433-4450.[4]EDWARDSSHꎬHASSINKMDꎬTAYLORKMꎬetal.To ̄bacco-SpecificNitrosaminesintheTobaccoandMain ̄streamSmokeofCommercialLittleCigars[J].ChemResToxicolꎬ2021ꎬ34(4):1034-1045.[5]ALHOOSHANIK.DeterminationofnitrosaminesinskincarecosmeticsusingCe-SBA-15basedstirbar-supportedmicro-solid-phaseextractioncoupledwithgaschromatographymassspectrometry[J].ArabJChemꎬ2020ꎬ13(1):2508-2516.[6]MALIHIFꎬWANGT.Animprovedanalyticalmethodforquantitationofnitrosamineimpuritiesinophthalmicsolu ̄tionsusingliquidchromatographywithtandemmassspec ̄trometry[J].JChromatogrOpenꎬ2022(2):100037. [7]FDA.UpdatesonAngiotensinIIReceptorBlocker(ARB)RecallsIncludingValsartanꎬLosartanandIrbesartan[EB/OL].[2022-12-24].http://www.fda.gov/Drugs/Drug ̄Safety/ucm613916.htm.[8]BHARATESS.CriticalAnalysisofDrugProductRecallsduetoNitrosamineImpurities[J].JMedChemꎬ2021ꎬ64(6):2923-2936.[9]GUNASEKARANPMꎬCHERTOWGMꎬBHALLAVꎬetal.CurrentStatusofAngiotensinReceptorBlockerRecalls[J].Hypertensionꎬ2019ꎬ74(6):1275-1278.[10]HERMANGAꎬSTEVENSCꎬVANDYCKKꎬetal.Phar ̄macokineticsandpharmacodynamicsofsitagliptinꎬanin ̄hibitorofdipeptidylpeptidaseIVꎬinhealthysubjects:Re ̄sultsfromtworandomizedꎬdouble-blindꎬplacebo-controlledstudieswithsingleoraldoses[J].ClinPharmacolTherꎬ2005ꎬ78(6):675-688.[11]HERMANGAꎬBERGMANAꎬLIUFꎬetal.Pharmacoki ̄neticsandpharmacodynamiceffectsoftheoralDPP-4in ̄hibitorsitagliptininmiddle-agedobesesubjects[J].JClinPharmacolꎬ2006ꎬ46(8):876-886.[12]JANANILꎬBAMEHRHꎬTANHAKꎬetal.EffectsofSita ̄gliptinasMonotherapyandAdd-OntoMetforminonWeightLossamongOverweightandObesePatientswithType2Diabetes:ASystematicReviewandMeta-Analysis[J].DrugRes(Stuttg)ꎬ2021ꎬ71(9):477-488.[13]HANSENKBꎬHSIAOYꎬXUFꎬetal.Highlyefficientasymmetricsynthesisofsitagliptin[J].JAmChemSocꎬ2009ꎬ131(25):8598-8804.[14]EuropeanMedicinesAgency.Questionsandanswersformarketingauthorizationholders/applicantsontheCHMPOpinionfortheArticle5(3)ofRegulation(EC)No726/2004referralonnitrosamineimpuritiesinhumanmedicinalproducts[EB/OL].[2022-12-24].https://www.ema.europa.eu/documents/referral/nitrosamines-emea-h-a53-1490-questions-answers-marketing-auth ̄orisation-holders/applicants-chmp-opinion-article-53-regulation-ec-no-726/2004-referral-nitrosamine-impu ̄rities-human-medicinal-products_en.pdf.[15]U.S.FoodandDrugAdministration.QFDAworkstoavoidshortageofsitagliptinfollowingdetectionofnitrosamineim ̄purity[EB/OL].[2022-12-24].https://www.fda.gov/drugs/drug-safety-and-availability/fda-works-avoid-shortage-sita ̄gliptin-following-detection-nitrosamine-impurity.(收稿日期:2023-02-25)。
磷酸西格列汀合成新技术
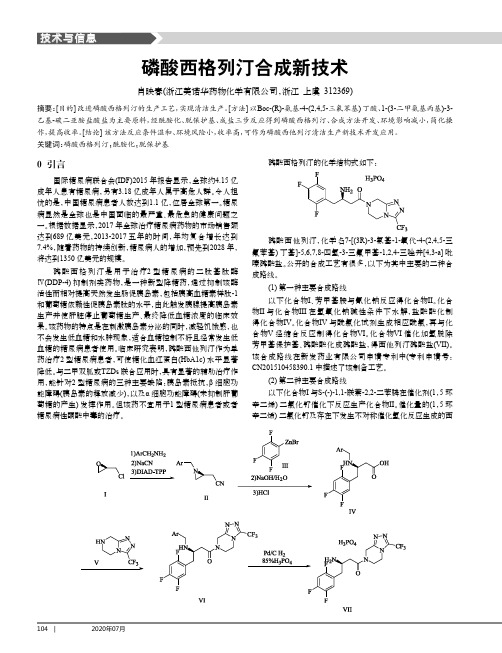
104 | 磷酸西格列汀的化学结构式如下:磷酸西他列汀,化学名7-[(3R)-3-氨基-1-氧代-4-(2,4,5-三氟苯基)丁基]-5,6,7,8-四氢-3-三氟甲基-1,2,4-三唑并[4,3-a]吡嗪磷酸盐。
公开的合成工艺有很多,以下为其中主要的二种合成路线。
(1)第一种主要合成路线以下化合物I 、芳甲基胺与氰化钠反应得化合物II 。
化合物II 与化合物III 在氢氧化钠碱性条件下水解,盐酸酸化制得化合物IV 。
化合物IV 与酰氯化试剂生成相应酰氯,再与化合物V 经缩合反应制得化合物VI 。
化合物VI 催化加氢脱除芳甲基保护基、磷酸酸化成磷酸盐,得西他列汀磷酸盐(VII)。
该合成路线在新发药业有限公司申请专利中(专利申请号:CN201510458390.1中描述了该制备工艺。
(2)第二种主要合成路线以下化合物I 与S-(-)-1,1-联萘-2,2-二苯膦在催化剂(1,5环辛二烯)二氯化钌催化下反应生产化合物II 。
催化量的(1,5环辛二烯)二氯化钌及存在下发生不对称催化氢化反应生成的西0 引言国际糖尿病联合会(IDF)2015年报告显示,全球约4.15亿成年人患有糖尿病,另有3.18亿成年人属于高危人群。
令人担忧的是,中国糖尿病患者人数达到1.1亿,位居全球第一。
糖尿病显然是全球也是中国面临的最严重、最危急的健康问题之一。
根据数据显示,2017年全球治疗糖尿病药物的市场销售额达到689亿美元,2013-2017五年的时间,年均复合增长达到7.4%,随着药物的持续创新,糖尿病人的增加,预先到2028年,将达到1350亿美元的规模。
磷酸西格列汀是用于治疗2型糖尿病的二肽基肽酶Ⅳ(DDP-4)抑制剂类药物,是一种新型降糖药,通过抑制该酶活性而相对提高天然发生肠促胰岛素,包括胰高血糖素样肽-1和葡萄糖依赖性促胰岛素肽的水平,由此触发胰腺提高胰岛素生产并使肝脏停止葡萄糖生产、最终降低血糖浓度的临床效果。
一种吡啶盐酸盐的合成方法[发明专利]
![一种吡啶盐酸盐的合成方法[发明专利]](https://img.taocdn.com/s3/m/78c54433a7c30c22590102020740be1e640ecc5c.png)
(19)中华人民共和国国家知识产权局(12)发明专利申请(10)申请公布号 (43)申请公布日 (21)申请号 201710496639.7(22)申请日 2017.06.26(71)申请人 蒋潇地址 213200 江苏省徐州市沛县沛城东风东路东关新村212号(72)发明人 蒋潇 (51)Int.Cl.C07D 213/18(2006.01)C07D 213/127(2006.01)(54)发明名称一种吡啶盐酸盐的合成方法(57)摘要本发明涉及有机化学领域,特别是一种吡啶盐酸盐的合成方法,包括以下步骤:(a )在室温下,将浓盐酸滴入到氯化钙颗粒中,可获得氯化氢,将氯化氢气体经过浓硫酸洗气瓶和缓冲瓶得到干燥的氯化氢气体;(b)把装有搅拌、温度计、回流冷凝器和进气管的四口烧瓶置于冷水浴锅中,在烧瓶中加入吡啶和二氯甲烷,开始搅拌并通入干燥氯化氢气体;保持反应温度30℃-40℃,生成白色吡啶盐酸盐;(c)停止搅拌,过滤吡啶盐酸盐和二氯甲烷,滤液为二氯甲烷,滤饼为吡啶盐酸盐,将滤饼放入真空干燥器中干燥1-3h,得到成品吡啶盐酸盐。
本发明的合成方法总收率高、原料价格便宜、反应时间短、条件温和、工艺操作简单。
权利要求书1页 说明书2页CN 107056683 A 2017.08.18C N 107056683A1.一种吡啶盐酸盐的合成方法,其特征在于,包括以下步骤:(a)在室温下,将浓盐酸滴入到氯化钙颗粒中,可获得氯化氢,将氯化氢气体经过浓硫酸洗气瓶和缓冲瓶得到干燥的氯化氢气体;(b)把装有搅拌、温度计、回流冷凝器和进气管的四口烧瓶置于冷水浴锅中,在烧瓶中加入吡啶和二氯甲烷,开始搅拌并通入干燥氯化氢气体;保持反应温度30℃-40℃,生成白色吡啶盐酸盐;(c)停止搅拌,过滤吡啶盐酸盐和二氯甲烷,滤液为二氯甲烷,滤饼为吡啶盐酸盐,将滤饼放入真空干燥器中干燥1-3h,得到成品吡啶盐酸盐。
2.按照权利要求1所述的一种吡啶盐酸盐的合成方法,其特征在于:将步骤(c)中的滤液二氯甲烷二次回用到步骤(b)中。
吡嗪盐酸盐的合成实验报告

吡嗪盐酸盐的合成实验报告吡嗪盐酸盐的合成实验报告实验目的•了解吡嗪盐酸盐的合成方法•掌握吡嗪盐酸盐的物理性质与化学性质实验原理1.吡啶:化学式为C5H5N,是一种重要的有机化合物,具有特殊的环结构和碱性特性。
2.盐酸盐:由盐酸和有机物质反应生成的盐。
实验步骤1.准备试剂:吡啶(C5H5N),盐酸(HCl),无水乙醚(C2H5OC2H5)。
2.取一定量的吡啶溶解于少量无水乙醚中,制备吡啶溶液。
3.在磁力搅拌器搅拌的条件下,将吡啶溶液缓慢滴加入盛有足够盐酸的容器中。
4.随着吡啶的滴加,会观察到产物的形成,即吡嗪盐酸盐。
5.滴加完成后,将产物沉淀出来,用无水乙醚进行洗涤和过滤。
6.最后,通过干燥,得到吡嗪盐酸盐的固体产物。
实验结果•成功合成吡嗪盐酸盐。
•吡嗪盐酸盐以白色晶体的形式得到。
实验讨论•吡嗪盐酸盐的合成实验基本顺利,得到了预期的产物。
•这个实验过程中需要注意滴加吡啶的速度,过快或过慢都可能导致产物的损失。
•实验中使用的无水乙醚可以有效地洗涤和分离产物。
总结通过本次实验,我们成功合成了吡嗪盐酸盐,并观察到了其物理性质。
该实验提供了理论和实践基础,帮助我们更深入地了解吡嗪盐酸盐的合成方法与特性。
在以后的研究与应用中,这将为我们提供重要的参考和指导。
实验改进•在本实验中,我们使用了无水乙醚进行洗涤和过滤。
然而,无水乙醚对环境和人体有一定的危害性。
可以尝试使用更环境友好的溶剂替代无水乙醚,如乙醇或醚类溶剂。
•在滴加吡啶的过程中,可以使用分布孔径调节器(Buret)进行较为精确的控制,以避免滴加速度过快或过慢。
•可以进一步对吡嗪盐酸盐进行物理性质和化学性质的研究,比如熔点、溶解度和反应性等方面的实验。
实验应用•吡嗪盐酸盐作为一种有机盐,在有机合成、医药学和农药学等领域有广泛的应用。
•吡嗪盐酸盐可以用于合成各种有机合成反应中的中间体或胺类化合物。
•在医药学中,吡嗪盐酸盐常用于合成治疗中枢神经系统疾病的药物。
吡嗪盐酸盐的合成实验报告(一)

吡嗪盐酸盐的合成实验报告(一)
吡嗪盐酸盐的合成实验报告
实验目的
•合成吡嗪盐酸盐
•研究吡嗪盐酸盐的性质及用途
材料与仪器
•吡嗪
•氯化氢
•氯仿
•烧杯
•试管
•恒温水浴器
•滤纸
•离心机
实验步骤
1.在烧杯中加入适量吡嗪粉末。
2.用滴管缓慢地向烧杯中加入氯化氢溶液,同时用玻璃杯隔热保护。
注意加入氯化氢溶液的速度要适当,以避免反应过快产生危险。
3.将烧杯置于恒温水浴器中,并控制温度在60°C左右。
保持这一
温度下反应2-3小时,直至反应结束。
4.将反应混合物转移至试管中,用氯仿洗涤混合物,使吡嗪盐酸盐
溶于氯仿中。
5.离心对溶液进行分离,得到含有吡嗪盐酸盐的氯仿溶液。
注意事项
•在实验过程中,要严格控制氯化氢的用量和反应温度,以确保实验安全和高效进行。
•实验完成后,应仔细清洗各个实验器材,避免反应物残留引发其他不必要的化学反应。
结果与讨论
•实验得到的吡嗪盐酸盐氯仿溶液可以用于后续实验或进一步分析。
•吡嗪盐酸盐是一种常用的药物中间体,具有广泛的应用领域,例如医学、农业和化学工业等。
结论
本实验成功合成了吡嗪盐酸盐,并研究了其性质及用途。
根据实
验结果,吡嗪盐酸盐可以作为药物中间体在多个领域中得到应用。
以上为本次实验的相关报告。
感谢您的关注和阅读!。
吡嗪盐酸盐含量测定方法

吡嗪盐酸盐含量测定方法
一、前言吡嗪盐酸盐是一种常用的药物成分,其含量测定方法对于
药品质量控制至关重要。
本文将介绍吡嗪盐酸盐含量测定方法。
二、
实验原理本实验采用紫外分光光度法进行吡嗪盐酸盐的含量测定。
在
特定波长下,吡嗪盐酸盐会发生最大吸收,通过比较样品和标准溶液
的吸光度差异来计算样品中吡嗪盐酸盐的含量。
三、实验步骤1. 样品
制备:取适量粉末状样品称入容器中,并加入少许去离子水悬浮均匀。
2. 制备标准曲线:取不同体积(0.5ml, 1ml, 1.5ml, 2ml)的10μg/ml 吡
嗪标准溶液置于4个25ml 容积瓶内,再加入足够数量去离子水稀释至
刻度线处得到四个不同浓度(2μg/ml, 4μg/ml, 6μg/ml,8 μg/ml)的标准
曲线溶液。
3. 测定样品和标准曲线:将上述制备好的样品和标准曲线
溶液放入紫外可见分光光度计中,在特定波长下记录其吸光度值,并
根据所建立起来的标准曲线计算出相应物质在待检测物质中所占百分
比或者摩尔数等信息。
四、注意事项1. 实验过程需保持干燥清洁,避
免灰尘进入仪器影响结果;2. 操作时需佩戴手套及口罩以防止污染; 3. 实验操作需要严格按照相关规范进行; 4. 所使用试剂必须为优质纯化
试剂并符合国家相关规范; 5.所有玻璃仪器都要经过彻底清洁后方可使用。
农药设计合成实验报告(3篇)

第1篇一、实验目的1. 了解农药分子的设计原理和合成方法。
2. 掌握农药活性成分的筛选和活性测定技术。
3. 学习实验操作技能,提高实验设计能力。
二、实验原理农药设计合成是指根据植物病害的发生机理和农药的作用机制,通过化学合成方法设计并合成具有特定药理活性的化合物。
本实验以设计合成一种新型除草剂为目标,采用活性基团拼接和骨架跃迁的方法,设计并合成了一系列吡唑酰胺类衍生物,并对其除草活性进行了筛选和测定。
三、实验材料与仪器1. 实验材料:- 吡唑类化合物- 酰胺类化合物- 反应溶剂- 标准农药- 草本植物样本2. 实验仪器:- 热水浴- 磁力搅拌器- 反应釜- 红外光谱仪- 核磁共振波谱仪- 高效液相色谱仪四、实验步骤1. 设计阶段:- 分析除草剂的活性成分,确定目标化合物类型。
- 根据活性成分的化学结构,设计合成路线。
- 选择合适的活性基团和骨架结构,进行结构优化。
2. 合成阶段:- 按照设计好的合成路线,进行反应物选择和反应条件优化。
- 通过加热、搅拌等操作,进行反应物的混合和反应。
- 反应结束后,进行分离纯化,得到目标化合物。
3. 活性筛选阶段:- 将合成的化合物与标准农药进行对比,确定其活性。
- 通过草本植物样本,测定化合物的除草活性。
- 分析化合物的构效关系,优化结构。
4. 活性测定阶段:- 利用高效液相色谱仪,测定化合物的纯度和含量。
- 利用红外光谱仪和核磁共振波谱仪,分析化合物的结构。
- 对比标准农药,确定化合物的活性。
五、实验结果与分析1. 合成结果:- 按照设计路线,成功合成了目标化合物。
- 通过红外光谱和核磁共振波谱分析,确定了化合物的结构。
2. 活性筛选结果:- 与标准农药相比,目标化合物具有较好的除草活性。
- 通过草本植物样本的测定,发现目标化合物对多种杂草具有显著的抑制作用。
3. 构效关系分析:- 通过对比不同结构的目标化合物,发现活性基团和骨架结构对活性有显著影响。
- 优化结构后,目标化合物的活性得到提高。
吡嗪盐酸盐的合成实验报告

吡嗪盐酸盐的合成实验报告背景吡嗪盐酸盐是一种重要的有机化合物,具有广泛的应用领域,包括药物、染料、农药等。
本实验旨在通过合成吡嗪盐酸盐的方法,探究其合成过程及条件对产率的影响,为吡嗪盐酸盐的合成提供参考。
实验目的1.合成吡嗪盐酸盐;2.探究合成过程中反应条件对产率的影响;3.分析合成产物的质量和纯度。
实验原理吡嗪盐酸盐的合成可通过对吡嗪和盐酸进行反应得到。
吡嗪是一种含有氮原子的芳香化合物,其与酸反应能够形成相应的盐酸盐。
反应方程式如下所示:吡嗪 + 盐酸→ 吡嗪盐酸盐在实验中,我们将通过控制反应条件,如温度、溶剂和反应时间等,来优化吡嗪盐酸盐的合成产率。
实验步骤1.准备实验室所需的器材和试剂,包括吡嗪、盐酸、溶剂等;2.在反应容器中加入适量的吡嗪,并慢慢滴加盐酸溶液,同时控制反应温度在适宜的范围内;3.在反应过程中,观察反应物的溶解情况和反应的进行程度;4.反应完成后,将产物进行过滤、洗涤和干燥,得到吡嗪盐酸盐;5.对合成产物进行质量和纯度分析,包括使用红外光谱仪进行谱图分析和使用熔点仪测定熔点。
实验结果与分析根据实验步骤所述方法,我们成功合成了吡嗪盐酸盐。
合成产物经过过滤、洗涤和干燥后,得到了白色结晶固体。
通过红外光谱仪对产物进行谱图分析,发现产物的红外光谱与吡嗪盐酸盐的标准谱图吻合度较高,表明合成产物的结构与目标产物一致。
在合成过程中,我们还探究了反应条件对产物产率的影响。
结果显示,在较低的温度和适宜的反应时间下,产物的产率较高。
这表明反应温度和反应时间对反应的进行和产物的生成起到重要的影响作用。
通过熔点仪测定,我们得到了合成产物的熔点。
与吡嗪盐酸盐的标准熔点相比,合成产物的熔点在一定范围内,表明合成产物的纯度较高。
结论与建议通过本实验,我们成功合成了吡嗪盐酸盐,并通过红外光谱和熔点测定对产物的质量和纯度进行了分析。
在合成过程中,我们发现反应温度和时间对产物的产率有较大的影响。
在以后的实验中,可以进一步优化反应条件,如调整反应温度和时间,以提高产物的产率。
药物合成实验

实验一 盐酸苯海索(Benzhexol Hydrochloride )的合成 一、目的要求1. 了解Grignard 反应、Mannich 反应机理以及在药物合成上的应用。
2. 通过Grignard 反应掌握无水反应基本操作。
3. 了解无水乙醚的制备及操作注意点。
4. 正确掌握搅拌、回流、蒸馏、重结晶等基本单元操作。
二、实验原理盐酸苯海索又名安坦(Antane Hydrochloride ),化学名为1-环已基-1-苯基-3-哌啶基丙醇盐酸盐(1-Cyclohexyl-1-phenyl-3-piperdino- propanol hydrochloride )。
本品能阻断中枢神经系统和周围神经系统的毒蕈碱样胆碱受体。
临床上用于治疗震颤麻痹综合症,也用于斜颈、颜面痉挛等症的治疗。
盐酸苯海索大多以苯乙酮为原料与甲醛、哌啶盐酸盐进行Mannich 反应制得β-哌啶基苯丙酮盐酸盐中间体,再与氯代环已烷、金属镁作用制得的Grignard 试剂反应,得到盐酸苯海索。
反应如下:COCH3HCOH N H HCl COCH2CH2N HCl Cl Mg I 2无水乙醚MgCl COCH2CH2HCl MgCl C CH 2CH 2OMgClN HCl2CH2HCl无水乙醚三、实验方法(一)β-哌啶基苯丙酮盐酸盐1. 原料与试剂苯乙酮 18.1g(0.15mol)多聚甲醛 7.6g(0.25mol)哌啶(六氢吡啶) 30g(0.35mol ,约37.5 ml)浓盐酸 30~40ml 95%乙醇 96ml2. 实验步骤(1)哌啶盐酸盐的制备在分别装有搅拌器、恒压滴液漏斗(事先用95%乙醇检漏)、回流冷凝管及干燥管(如何安装)的250 mL三颈瓶中,加入30 g(约37.5 mL)的哌啶,60mL 95%乙醇。
在搅拌下从恒压滴液漏斗向反应瓶中滴入35mL浓盐酸,搅拌至反应液pH值约为1,约1小时左右。
然后拆除搅拌器、恒压滴液漏斗、回流冷凝管及干燥管,改装成蒸馏装置,加热蒸去乙醇和水,当反应物呈稀糊状时[1]停止蒸馏。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
吡嗪盐酸盐的合成实验报告
实验目的:
本实验旨在通过反应合成吡嗪盐酸盐,并利用物理方法对合成产物进行表征。
实验原理:
吡嗪(Pyrimidine)是一种六元杂环化合物,化学式为C4H4N2、吡嗪盐酸盐是吡嗪的盐酸盐形式,由于其中的阳离子是盐酸根,因此会增加溶解度和稳定性。
吡嗪盐酸盐在医药领域具有广泛的应用,因此其合成具有重要意义。
实验步骤:
1. 在一个干燥的圆底烧瓶中加入吡嗪(0.1 mol)。
2. 缓慢加入氯化氢气(稀释到1 mol/L),并同时加热至回流。
3.在反应进行的同时,将烧瓶表面进行冷却以充分回收氯化氢气。
4.反应持续3小时后,关闭加热,将混合溶液冷却至室温。
5.用去离子水稀释混合溶液,直至氨水添加后PH值大于7
6.过滤得到橙色的沉淀产物。
7.将沉淀产物在抽滤干燥器中干燥至恒定质量。
8.对产物进行红外光谱、质谱和元素分析进行表征。
实验结果与讨论:
形成的吡嗪盐酸盐产物以橙色的沉淀形式出现,在干燥后呈现黄色粉
末状。
通过元素分析,发现样品中含有C、H和N元素,符合吡嗪盐酸盐
的化学式C4H4N2·HCl。
红外光谱显示样品中有特征的C-N、C=O和C-H
官能团的伸缩振动吸收峰,进一步验证了样品的结构。
质谱分析进一步证
实了产物的分子质量以及分子离子峰。
结论:
本实验成功合成了吡嗪盐酸盐,并通过红外光谱、质谱和元素分析对
其进行了表征。
实验结果表明所得产物为吡嗪盐酸盐,并与理论值符合较好。
本实验为吡嗪盐酸盐的合成提供了一个简单有效的方法,对于相关领
域的研究具有重要意义。
实验中可能存在的问题和改进方法:
1.实验过程中需要注意溶剂的干燥和质量控制,以避免水分对反应的
干扰。
2.实验过程中可以考虑加入催化剂或调整反应条件,以提高反应速率
和产率。
3.为了更好地对产物进行分析和表征,可以增加其他实验技术的应用,如核磁共振和X射线衍射分析。