药材检验原始记录样本
何首乌药材检验记录-模板

药材检验记录文件编码:RC-8010品名: 何首乌批号:规格: 来源:取样量: 检验日期:代表量: 完成日期:检验依据:《中国药典》2010年版一部【性状】本品呈团块状或不规则纺捶形,长,,,,cm,直径,,,,,m。
表面红棕色或红褐色,皱缩不平,有浅沟,并有横长皮孔样突起和细根痕。
体重,质坚实,不易折断,断面浅黄棕色或浅红棕色,显粉性,皮部有,,,,,个类圆形异型维管束环列,形成云锦状花纹,中央木部较大,有的呈木心。
气微,味微苦而甘涩。
【鉴别】(1)本品横切面:木栓层为数列细胞,充满棕色物。
韧皮部较宽,散有类圆形异型维管束,,,,,个,为外韧型,导管稀少。
根的中央形成层成环;木质部导管较少,周围有管胞和少数木纤维。
薄壁细胞含草酸钙簇晶和淀粉粒。
粉末黄棕色。
淀粉粒单粒类圆形,直径,,,,,μm,脐点人字形、星状或三叉状,大粒者隐约可见层纹;复粒由2〜9分粒组成。
草酸钙簇晶直径,,,,,μm,偶见簇晶与较大的方形结晶合生。
棕色细胞类圆形或椭圆形,壁稍厚,胞腔内充满淡黄棕色、棕色或红棕色物质,并含淀粉粒。
具缘紋孔导管直径,,,,,μm。
棕色块散在,形状、大小及颜色深浅不一。
(2)取本品粉末g,加乙醇50ml,加热回流1小时,滤过,滤液浓缩至3ml,作为供试品溶液。
另取何首乌对照药材g,同法制成对照药材溶液。
照薄层色谱法(附录VI B)试验,吸取上述两种溶液各2μl,分别点于同一以羧甲基纤维素钠为黏合剂的硅胶H薄层板上使成条状,以三氯甲烷-甲醇(7 :3)为展开剂,展至约3. 5cm ,取出,晾干,再以三氯甲烷-甲醇(20 :1)为展开剂,展至约7cm ,取出,晾干,置紫外光灯(365nm)下检视。
供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点。
1:2:何首乌对照药材批号:来源:检验结果:()【检查】1.水分标准规定本品含水分不得过10.0% 。
(附录IX H 第一法)仪器:FA2004电子天平JC-101电热干燥箱单位:g1# 2#称量瓶烘2小时重量 W 1 称量瓶烘2小时重量 W 1称量瓶再烘0.5小时重量 W 2 称量瓶再烘0.5小时重量 W 2样品+称量瓶重 W 3 样品+称量瓶重 W 3样品+称量瓶烘5小时重量 W 4 样品+称量瓶烘5小时重量 W 4样品+称量瓶再烘1小时重量 W 5 样品+称量瓶烘1小时重量 W 5计算公式:P= W 3- W 5 W 3- W 2×100% P1= ×100% = P2=×100%= P=相对偏差=P1- P2 ×100%= 检验结果:( )2.总灰分 标准不得过5.0% (附录IX K)仪器: FA2004电子天平 4—10型 箱式电阻炉 单位:g1# 2#坩埚500℃炽灼2小时称重 W1 坩埚500℃炽灼2小时称重 W1坩埚500℃ 再炽灼1小时称重 W2 坩埚500℃ 再炽灼1小时称重 W2样品重 W3 样品重 W3样品+坩埚 550℃炽灼5小时 W4 样品+坩埚 550℃炽灼2小时 W4样品+坩埚 550℃ 再灰炽灼1小时 W5 样品+坩埚 550℃ 再炽灼1小时 W5计算公式:P= W5- W2 W3×100% P1= ×100% = P2=×100%=P=相对偏差=P1- P2 P1+ P2×100%= 检验结果:( ) 【含量测定】二苯乙烯苷避光操作。
药材检验原始记录样本

XXXXX药业(饮片)有限公司原药材检验报告单XXXXX药业(饮片)有限公司原药材检验记录【性状】结果:【鉴别】(1)显微鉴别横截面:结果:粉末:结果:(2)薄层鉴别供试品溶液的制备:取粉末1g,加乙醇15ml,加热回流1小时,放冷,滤过,滤液蒸干,残渣加乙醇5ml使溶解。
对照药材、对照品溶液配制:取菊花对照药材1g,同法制成对照药材溶液。
再取绿原酸对照品,加乙醇制成每1ml含O.5mg的溶液。
温度:(℃)相对湿度:(%)展开剂:三氯甲烷-丙酮-甲醇-5%浓氨试液(6:1:1:0.1)薄层板:硅胶G显色剂:稀碘化铋钾试液灯光:白光、紫外光灯(365nm)展距:(cm)供试品色谱中,在与对照药材色谱相对应的位置上,显相同颜色的荧光斑点。
S1为对照药材(对照品为中检所提供编号为)S2为对照品(对照品为中检所提供编号为) T为样品结果:【检查】杂质不得过 XX % (附录IX A)杂质称重: g杂质计算结果为: % (标准规定不得过 XX %)结果:膨胀度应不低于4.0(附录IX O)温度:(℃)相对湿度:(%)电子天平型号:CP214 溶剂:水样品编号 1# 2# 3#干燥品称重: g g g第一次样品膨胀后体积: ml ml ml第二次样品膨胀后体积: ml ml ml(两次差异不超过0.1ml)膨胀度计算结果为:(标准规定不低于4.0)结果:水分不得过12.0% (附录Ⅸ H 第一法)。
温度:(℃)相对湿度:(%)烘箱型号:DHG-91012SA型电子天平型号:CP214样品编号 1# 2#第一次称量瓶干燥(105℃ 3h) (g)(g)第二次称量瓶恒重(105℃ 1h) (g)(g)样品称重(g)(g)第一次称量瓶+样品干燥(105℃ 5h) (g)(g)第二次称量瓶+样品恒重(105℃ 1h) (g)(g)水分计算结果为:(%)(标准规定不得过12.0%)结果:总灰分不得过4.0%(附录Ⅸ K)温度:(℃)相对湿度:(%)马福炉型号:SX2.5-10 电子天平型号:CP214样品编号 1# 2#第一次坩锅称重(600℃ 3h) (g)(g)第二次坩锅恒重(600℃ 0.5h) (g)(g)样品称重(g)(g)第一次坩锅+残渣称重(600℃ 3h) (g)(g)第二次坩锅+残渣恒重(600℃ 0.5h) (g)(g)总灰分计算结果为:(%)(标准规定不得过4.0%)结果:酸不溶性灰分不得过3.0%(附录Ⅸ K)。
复方丹参片成品检验原始记录

复方丹参片成品检验原始记录
一、检验目的:
1.确定复方丹参片的各项质量指标是否符合药典要求;
2.检测复方丹参片中是否存在有害物质。
二、检验样品:
三、检验仪器和试剂:
1.恒温槽;
2.紫外分光光度计;
3.高效液相色谱仪;
4.显微镜;
5.乙醇;
6.氢氧化钠溶液;
7.硝酸银溶液;
8.碘液;
9.铁氯化物溶液。
四、检验项目和方法:
1.外观检查:
根据药典规定,检查复方丹参片的色泽、形状、气味等特征,判断是否符合标准。
2.含量测定:
采用高效液相色谱法测定复方丹参片中丹参酮酸B的含量。
3.汞、铅、镉、砷的含量测定:
采用草酸法测定复方丹参片中重金属的含量。
4.色谱指纹图谱分析:
采用高效液相色谱法,建立复方丹参片的色谱指纹图谱,比较样品与对照品的相似度。
5.微生物限度测试:
根据药典规定,采用菌落总数限度法和霉菌和酵母菌限度法,检测复方丹参片中的微生物限度。
五、检验结果记录:
1.外观检查:
2.含量测定:
3.汞、铅、镉、砷的含量测定:
4.色谱指纹图谱分析:
与对照品相比,复方丹参片的色谱指纹图谱相似度为98%,符合药典要求。
5.微生物限度测试:
六、检验结论:
根据上述检验结果,复方丹参片的各项质量指标均符合药典要求,未检出有害物质,微生物限度也在合理范围内,可以确认该批复方丹参片合格。
1113厚朴成品检验原始记录
标准规定
检 验 结 果
检验结论
检验人
复核人
检验日期
供试品与对照品色谱相应的位置上,显相同颜色的斑点。
显相同颜色的
斑点□
3。水分不得过12.0%(附录ⅨH第一法).
3.1。1主要检验仪器:电子分析天平, 型号,编号
鼓风干燥箱, 型号,编号
(1)恒重称量瓶重W瓶1:
(2)恒重称量瓶重W瓶1:
7。1.4对照品溶液的制备取盐酸小檗碱对照品适量,精密称定,加流动相制成每1ml含0.1mg的溶液,即得。
7.1。5供试品溶液的制备取本品粉末(过三号筛)(样1)g(样2)g(0。1g),精密称定,置100ml量瓶中,加流动相80ml,超声处理(功率250W,频率40kHz)40分钟,放冷,用流动相稀释至刻度,摇匀,滤过,取续滤液,即得.
WX(1-水分%)X25/100
浸出物(%)1=×100%=
浸出物(%)2=×100%=
平均浸出物(%)= = =
6.3结果判定:T:℃,RH:%
项目
标准规定
检验结果
检验结论
检验人
复核人
检验时间
浸出物
≥14%
%
7【含量测定】照高效液相色谱法(附录Ⅵ D)测定。
7。1小檗碱
7.1.1主要检验仪器:电子分析天平, 型号,编号
恒重一次总重W1: 恒重一次总重W1:
恒重二次总重W2: 恒重二次总重W2:
总灰分(%)= ×100%
总灰分(%)1=×100%=
总灰分(%)2=×100%=
平均总灰分(%)= = =
4.1。2结果判定:T:℃,RH:%
项目
标准规定
药材检验原始记录样本

XXXXX药业(饮片)有限公司原药材检验报告单XXXXX药业(饮片)有限公司原药材检验记录【性状】结果:【鉴别】(1)显微鉴别横截面:结果:粉末:结果:(2)薄层鉴别供试品溶液的制备:取粉末1g,加乙醇15ml,加热回流1小时,放冷,滤过,滤液蒸干,残渣加乙醇5ml使溶解。
对照药材、对照品溶液配制:取菊花对照药材1g,同法制成对照药材溶液。
再取绿原酸对照品,加乙醇制成每1ml含O.5mg的溶液。
温度:(℃)展开剂:三氯甲烷-丙酮-甲醇-5%浓氨试液(6:1:1:0.1)薄层板:硅胶G显色剂:稀碘化铋钾试液灯光:白光、紫外光灯(365nm)展距:(cm)供试品色谱中,在与对照药材色谱相对应的位置上,显相同颜色的荧光斑点。
S1为对照药材(对照品为中检所提供编号为)S2为对照品(对照品为中检所提供编号为)T为样品结果:【检查】杂质不得过XX % (附录IX A)杂质称重: g杂质计算结果为:% (标准规定不得过XX %)结果:膨胀度应不低于4.0(附录IX O)温度:(℃)相对湿度:(%)电子天平型号:CP214 溶剂:水样品编号1# 2# 3#干燥品称重:g g g第一次样品膨胀后体积:ml ml ml第二次样品膨胀后体积:ml ml ml(两次差异不超过0.1ml)膨胀度计算结果为:(标准规定不低于4.0)结果:水分不得过12.0% (附录ⅨH 第一法)。
温度:(℃)相对湿度:(%)烘箱型号:DHG-91012SA型电子天平型号:CP214样品编号1# 2#第一次称量瓶干燥(105℃3h) (g)(g)第二次称量瓶恒重(105℃1h) (g)(g)样品称重(g)(g)第一次称量瓶+样品干燥(105℃5h) (g)(g)第二次称量瓶+样品恒重(105℃1h) (g)(g)水分计算结果为:(%)(标准规定不得过12.0%)结果:总灰分不得过4.0%(附录ⅨK)温度:(℃)相对湿度:(%)马福炉型号:SX2.5-10 电子天平型号:CP214样品编号1# 2#第一次坩锅称重(600℃3h) (g)(g)第二次坩锅恒重(600℃0.5h) (g)(g)样品称重(g)(g)第一次坩锅+残渣称重(600℃3h) (g)(g)第二次坩锅+残渣恒重(600℃0.5h) (g)(g)总灰分计算结果为:(%)(标准规定不得过4.0%)结果:酸不溶性灰分不得过3.0%(附录ⅨK)。
中药饮片——薄层扫描检验原始记录

中药饮片——薄层扫描检验原始记录XXXXXXXXX有限企业薄层扫描原始记录编号:品名规格温度编码(批号)请验日期湿度甜菜碱取本品剪碎,取约g,精细称定,加80%甲醇 50ml,加热回流 1 小时,放冷,滤过,用80%甲醇 30ml 分次清洗残渣和滤器,归并洗液与滤液,浓缩至10ml ,用盐酸调节 pH 值至 1,加人活性炭lg ,加热煮沸,放冷,滤过,用水15ml 分次清洗,归并洗液与滤液,加人新配制的 2.5%硫氰酸铬铵溶液20ml,搅匀, 100C 以下搁置 3 小时。
用G4垂熔漏斗滤过,积淀用少许冰水清洗,抽干,残渣加丙酮溶解,转移至5ml 量瓶中,加丙酮至刻度,摇匀,作为供试品溶液。
另取甜菜碱比较品适当,精细称定,加盐酸甲醇溶液(0. 5— 100)制成每 l m l 含 4 mg 的溶液,作为比较品溶液。
照薄层色谱法(公则 0502)试验。
电子天平型号:电子天平编号:薄层色谱扫描仪型号:薄层色谱扫描仪编号:(1)点样:精细汲取供试品溶液5μl、比较品溶液3μ1与6μl(2)薄层板: G 薄层板上(3)睁开剂:以丙酮 -无水乙醇 -盐酸( 10 :6 : 1)为睁开剂(4)睁开:预饱和 30 分钟,睁开,拿出,挥干溶剂,立刻喷以新配制的改进碘化铋钾试液,搁置1~3 小时至斑点清楚照(5)测试条件:波长:;λS = 515nm, λR= 590nm,丈量供试品吸光度积分值与比较品吸光度积分值 ,计算,即得。
计算:结果:(本品按干燥品计算,含甜菜碱(C5112)不得少于0. 30%)H NORSD=□切合规定□不切合规定□仅作数据累积查验人:复核人:日期:日期:。
黄芪原药材检验记录
检验结果:
检验结论:
2理化鉴别:取本品粉末3g,加甲醇20ml,加热回流1小时,滤过,滤液加于中性氧化铝柱(100~120目,5g,内径为10~15mm)上,用40%甲醇100ml洗脱,收集洗脱液,蒸干,残渣加水30ml使溶解,用水饱和的正丁醇振摇提取2次,每次20ml,合并正丁醇液,用水洗涤2次,每次20ml,弃去水液,正丁醇液蒸干,残渣加甲醇0.5ml使溶解,作为供试品溶液。另取黄芪甲苷对照品,加甲醇制成每1ml含1mg的溶液,作为对照品溶液。照薄层色谱法(通则0502)试验,吸取上述两种溶液各2μl,分别点于同一硅胶G薄层板上,以三氯甲烷-甲醇-水(13:7:2)的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,日光下显相同的棕褐色斑点;紫外光灯(365nm)下显相同的橙黄色荧光斑点。
T1
T2
减失重量(%)T=(W2-W3)/(W2-W1)
T1= T2 平均值:
检验结论:
2总灰分:标准规定:不得过5.0%
温度:℃,相对湿度%
方法:炽灼法
炽灼条件:在℃炽灼小时,冷却分钟,再在℃下炽灼小时,冷却分钟
空坩埚 样品+坩埚 灰分+坩埚
T1
T2
总灰分重量(%)T=(灰分及坩埚重量-空坩埚)/供试品重量*100%
4有机氯农药残留量 照农药残留量测定法(通则2341有机氯类农药残留量测定法一第一法)测定。 含总六六六(α-BHC、β-BHC、γ-BHC、δ-BHC之和)不得过0.2mg/kg;总滴滴涕(pp'-DDE、pp'-DDD、op'-DDT,pp-DDT之和)不得过0.2mg/kg;五氯硝基苯不得过0.1mg/kg。
00-002川芎检验原始记录
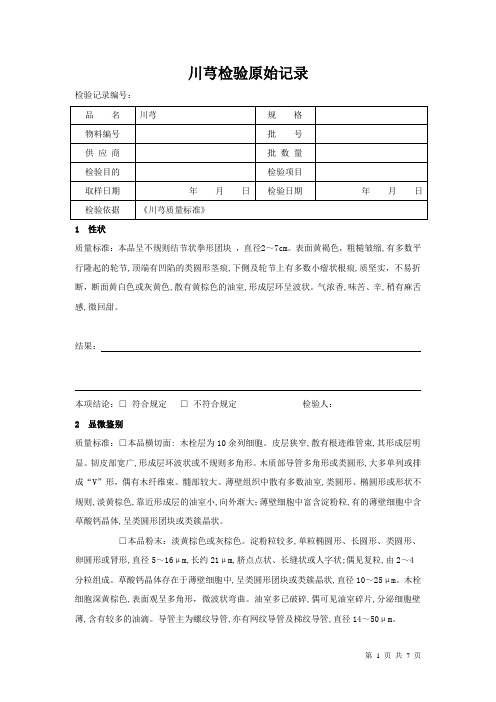
川芎检验原始记录检验记录编号:1 性状质量标准:本品呈不规则结节状拳形团块,直径2~7cm。
表面黄褐色,粗糙皱缩,有多数平行隆起的轮节,顶端有凹陷的类圆形茎痕,下侧及轮节上有多数小瘤状根痕,质坚实,不易折断,断面黄白色或灰黄色,散有黄棕色的油室,形成层环呈波状。
气浓香,味苦、辛,稍有麻舌感,微回甜。
结果:本项结论:□符合规定□不符合规定检验人:2 显微鉴别质量标准:□本品横切面: 木栓层为10余列细胞。
皮层狭窄,散有根迹维管束,其形成层明显。
韧皮部宽广,形成层环波状或不规则多角形。
木质部导管多角形或类圆形,大多单列或排成“V”形,偶有木纤维束。
髓部较大。
薄壁组织中散有多数油室,类圆形、椭圆形或形状不规则,淡黄棕色,靠近形成层的油室小,向外渐大;薄壁细胞中富含淀粉粒,有的薄壁细胞中含草酸钙晶体,呈类圆形团块或类簇晶状。
□本品粉末:淡黄棕色或灰棕色。
淀粉粒较多,单粒椭圆形、长圆形、类圆形、卵圆形或肾形,直径5~16μm,长约21μm,脐点点状、长缝状或人字状;偶见复粒,由2~4分粒组成。
草酸钙晶体存在于薄壁细胞中,呈类圆形团块或类簇晶状,直径10~25μm。
木栓细胞深黄棕色,表面观呈多角形,微波状弯曲。
油室多已破碎,偶可见油室碎片,分泌细胞壁薄,含有较多的油滴。
导管主为螺纹导管,亦有网纹导管及梯纹导管,直径14~50μm。
仪器及型号:□X2型双目显微镜□XSP-2C型生物显微镜结果:本项结论:□符合规定□不符合规定检验人:3 化学反应质量标准:应显红紫色取本品粉末[1g] g,加石油醚(30~60℃)5ml,放置10小时,时时振摇,静置,取上清液1ml,挥干后,残渣加甲醇1ml使溶解,再加2﹪3,5-二硝基苯甲酸的甲醇溶液2~3滴与甲醇饱和的氢氧化钾溶液2滴,观察。
本项结论:□符合规定□不符合规定检验人:4 薄层鉴别质量标准:供试品色谱中,在与川芎对照药材色谱相应的位置上,显相同颜色的荧光斑点。
仪器及型号:天平:□AB204-N电子天平□ESJ-1823电子天平□ZF—90型暗箱式紫外透射仪对照药材溶液制备:取川芎对照药材1g,同法制成对照药材溶液;对照品溶液制备:取欧当归内酯A对照品,加乙酸乙酯制成每1ml含0.1mg的溶液(置棕色量瓶中),作为对照品溶液。
鱼腥草检验原始记录干

浙江泰康药业集团有限公司鱼腥草检验原始记录检验内容1、【性状】:茎呈 ,扭曲,表面 ,具纵棱数条;质脆,易折断。
叶片卷折皱缩,展平后呈心形,上表面暗黄绿色至暗棕色,下表面灰绿色或灰棕色。
穗状花序黄棕色。
结果:2、【鉴别】2.1、 取本品粉末适量,置小试管中,用玻棒压紧,滴加品红亚硫酸试液少量至上层粉末湿润,放置片刻,自侧壁观察,湿粉末显 色。
结果:2.2、取干品25g 剪碎,照挥发油测定法加乙酸乙酯1ml , 缓缓加热至沸,并保持微沸4小时,放置半小时,取乙酸 乙酯液作为供试品溶液。
另取甲基正壬酮对照品,加乙酸 乙酯制成每1ml 含10μl 的溶液,作为对照品溶液。
照薄 层色谱法试验。
吸取上述供试品溶液5μl 、对照品溶液2μl ,分别点于同一硅胶G 薄层板上,以环乙烷-乙酸乙酯(9:1)为展开剂,展开,取出,晾干,喷以二硝基苯肼试液。
供试品色谱中与对照品色谱相应的位置上显色斑点。
结果:3、【检查】3.1、水分:照水分测定法依法测定,样品1:干燥的空称量瓶重M称量瓶= 1# 2# 3# 4# gM称量瓶+样品= g,干燥后M′称量瓶+样品= 1# 2# 3# 4# g水分%=M称量瓶+样品-M′称量瓶+样品M称量瓶+样品- M称量瓶×100%=样品2:干燥的空称量瓶重M称量瓶= 1# 2# 3# 4# gM称量瓶+样品= g,干燥后M′称量瓶+样品= 1# 2# 3# 4# g水分%=M称量瓶+样品-M′称量瓶+样品M称量瓶+样品- M称量瓶×100%=结果:3.2、酸不溶性灰分:照灰分测定法依法测定,样品1:空坩埚重量W1′= 1# 2# 3# 4# g残渣和坩埚重量W3′= 1# 2# 3# 4# g总灰分项下空坩埚重W1= 1# 2# 3# 4# g总灰分项下供试品和坩埚重W2= g酸不溶性灰分% = W3′- W1′W2-W1×100% =样品2:空坩埚重量W1′= 1# 2# 3# 4# g残渣和坩埚重量W3′= 1# 2# 3# 4# g总灰分项下空坩埚重W1= 1# 2# 3# 4# g总灰分项下供试品和坩埚重W2= g酸不溶性灰分% = W3′- W1′W2-W1×100% =结果:【二氧化硫残留量】1、精密称取中药材或饮片细粉10g,置1000ml两颈圆底烧瓶A中,加水300-400ml(没过刻度分液漏斗下端),在刻度分液漏斗C中加入盐酸溶液(6mol/L)适量备用;2、打开与冷凝水连接的回流冷凝管开关给水,冷凝管的上端E口处连接一橡胶导气管置于三角烧瓶内,加入水125ml和淀粉指示液1ml作为吸收液;3、开通氮气,调节适宜的气体流量(参考流量0.2/min),打开分液漏斗活塞,加入盐酸(6mol/L)10ml,给两颈烧瓶内的溶液加热至沸,并保持微沸约3min后开始用碘滴定液滴定,吸收液至磁力搅拌器上不断搅拌,至吸收液显蓝色且在20秒内蓝色或蓝紫色持续不褪,并将滴定结果用空白试验校正;每1ml碘滴定液(1mol/L)相当于0.032g的二氧化硫。
吴茱萸检验原始记录

XXX药业有限公司吴茱萸检验记录【性状】本品呈形,直径mm。
表面色至褐色,粗糙,有多数点状突起或凹下的。
顶端有的裂隙,基部残留被有的果梗。
质而脆,横切面可见子房室,每室有种子1 粒。
气,味。
结论:□符合规定□不符合规定检验人:日期:年月日复核人:日期:年月日【鉴别】(1)本品粉末。
非腺毛细胞,长μm,明显,有的胞腔内含棕黄色至物。
腺毛头部细胞,,常含黄棕色内含物;柄细胞。
较多,直径μm;偶有方晶。
石细胞,直径μm,胞腔。
油室碎片有时可见,。
结论:□符合规定□不符合规定检验人:日期:年月日复核人:日期:年月日(2)薄层鉴别供试品溶液的制备:取本品粉末0.4g,加乙醇10ml,静置30分钟,超声处理30分钟,滤过。
对照品溶液配制:取吴茱萸次碱对照品(中国食品药品检定研究院批号含量),g,吴茱萸碱对照品(中国食品药品检定研究院批号含量),g,加乙醇分别制成mg/ml和mg/ml的溶液。
温度:(℃)相对湿度:(%)展开剂:石油醚(60~90℃)-乙酸乙酯-三乙胺(7:3:0.1)薄层板:硅胶G点样量:各2ul显色剂:磷钼酸硫酸溶液烘烤温度:105℃灯光:紫外光灯(365nm)展距:(cm)标准:供试品色谱中,在与对照品色谱相对应的位置上,显相同颜色的荧光斑点。
S1:吴茱萸次碱对照品溶液S2:吴茱萸碱对照品溶液T:供试品溶液检验结果:供试品色谱中,在与对照品色谱的位置上,显颜色的荧光斑点。
结论:□符合规定□不符合规定检验人:日期:年月日复核人:日期:年月日【检查】杂质不得过7%(通则2301)。
分析天平:(AUW120D)YHJ016样品重量m:g 杂质质量m杂:g ;计算公式:W(%)= m杂m× 100%检验结果:杂质为%结论:□符合规定□不符合规定检验人:日期:年月日复核人:日期:年月日水分不得过15.0%(通则0832 第二法)。
温度:(℃)相对湿度:(%)分析天平:(AUW120D)YHJ016 电热鼓风干燥箱:(101-1A)YHJ005编号1# 2#第一次称量瓶干燥(105℃ 5h) (g)(g)第二次称量瓶恒重m p(105℃ 1h) (g)(g)样品称重m y(g)(g)第一次称量瓶+样品干燥(105℃ 5h) (g)(g)第二次称量瓶+样品恒重m z(105℃ 1h) (g)(g)计算公式:水分(%)= m p+ m y - m zm y×100%检验结果:水分1# = 2# = 平均值= %结论:□符合规定□不符合规定检验人:日期:年月日复核人:日期:年月日总灰分不得过10.0%(通则2302)温度:(℃)相对湿度:(%)分析天平:(AUW120D)YHJ016 箱式电阻炉:(SX-2.5-10)YHJ006 编号1# 2# 第一次坩锅称重(600℃ 3h) (g)(g)第二次坩锅恒重m p(600℃ 0.5h) (g)(g)样品称重m y(g)(g)第一次坩锅+残渣称重(600℃ 3h) (g)(g)第二次坩锅+残渣恒重m z(600℃ 0.5h) (g)(g)计算公式:总灰分(%)= m z-m pm y×100%检验结果:总灰分1# = 2# = 平均值= %结论:□符合规定□不符合规定检验人:日期:年月日复核人:日期:年月日浸出物照醇溶性浸出物测定法(通则2201)项下的热浸法测定,用稀乙醇作溶剂,不得少于30.0%。
332甘草原始检验记录

浙江泰康药业集团有限公司甘草检验原始记录检验内容1、【性状】:根呈,长25~100cm,直径0.6~3.5cm。
外皮松紧不一。
表面红棕色或灰棕色,具显著的纵皱纹、沟纹、皮孔及稀疏的细根痕。
质坚实,断面略显纤维性,黄白色,粉性,形成层环明显,射线放射状,有的有裂隙。
根茎呈圆柱形,表面有芽痕,断面中部有髓。
气微,味甜而特殊。
结果:2、【鉴别】2.1、本品横切面:木栓层为数列棕色细胞。
栓内层较窄。
韧皮部射线宽广,多弯曲,常现裂隙;纤维多成束,非木化或微木化,周围壁薄细胞常含草酸钙方晶;筛管群常因压缩而变形。
束内形成层明显。
木质部射线宽3~5列细胞;导管较多,直径约至160µm;木纤维成束,周围薄壁细胞亦含草酸钙方晶。
根中心无髓;根茎中心有髓。
粉末淡棕色。
纤维成束,直径8~14µm,壁厚,微木化,周围薄壁细胞含草酸钙方晶多见。
具缘纹孔导管较大,稀有网纹导管。
木栓细胞红棕色,多角形,微木化。
结果:2.2、取本品粉末1g,加乙醚40ml,加热回流1小时,滤过,药渣加甲醇30ml,加热回流1小时,滤过,滤液蒸干,残渣加水40ml使溶解,用正丁醇提取3次,每次20ml,合并正丁醇液,用水洗涤3次,蒸干,残渣加甲醇5ml使溶解,作为供试品溶液。
另取甘草对照药材1g,同法制成对照药材溶液。
再取甘草酸铵对照品,加甲醇制成每1ml含2mg的溶液,作为对照品溶液。
照薄层色谱法(附录VI B)试验,吸取上述三种溶液各1~2µl,分别点于同一用1%氢氧化钠溶液制备的硅胶G薄层板上,以醋酸乙酯-甲酸-冰醋酸–水(15:1:1:2)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,置紫外灯下(365nm)下检视。
供试品色谱中,在与对照药材色谱相应的位置上,显荧光斑点;在与对照品色谱相应的位置上显相的荧光斑点。
结果:3、【检查】3.1、水分:照水分测定法(附录IX H第一法)测定。
黄芪药材检验记录-模板

H 第一法) 。 JC-101 电热干燥箱 2# W1 W2 W3 W4 W5 称量瓶烘 2 小时重量 称量瓶再烘 0.5 小时重量 样品+称量瓶重 样品+称量瓶烘 5 小时重量 样品+称量瓶烘 1 小时重量 W1 W2 W3 W4 W5 单位:g
×100% =
P2=
×100%=
P=
相对偏差=
P1- P2 ×100%= P1+ P2 K)。 4—10 型 箱式电阻炉 2# W1 W2 W3 W4
共4页
1
紫外光灯(365nm)下检视。
1: 黄芪对照药材批号:
2: 来源: 检验结果: ( )
【检查】 1.水分 不得过 10.0% (附录 IX 仪器:FA2004 电子天平 1# 称量瓶烘 2 小时重量 称量瓶再烘 0.5 小时重量 样品+称量瓶重 样品+称量瓶烘 5 小时重量 样品+称量瓶再烘 1 小时重量 计算公式:P= P1= W3- W5 ×100% W3- W2
检验结果: (
)
2.总灰分 不得过 5 . 0% (附录 IX 仪器: FA2004 电子天平 1# 坩埚 500℃炽灼 2 小时称重 坩埚 500℃ 再炽灼 1 小时称重 样品重 样品+坩埚 550℃炽灼 5 小时 检验人:
单位:g
坩埚 500℃炽灼 2 小时称重 坩埚 500℃ 再炽灼 1 小时称重 样品重 样品+坩埚 550℃炽灼 2 小时 质量负责人:
1: 黄芪甲苷对照品批号:
2: 来源: 检验结果: ( )
(3)取本品粉末 g,加乙醇 30ml,加热回流 20 分钟,滤过,滤液蒸干,残渣加 0. 3%氢氧化钠溶液 15ml 使溶解,滤过,滤液用稀盐酸调节 pH 值至 5~6,用乙酸乙酯 15ml 振摇提取,分取乙酸乙酯液,用铺 有适量无水硫酸钠的滤纸滤过,滤液蒸干。残渣加乙酸乙酯 lml 使溶解,作为供试品溶液。另取黄芪对照 药材 g,同法制成对照药材溶液。照薄层色谱法(附录 VI B)试验,吸取上述两种溶液各 10μ l,分别 点于同一硅胶 G 薄层板上,以三氯甲烷-甲醇(10:1)为展开剂,展开,取出,晾干,置氨蒸气中熏后,置 检验人: 复核人: 质量负责人:
3.山药成品检验原始记录

山药成品检验原始记录品名麸炒山药规格编号/批号生产商(供应商)取样日期年月日检验依据:《中国药典》2010年版一部质量标准操作规程:麸炒山药检验操作规程性状本品结论:检验人:检验日期:显微鉴别显微镜的型号XSZ -107BA粉末观察:取干燥饮片,磨成细粉(过四号筛)。
用解剖针挑取样品粉末少许,置载玻片中央,加试液1滴,用针搅匀,待液体渗入粉末后,盖好盖玻片,于显微镜下观察。
本品粉末类白色。
淀粉粒单粒扁卵形、三角状卵形、类圆形或矩圆形,直径8~35µm,脐点点状、人字状、十字状或短缝状,可见层纹;复粒稀少,由2~3分粒组成。
草酸钙针晶束存在于黏液细胞中,长约至240µm,针晶粗2~5µm。
具缘纹孔导管、网纹导管、螺纹导管及环纹导管直径12~48µm。
观察结果:。
结论:检验人:检验日期:薄层鉴别电子天平型号AUW120D山药对照药材来源中检所批号供试品溶液制备:取本品粉末5g,加二氯甲烷30ml,加热回流2小时,滤过,滤液蒸干,残渣加二氯甲烷1ml使溶解,作为供试品溶液。
另取山药对照药材5g,同法制成对照药材溶液。
照薄层色谱法(附录Ⅵ B)试验,吸取上述两种溶液各4µl,分别点于同一硅胶G薄层板上,以乙酸乙酯-甲醇-浓氨试液(9:1:0.5)为展开剂,展开,取出,晾干,喷以10%磷钼酸乙醇溶液,在105℃加热至斑点显色清晰。
供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点。
1.山药对照药材供试品溶液所显斑点的位置与对照药材溶液的斑点, 2. 样品颜色。
复核人:复核日期:结论:检验人: 检验日期: 杂质取10-20g 供试品,摊开,用肉眼或放大镜(5-10倍)观察,将杂质拣出,过筛,过 号筛筛除药屑、灰屑。
将杂质、药屑和灰屑分出并称重,计算其在供试品中的含量(%)。
杂质限度:不得过3.0%。
供试品取样量W : g ;杂质、药屑、灰屑重m : g 计算公式:药屑杂质(%)=%100wm× 计算结果:结论:检验人: 检验日期:水分烘箱型号: ;电子天平型号: AUW120D取本品2-5g ,平铺于干燥至恒重的扁形称量瓶中,厚度不超过5mm ,疏松供试品不超过10mm ,精密称定,在100-105℃干燥5小时,冷却称重,再在上述温度干燥1小时,冷却称重,至连续两次称重的差异不超过5mg 为止。
复方丹参片成品检验原始记录

HB-09-OR-083/R02复方丹参片成品检验原始记录(共 4 页)品 名: 规 格: 批 号: 请检单号: 数 量: 取样日期: 来 源: 取样量: 检验日期: 检验依据:《中国药典》2010版一部【性状】 本品应为糖衣片或薄膜衣片,除去包衣后显棕色至棕褐色;气芳香,味微苦。
记录: 。
结论: 。
【鉴别】 (1)取本品,置显微镜下观察:树脂道碎片含黄色分泌物。
(三七)记录: 。
结论: 。
(2)取本品1.6g ,研细,加乙醚10ml ,超声处理5分钟,滤过,药渣备用,滤液挥干,残渣加醋酸乙酯2ml 使溶解,作为供试品溶液。
另取丹参酮ⅡA 、冰片对照品,分别加醋酸乙酯制成每1ml 含0.5mg 的溶液,作为对照品溶液。
照薄层色谱法(附录Ⅵ B )试验,吸取上述三种溶液各4ul ,分别点于同一硅胶G 薄层板上,以苯-醋酸乙酯(19:1)为展开剂,展开,取出,晾干。
供试品色谱中,在与丹参酮ⅡA 对照品色谱相应的位置上,应显相同颜色的斑点;喷以1%香草醛硫酸溶液,在110℃加热数分钟,在与冰片对照品色谱的位置上,应显相同颜色的斑点。
结论: 。
(3)取〔鉴别〕(2)项下的药渣,加甲醇25ml ,加热回流15分钟,放冷,滤过,滤液蒸干,残渣加水25ml ,微热使溶解,加水饱和的正丁醇25ml ,振摇提取,取正丁醇提取液,用氨试液25ml 洗涤,弃去氨溶液,再用正丁醇饱和的水洗涤2次,每次25ml ,正丁醇液浓缩至干,残渣加甲醇1ml 使溶解,作为供试品溶液。
另取三七对照药材0.5g ,同法制成对照药材溶液,再取三七皂苷R1对照品及人参皂苷Rb1、Rg1对照品,分别加甲醇制成每1ml 含1mg 的溶液,作为对照品溶液。
照薄层色谱法(附录Ⅵ B )试验,吸取上述四种溶液各1ul ,分别点于同一硅胶G 薄层板上,以三氯甲烷-甲醇-水(13:7:2)10℃以下放置分层的下层溶液为展开剂,展开,取出,晾干,喷以硫酸乙醇溶液(1→10),在110℃加热至斑点显色清晰。
甘草原始记录模板

检验原始记录检品编号:YPW20130187外观性状SYYJ-YSJL-001A检验者:年月日核对者:年月日原子吸收分光光度法SYYJ-YPJL-011D检验者:年月日核对者:年月日原子吸收分光光度法(续页)SYYJ-YPJL-011D检验者:年月日核对者:年月日原子吸收分光光度法(续页)SYYJ-YPJL-011D检验者:年月日核对者:年月日原子吸收分光光度法(续页)SYYJ-YPJL-011D检验者:年月日核对者:年月日原子吸收分光光度法(续页)SYYJ-YPJL-011D检验者:年月日核对者:年月日原子吸收分光光度法(续页)SYYJ-YPJL-011D检验者:年月日核对者:年月日检品名称甘草检品编号YPW20130187 批号20130203检验项目水分水分测定(烘干法)SYYJ-YPJL-009C 检查方法照水分测定法(中国药典年版部附录)天平型号档案编号仪器名称/型号档案编号干燥温度℃室温℃相对湿度 % 单位供试品处理计算公式测定与结果称量瓶恒重(W1)取样(W2)称瓶加供试品干燥(W3)结果% 修约值h 5hh 1hh 1h标准规定结论□符合规定□不符合规定检验者:年月日核对者:年月日有机氯农药残留量测定SYYJ-YPJL-049C检验者:年月日核对者:年月日有机氯农药残留量测定(续页)SYYJ-YPJL-049C检验者:年月日核对者:年月日有机氯农药残留量测定(续页)SYYJ-YPJL-049C检验者:年月日核对者:年月日高效液相色谱法(多个对照品,梯度)SYYJ-YPJL-049C检验者:年月日核对者:年月日高效液相色谱法续页(多个对照品,梯度)SYYJ-YPJL-007D检验者:年月日核对者:年月日检验者:年月日核对者:年月日。
中药饮片——薄层扫描检验原始记录

薄层色谱扫描仪型号:薄层色谱扫描仪编号:
(1)点样:精密吸取供试品溶液5µl、对照品溶液3µ1与6µl
(2)薄层板:G薄层板上
(3)展开剂:以丙酮-无水乙醇-盐酸(10:6:1)为展开剂
(4)展开:预饱和30分钟,展开,取出,挥干溶剂,立即喷以新配制的改良碘化铋钾试液,放置1~3小时至斑点清晰照
(5)测试条件:波长:;λS= 515nm,λR= 590nm,测量供试品吸光度积分值与对照品吸光度积分值,计算,即得。
计算:
结果:(本品按干燥品计算,含甜菜碱(C5H11NO2)不得少于0. 30%)
RSD=
□ 符合规定 □不符合规定 □ 仅作数据积累
XXXXXXXXX有限公司
薄层扫描原始记录
编号:
品 名
规格
温度
编码(批号)
请验日期
湿度
甜菜碱取本品剪碎,取约g,精密称定,加80%甲醇50ml,加热回流1小时,放冷,滤过,用80%甲醇30ml分次洗涤残渣和滤器,合并洗液与滤液,浓缩至10ml,用盐酸调节pH值至1,加人活性炭lห้องสมุดไป่ตู้,加热煮沸,放冷,滤过,用水15ml分次洗涤,合并洗液与滤液,加人新配制的2.5%硫氰酸铬铵溶液20ml,搅匀,100C以下放置3小时。用G4垂熔漏斗滤过,沉淀用少量冰水洗涤,抽干,残渣加丙酮溶解,转移至5ml量瓶中,加丙酮至刻度,摇匀,作为供试品溶液。另取甜菜碱对照品适量,精密称定,加盐酸甲醇溶液(0. 5—100)制成每l m l含4 mg的溶液,作为对照品溶液。照薄层色谱法(通则0502)试验。
人参药材检验记录-模板

1
共3页
1: 人参对照药材批号:
2:
3: 来源: 来源: 来源: 来源: 来源: 检验结果: ( )
人参皂苷 Rb1 对照品批号: 人参皂苷 Re 对照品批号: 人参皂苷 Rf 对照品批号: 人参皂苷 R gl 对照品批号:
【检查】 1.水分 标准规定本品水分不得过 12.0%(附录] X H 第一法) 。 仪器:FA2004 电子天平 1# 称量瓶烘 2 小时重量 称量瓶再烘 0.5 小时重量 样品+称量瓶重 样品+称量瓶烘 5 小时重量 样品+称量瓶再烘 1 小时重量 计算公式:P= P1= P= 相对偏差= P1- P2 ×100%= P1+ P2 检验结果: ( )
该批检验结论:
3
检验人:
复核人:
质量负责人:
共3页
对照品溶液的制备 精密称取人参皂苷 R gl 对照品、人参皂苷 Re 对照品及人参皂苷 Rb1 对照品,加甲 醇制成每 lm l 各含 0 . 2mg 的混合溶液,摇勾,即得。 供试品溶液的制备 取本品粉末(过四号筛)约 g,精密称定,置索氏提取器中,加三氯甲垸加热回 流 3 小时, 弃去三氯甲烷液, 药渣挥干溶剂, 连同滤纸筒移入 100ml 锥形瓶中, 精密加水饱和正丁醇 50ml, 密塞,放置过夜,超声处理(功率 250W,频率 50kHz)30 分钟,滤过,弃去初滤液,精密量取续滤液 25ml, 置蒸发皿中蒸干,残渣加甲醇溶解并转移至 5ml 量瓶中,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即 得。 测定法 分别精密吸取对照品溶液 10μ l 与供试品溶液 10〜20μ l,注人液相色谱仪,测定,即得。 本品按干燥品计算,含人参皂苷 Rg1, ( C4 2 H 7 2 C 1 4 )和人参皂苷 Re (C48 H8 2 C 1 8 )的总量不得少于 0.30%,人参皂苷 Rb1 (C5 4 H 9 2 C23 )不得少于 0 . 20% 。 检验结果: ( )
最新药材检验原始记录样本
XXXXX药业(饮片)有限公司1原药材检验报告单2检验单号:345XXXXX药业(饮片)有限公司6原药材检验记录7检验单号:8【性状】9101112结果:【鉴别】(1)显微鉴别1314横截面:15161718结果:粉末:192021结果:2223(2)薄层鉴别24供试品溶液的制备:取粉末1g,加乙醇15ml,加热回流1小时,放冷,滤过,滤液蒸干,残渣加乙醇5ml使溶解。
2526对照药材、对照品溶液配制:取菊花对照药材1g,同法制成对照药材溶液。
再取绿27原酸对照品,加乙醇制成每1ml含O.5mg的溶液。
28温度:(℃)29相对湿度:(%)30展开剂:三氯甲烷-丙酮-甲醇-5%浓氨试液31(6:1:1:0.1)32薄层板:硅胶G33显色剂:稀碘化铋钾试液34灯光:白光、紫外光灯(365nm)35展距:(cm)36供试品色谱中,在与对照药材色谱相对应的37位置上,显相同颜色的荧光斑点。
38S1为对照药材(对照品为中检所提供编号39为)40S2为对照品(对照品为中检所提供编号41为)42T为样品结果:434445【检查】杂质不得过 XX % (附录IX A)杂质称重: g4647杂质计算结果为: % (标准规定不得过 XX %)48结果:膨胀度应不低于4.0(附录IX O)4950温度:(℃)相对湿度:(%)51电子天平型号:CP214 溶剂:水52样品编号 1# 2# 3#53干燥品称重: g g g54第一次样品膨胀后体积: ml ml ml55第二次样品膨胀后体积: ml ml ml56(两次差异不超过0.1ml)57膨胀度计算结果为:(标准规定不低于4.0)58结果:59水分不得过12.0% (附录Ⅸ H 第一法)。
6061温度:(℃)相对湿度:(%)62烘箱型号:DHG-91012SA型电子天平型号:CP214样品编号 1# 2#6364第一次称量瓶干燥(105℃ 3h) (g)(g)第二次称量瓶恒重(105℃ 1h) (g)(g)6566样品称重(g)(g)67第一次称量瓶+样品干燥(105℃ 5h) (g)(g)68第二次称量瓶+样品恒重(105℃ 1h) (g)(g)69水分计算结果为:(%)(标准规定不得过12.0%)7071结果:7273总灰分不得过4.0%(附录Ⅸ K)74温度:(℃)相对湿度:(%)75马福炉型号:SX2.5-10 电子天平型号:CP21476样品编号 1# 2#77第一次坩锅称重(600℃ 3h) (g)(g)78第二次坩锅恒重(600℃ 0.5h) (g)(g)79样品称重(g)(g)80第一次坩锅+残渣称重(600℃ 3h) (g)(g)81第二次坩锅+残渣恒重(600℃ 0.5h) (g)(g)82总灰分计算结果为:(%)(标准规定不得过4.0%)83结果:8485酸不溶性灰分不得过3.0%(附录Ⅸ K)。
最新版大黄原始检验记录

大黄检验记录文件编号:第1页共10页名称大黄规格批号包装生产单位供应单位检验项目检验依据检验仪器【概述】大黄为蓼科植物掌叶大黄、唐古特大黄或药用大黄的干燥根及根茎。
秋末茎叶枯萎或次春发芽前采挖,除去细根,刮去外皮,切瓣或段,绳穿成串干燥或直接干燥。
【外观质量】本批项目结论:【性状】项目结论:【鉴别】(1) 本品横切面:根木栓层及皮层大多已除去。
韧皮部(筛管群)明显;(薄壁)组织发达。
形成层(成环),木质部射线(较密),宽(2~4)列细胞,内含(棕色物);导管(非木化),常1至数个相聚,稀疏排列。
薄壁细胞含(草酸钙簇晶),并含多数(淀粉粒)。
根茎髓部(宽广),其中常见(黏液腔),内有(红棕色物)。
异型维管束(散在),形成层(成环),木质部位于形成层(外方),韧皮部位于形成层(内方),射线呈(星状)射出。
检验者:年月日复核者:年月日粉末(黄棕色)。
草酸钙簇晶直径20~160μm,有的至190μm。
具缘纹孔、网纹、螺纹及环纹导管(非)木化。
淀粉粒甚多,单粒类球形或多角形,直径3~4.5μm,脐点(星)状,复粒由(2~8)分粒组成。
(附图1)项目结论:(2) 取本品粉末少量,进行微量升华,(应见菱状针晶或羽状结晶)。
项目结论:(3)取本品粉末0.1g,加甲醇20ml浸渍1小时,滤过,取滤液5ml,蒸干,残渣加水10ml使溶解,再加盐酸1ml,加热回流30分钟,立即冷却,用乙醚分两次提取,每次20ml,合并乙醚液,蒸干,残渣加三氯甲烷1ml 溶解,作为供试品溶液。
另取大黄对照药材0.1g(中检所,批号:),同法制成对照药材溶液(配制批号:)。
再取大黄酸对照品(中检所,批号:)g,加甲醇制成每1ml含1mg的溶液,作为对照溶液(配制批号:)。
照薄层色谱法(通则0502)试验,吸取上述三种溶液各4μl,分别点于同一以羧甲基纤维素钠为黏合剂的硅胶H薄层板上,以石油醚(30~60℃)-甲酸乙酯-甲酸(15:5:1)的上层溶液为展开剂,展开,取出,晾干,置紫外光灯(365nm)下检视。
药材检验原始记录样本

药材检验原始记录样本
检验日期:XXXX年XX月XX日
检验单位:XXXX药材检验中心
被检药材名称:XXXX草
产地:XXXX省XXX市
取样地点:XXXX县农贸市场
取样人员:XXX
取样时间:XXXX年XX月XX日
检验项目及方法:
项目1:外观质量检验,按《药材鉴定》(国家药典委员会药材分会
制定)进行
项目2:水分含量检验,按《药材鉴定》(国家药典委员会药材分会
制定)进行
项目3:含量测定,按《药材鉴定》(国家药典委员会药材分会制定)进行
检验结果及评价:
外观质量检验结果:外观形态:茎粗壮,质硬;表面颜色为浅黄色,
有少许褐斑,无明显异味。
根和叶均具有特征的气味。
评价:符合《药材鉴定》要求。
水分含量检验结果:水分含量为XX.XX%(空气干燥法测定)。
评价:符合《药材鉴定》要求。
含量测定结果:
有效成分1:XX.XX%
有效成分2:XX.XX%
评价:有效成分含量达到或超过国家标准要求,品质良好。
总结:
本次药材检验结果显示,被检药材外观质量良好,符合药材鉴定要求。
水分含量和有效成分含量也均符合国家标准要求,品质良好。
建议继续采
样检验,并严格按照标准操作规程进行检测,以确保药材的质量。
检验人员签名:______。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
XXXXX药业(饮片)有限公司原药材检验报告单检验单号:XXXXX药业(饮片)有限公司原药材检验记录检验单号:【性状】结果:【鉴别】(1)显微鉴别横截面:结果:粉末:结果:(2)薄层鉴别供试品溶液的制备:取粉末1g,加乙醇15ml,加热回流1小时,放冷,滤过,滤液蒸干,残渣加乙醇5ml使溶解。
对照药材、对照品溶液配制:取菊花对照药材1g,同法制成对照药材溶液。
再取绿原酸对照品,加乙醇制成每1ml含O.5mg的溶液。
温度:(℃)相对湿度:(%)展开剂:三氯甲烷-丙酮-甲醇-5%浓氨试液(6:1:1:0.1)薄层板:硅胶G显色剂:稀碘化铋钾试液灯光:白光、紫外光灯(365nm)展距:(cm)供试品色谱中,在与对照药材色谱相对应的位置上,显相同颜色的荧光斑点。
S1为对照药材(对照品为中检所提供编号为)S2为对照品(对照品为中检所提供编号为)T为样品结果:【检查】杂质不得过XX % (附录IX A)杂质称重: g杂质计算结果为:% (标准规定不得过XX %)结果:膨胀度应不低于4.0(附录IX O)温度:(℃)相对湿度:(%)电子天平型号:CP214 溶剂:水样品编号1# 2# 3#干燥品称重:g g g第一次样品膨胀后体积:ml ml ml第二次样品膨胀后体积:ml ml ml(两次差异不超过0.1ml)膨胀度计算结果为:(标准规定不低于4.0)结果:水分不得过12.0% (附录ⅨH 第一法)。
温度:(℃)相对湿度:(%)烘箱型号:DHG-91012SA型电子天平型号:CP214样品编号1# 2#第一次称量瓶干燥(105℃3h) (g)(g)第二次称量瓶恒重(105℃1h) (g)(g)样品称重(g)(g)第一次称量瓶+样品干燥(105℃5h) (g)(g)第二次称量瓶+样品恒重(105℃1h) (g)(g)水分计算结果为:(%)(标准规定不得过12.0%)结果:总灰分不得过4.0%(附录ⅨK)温度:(℃)相对湿度:(%)马福炉型号:SX2.5-10 电子天平型号:CP214样品编号1# 2#第一次坩锅称重(600℃3h) (g)(g)第二次坩锅恒重(600℃0.5h) (g)(g)样品称重(g)(g)第一次坩锅+残渣称重(600℃3h) (g)(g)第二次坩锅+残渣恒重(600℃0.5h) (g)(g)总灰分计算结果为:(%)(标准规定不得过4.0%)结果:酸不溶性灰分不得过3.0%(附录ⅨK)。
温度:(℃)相对湿度:(%)马福炉型号SX2.5-10电子天平型号CP214样品编号1# 2#第一次坩埚+滤渣称重(600℃3h) (g)(g)第二次坩锅+滤渣称重(600℃0.5h) (g)(g)酸不溶性灰分计算结果为(标准规定不得过3.0%)结果:【浸出物】醇溶性浸出物测定法(附录X A)项下的热浸法取供试品g,加乙醇100ml,静置1小时,回流1小时,精密滤取25ml,105℃烘3小时,置干燥器中冷却30分钟,迅速精密称重。
温度:(℃)相对湿度:(%)烘箱型号:DHG-91012SA型电子天平型号:CP214样品编号1# 2#第一次蒸发皿称重(105℃3h) (g)(g)第二次蒸发皿恒重(105℃1h) (g)(g)样品称重(g)(g)蒸发皿+浸出物称重(105℃3h) (g)(g)本品浸出物计算公式:浸出物(%)=[(m2-m0)×100] / (25×m1)×100%式中:m0——蒸发皿重量(g);m1——样品重(g);m2——干燥后蒸发皿+浸出物重(g)。
1#浸出物=(%)2#浸出物=(%)浸出物平均值为:(%)(标准规定不得少于5.0%)结果:【含量测定】挥发油测定法(附录ⅩD)称取样品:(g)加水:ml加热时间:保持微沸5小时,至测定器中挥发油不再增加。
收集挥发油:ml挥发油含量:%(ml/g)挥发油含量=收集挥发油/称样量×100%(标准规定:挥发油不得少于1.0%(ml/g))结果:【含量测定】高效液相色谱法(附录ⅥD)1 仪器与测定条件天平室温度:(℃)天平室相对湿度:(%)仪器室温度:(℃)仪器室相对湿度:(%)仪器型号:岛津LC-10Avp高效液相色谱仪电子天平型号:CP214 超声仪型号:KQ3200E所用试剂:XXXXX色谱柱填充剂:十八烷基硅烷键合硅胶色谱柱长度:250 (mm)柱温:常温(℃)检测器:紫外检测器检测器波长:XCX(nm)流动相:XXXX流速: 1.0 (ml/min)2 溶液的制备2.1对照品溶液的制备取粉防己碱、防己诺林碱对照品精密称定g、g,加甲醇醇使溶解并稀释至刻度,摇匀,即得。
2.2供试品溶液的制备取供试品(过三号筛)约0.5g,精密称定,精密加入2%盐酸-甲醇溶液25ml,称定重量,加热回流30分钟,放冷,再称定重量,用2%盐酸-甲醇溶液补足减失的重量,摇匀,滤过,精密量取续滤液5ml,置10 ml量瓶中,加流动相至刻度,摇匀,即得。
3 测定方法精密吸取对照品溶液与供试品溶液各μl,注入液相色谱仪,测定,即得。
4 结果与计算4.1 系统适用性试验重复性:取对照品溶液连续进样针,主峰面积的RSD为%(标准规定应不大于2.0%);理论板数:(标准规定应不低于4000 );分离度:见附页图谱,(标准规定应不低于1.5),。
4.2 响应因子计算对照品名称:粉防己碱、防己诺林碱对照品来源:中检所对照品批号:干燥条件:对照品称重W R:(g)对照品稀释过程:对照品纯度:(%)对照品浓度C R:(mg/ml)进样量:(μl)对照品峰面积A R :平均峰面积A:R相对标准偏差RSD:(%)响应因子 F =计算公式:F=(A/ 进样量(μl))/ C RR4.3 样品测定实验编号1# 2#称样量Wx:(g)(g)稀释倍数f:样品进样量:(μl)(μl)样品峰面积A X:平均峰面积Ax:含量:(mg /g)(mg /g)含量平均值:(mg /g)相对标准偏差RSD:(%)计算公式:含量(mg /g)=(Ax/进样量)/F×f×1/Wx标准规定:本品按干燥品计算,含粉防己碱(C38H42N2O6)和防己诺林碱(C37H40N2O6)的总量不得少于1.6% 。
结果:【含量测定】枸杞多糖紫外-可见分光光度法(附录V A)1 仪器与测定条件天平室温度:(℃)天平室相对湿度:(%)仪器室温度:(℃)仪器室相对湿度:(%)仪器型号:紫外可见分光光度计WFZUV-3802电子天平型号:CP2142 溶液的制备2.1 对照品溶液的制备取无水葡萄糖对照品25mg,精密称定,置250ml量瓶中,加水适量溶解,稀释至刻度,摇匀,即得(每1mI中含无水葡萄糖0.1mg)。
2.2 标准曲线的制备精密量取对照品溶液0.2ml、0.4ml、0.6ml、0.8ml、1.0ml,分别置具塞试管中,分别加水补至2.0ml,各精密加入5%苯酚溶液1m1,摇匀,迅速精密加入硫酸5ml,摇匀,放置10分钟,置40℃水浴中保温15分钟,取出,迅速冷却至室温,以相应的试剂为空白,照紫外-可见分光光度法(附录V A),在490nm的波长处测定吸光度,以吸光度为纵坐标,浓度为横坐标,绘制标准曲线。
2.3 供试品溶液的制备取本品粗粉约0.5g,精密称定,加乙醚100ml。
加热回流1小时,静置,放冷,小心弃去乙醚液,残渣置水浴上挥尽乙醚。
加入80%乙醇100ml,加热回流l小时.趁热滤过,滤渣与滤器用热80%乙醇30ml分次洗涤,滤渣连同滤纸置烧瓶中,加水150ml,加热回流2小时。
趁热滤过,用少量热水洗涤滤器,合并滤液与洗液,放冷,移至250ml 量瓶中,用水稀释至刻度,摇匀,精密量取1ml,置具塞试管中,加水1.0ml。
3 测定法照标准曲线的制备项下的方法,自“各精密加入5%苯酚溶液1m1”起,依法测定吸光,从标准曲线上读出供试品溶液中含葡萄糖的重量(mg),计算,即得。
标准规定:本品按干燥品计算,含枸杞多糖以葡萄糖(C6H12O6)计,不得少于1.8%。
结果:【含量测定】甜菜碱薄层色谱法(附录ⅥB薄层色谱扫描法)1 仪器与测定条件天平室温度:(℃)天平室相对湿度:(%)仪器室温度:(℃)仪器室相对湿度:(%)仪器型号:电子天平型号:CP214展开剂:丙酮-无水乙醇-盐酸(10:6:1)薄层板:硅胶G显色剂:新配制的改良碘化铋钾试液,放置1~3小时。
2.1 对照品溶液的制备供试品溶液的制备:取本品剪碎,取约2g,精密称定,加80%甲醇50ml,加热回流1小时,放冷,滤过,用80%甲醇:30m1分次洗涤残渣和滤器,合并洗液与滤液,浓缩至1Oml,用盐酸调节pH值至1,加入活性炭1g,加热煮沸,放冷,滤过,用水15m1分次洗涤,合并洗液与滤液,加入新配制的2.5%硫氰酸铬铵溶液20ml,搅匀,10℃以下放置3小时。
用G4垂熔漏斗滤过,沉淀用少量冰水洗涤,抽干,残渣加丙酮溶解,转移至5ml量瓶中,加丙酮至刻度,摇匀。
2.2 供试品溶液的制备对照品溶液配制:取甜菜碱对照品适量,精密称定,加盐酸甲醇溶液(O.5→100)制成每1m1含4mg的溶液。
3 测定法照薄层色谱法(附录ⅥB薄层色谱扫描法)进行扫描,波长:入s=515nm,入R=590nm,测量供试品吸光度积分值与对照品吸光度积分值,计算,即得。
标准规定:本品按干燥品计算,含甜菜碱(C5H11NO2)不得少于0.30%。
结果:【含量测定】化学滴定法1 仪器与测定条件天平室温度:(℃)天平室相对湿度:(%)电子天平型号:CP2142 测定方法取本品细粉约1g,精密称定,精密加入水1OOml,室温下浸泡4小时,时时振摇,滤过。
精密量取续滤液25ml,加水50ml,加酚酞指示液2滴,用氢氧化钠滴定液(0.1mol /L) 滴定,即得。
每1ml氢氧化钠滴定液(0.1mol/L)相当于6.404mg的枸橼酸(C6H807)。
3 结果与计算氢氧化钠滴定液的浓度:(mol/L)样品编号1# 2#样品称重:(g)(g)消耗氢氧化钠滴定液:(ml)(ml)有机酸含量:(g)(g)百分含量:(%)(%)百分含量平均值:(%)相对标准偏差RSD:(%)计算公式:百分含量(%)=(m2×100)/ (25×m1)×100%式中:m1——样品重(g);m2——有机酸含量(g)。
标准规定:本品按干燥品计算,含有机酸以枸橼酸(C6H807)计,不得少于5.0%。
结果:结论:本品按《中华人民共和国药典》2010版一部标准检验上述项目。
结果:。