青霉素的结构改造.
青霉素的研究发展

青霉素的研究发展一、青霉素的发展1、青霉素的发现青霉素是人类发现的第一种毒性很小又能有效杀菌的抗生素,从其发现到量产经历了14年。
1928年,英国人亚历山大·弗莱明意外地发现了一种能够“溶解”葡萄球菌的霉菌,他把这种霉菌命名为青霉素。
1939年,他将历时10年培养的菌种提供给牛津大学澳大利亚病理学家弗洛里和英国生物化学家钱恩。
1940年,他们完成了制备青霉素结晶体和动物实验。
辉瑞公司第一个盯上青霉素的人叫约翰·史密斯,他1906年加入辉瑞实验室,一直致力于把辉瑞从化学品提供商转型为主要的以研究为基础的制药企业。
1914年,他曾经一度离开辉瑞,加入施贵宝公司负责研发,1919年回到辉瑞。
1930年后,他了解到弗莱明对青霉素的早期研究之后,对其疗效做了进一步的调查。
1941年,第二次世界大战爆发,史密斯接受了美国政府下达的艰巨任务:大规模量产青霉素,以供战时之需。
辉瑞采用其特有的深罐发酵技术完成了任务(由约翰·麦基具体领导),并同时成为世界上首个生产青霉素的公司。
1945年,辉瑞生产的青霉素已经占到全球产量的一半(我国从1953年开始生产青霉素,从当时看,也是紧跟世界的脚步了,到2001年,我国生产的青霉素也超过了全球产量一半,可是辉瑞已经准备关闭其抗生素工厂了),无数在战时负伤感染的人得到拯救。
2.1、青霉素的发展自1940年青霉素投入使用以来,该类抗生素以其疗效确切、对人体细胞毒性小且价格低廉而广泛应用,临床首选于G+球菌所致的感染。
目前,青霉素类抗生素已从抗阳性窄谱品种发展到广谱的品种,按其抗菌作用可分为:①主要抗G+菌的窄谱青霉素,如天然青霉素G、青霉素V,耐青霉素酶的半合成青霉素甲氧西林、氯唑西林、氟氯西林。
②主要作用于G-菌的窄谱青霉素,如美西林、替莫西林。
③抗一般G-杆菌的普青霉素,如氨苄西林、阿莫西林、仓氨西林。
④抗绿脓杆菌的广谱青霉素,如羧苄西林、替卡西林、哌拉西林、阿洛西林、阿扑西林等。
药物化学问答题1

1.药物化学的研究内容和任务包括哪些?答:药物化学的内容是研究药物,包括发现药物①发现药物②合成化学药物③阐明药物的化学④.研究药物分子与机体之间的相互作用规律主要任务:为药物的使用提供化学基础,研究药物生产的工艺,创制新药,发现和发展新药2.药物的杂质指的是哪些物质?答:杂质是指生产、贮存过程中引进或产生的药物以外的其他化学物质。
3.新药的研发过程?答:新药是指未曾在中国境内上市销售的药品,新药的研发过程:①临床前研究包括药物化学、工艺路线、理化性质、质量标准、稳定性、毒性、药效、药动学关系的研究②.临床研究包括123期临床试验,观察人对新药的耐受性程度和药物代谢动力学,为制定给药方案提供依据,对安全性做出评价③.售后调研,上市后的监测,在广泛使用条件下考察疗效和不良反应。
4.简述局麻药的构效关系。
答:局部麻醉药的化学结构可概括为三部分:亲脂部分、中问连接链和亲水部分。
亲脂部分为取代的苯环或芳杂环,以苯环作用较强。
(初级药士药物化学辅导精华)中间连接链与局部麻醉药作用持效时间及强度有关。
其麻醉作用时间顺序为-CH2->-NH->-S->-O-,麻醉作用强度顺序为-S->-O->-CH2->-NH-。
亲水部分一般为叔胺,医学教育网|收集整理烷基以3~4个碳原子时作用最强。
局部麻醉药结构中的亲水部分和亲脂部分必须有适当的平衡,即有合适的脂水分配系数,才有利于发挥其麻醉活性。
一般来说,具有较高的脂溶性和较低的pKa的局麻药通常具有较快的麻醉作用和较低的毒性。
5.分析盐酸普鲁卡因的结构,讨论化学性质和其结构关系。
答:盐酸普鲁卡因的化学式是:结构式是:H2NONO. HCl化学名:4-氨基苯甲酸-2-(二乙氨基)乙酯盐酸盐,本品为白色结晶或结晶性粉末,无臭,味微苦,随后有麻痹感,易溶于水,略溶于乙醇,微容易三氯甲烷,几乎不容于乙醚,在空气中稳定,但对光敏感应避光保存。
试述半合成青霉素的结构改造方法

试述半合成青霉素的结构改造方法一、前言半合成青霉素是一种抗生素,广泛应用于医药领域。
为了提高其药效和稳定性,需要对其进行结构改造。
本文将详细介绍半合成青霉素的结构改造方法。
二、半合成青霉素的结构半合成青霉素的分子结构由苯甲酰基、侧链、吡啶环和β-内酰胺环组成。
其中,苯甲酰基和侧链决定了其抗菌活性,吡啶环和β-内酰胺环则是其核心结构。
三、半合成青霉素的结构改造方法1. 苯甲酰基的改造苯甲酰基是半合成青霉素分子中最容易被替换的部分。
常见的替换基团有氨基、羟基等。
将苯甲酰基替换为氨基后得到氨苄青霉素,其抗菌活性比原来的半合成青霉素更强。
2. 侧链的改造侧链也是影响半合成青霉素抗菌活性的重要因素。
常见的改造方法包括延长侧链、改变侧链的位置等。
将侧链延长为2-羟乙基丙酸基后得到氨苄西林,其抗菌活性比氨苄青霉素更强。
3. 吡啶环的改造吡啶环是半合成青霉素分子中不可替代的部分,因此对其进行改造相对困难。
但是,通过在吡啶环上引入新的基团可以提高半合成青霉素的药效和稳定性。
在吡啶环上引入双氢吡啶基后得到噻唑西林,其抗菌活性比半合成青霉素更强。
4. β-内酰胺环的改造β-内酰胺环也是半合成青霉素分子中不可替代的部分。
但是,在β-内酰胺环上引入新的基团可以提高其稳定性和抗菌活性。
在β-内酰胺环上引入硫代甲基后得到甲硫唑林,其抗菌活性比半合成青霉素更强。
四、总结通过对半合成青霉素结构进行改造,可以提高其药效和稳定性。
常见的改造方法包括替换苯甲酰基、延长侧链、在吡啶环上引入新的基团和在β-内酰胺环上引入新的基团等。
这些改造方法为半合成青霉素的应用提供了更多可能性。
青霉素结构的探究

青霉素结构的探究摘要青霉素是人类抗菌历史上最伟大的产物。
在极其简陋的实验条件下,正是由于科学家不懈地探索,青霉素神秘的结构才逐渐展现在人类面前。
现在广泛用于临床上的β-内酰胺抗生素,大都是在青霉素原有结构基础上修饰改造而来。
关键词青霉素立体构型结构改造青霉素(Penicillin),音译名盘尼西林,人类历史上最负盛名的抗生素,它的研制成功大大增强了人类抵抗细菌感染的能力,带动了抗生素家族的诞生。
由于分子中含有4个原子构成的β-内酰胺结构(图1),故统称为β-内酰胺抗生素。
青霉素分子由氢化噻唑环与β-内酰胺环并和而成,二者构成青霉素分子的母核,在母核上分别连有羧基和酰氨基侧链。
β-内酰胺环为一个平面结构,但2个稠和环不共平面。
青霉素分子中含有3个手性碳原子,只有3个碳原子绝对构型为2S,5R,6R的具有抗菌活性。
从青霉菌培养液中得到6种天然青霉素,现已证实为侧链不同的青霉素(见表1)。
其中以青霉素G的含量最高,效用最好,故在临床上广泛使用。
1 青霉素结构的探索对青霉素结构工作的探索是极其曲折的。
在那个设备粗糙、条件简陋的年代,科学家对青霉素研究的困难程度是现代科学家所无法想象的。
1.1 分子式的确定早期实验曾指出青霉素分子中不含S原子,这个错误的结论直到1943年7月才被纠正。
不同的青霉素水解都可以得到一种氨基酸——青霉胺,其分子式是C5H11NO2S,除此之外还有不同的青霉醛和二氧化碳。
从反应的产物可看出,青霉素分子中含有2个氮原子,4个氧原子和1个硫原子。
再后来研究发现2-戊烯基青霉素的钠盐分子式为C14H19N2O4SNa,苄基青霉素的钠盐分子式为C16H17N2O4SNa。
1.2 6种不同的青霉素化学家们在刚着手研究青霉素时就遇到了很大的困难,在自然界中不止存在一种天然的青霉素。
在英国,采用弗莱明发现青霉素时的表面培养法获得的青霉素与在美国采用玉米浸渍液培养出来的青霉素不一致,后来又陆续发现了另外一些共6种天然的青霉素(表1)。
01759药物化学(二)-简答题

01759药物化学(二)简答题1、先导化合物进行前药修饰的目的是什么?(1)增加脂溶性以提高吸收性能;(2)部位特异性;(3)增加药物的化学稳定性:(4)消除不适宜的制剂性质;(5)延长作用时间。
2、简述发现先导物的主要途径。
(1)由天然有效成分获得,包括植物、微生物和内源性活性物质;(2)反义核普酸;(3)基于生物大分子结构和作用机理设计;(4)组合化学;(5)基于生物转化发现。
3、利用前药原理对药物进行结构修饰,可以改变药物的哪些性质?(1)提高药物的组织选择性;(2)提高药物的稳定性;(3)延长药物作用时间;(4)改善药物的吸收;(5)改善药物的溶解度;(6)消除药物的不良味觉;(7)发挥药物的配伍作用。
4、前药的主要特征是什么?(1)原药与暂时转运基团以共价键连接,并且在体内可断裂,形成原药;(2)前药无活性或活性低于原药;(3)前药与暂时转运基团无毒性;(4)前药在体内产生原药的速率是快速的,以保障原药在作用部位有足够的药物浓度,并且应尽量减低前药的直接代谢。
5、叙述前药与软药设计的区别。
(1)前药是指用化学方法由有活性原药转变的无活性衍生物,后者在体内经酶或非酶解作用释放出原药而发挥疗效。
(2)软药系本身其有活性,在体内产生药理作用后可按预知方式和可控速率经进一步代谢转化成无活性产物的药物。
6、药物的第Ⅰ相生物转化的主要目的是什么?第II相生物转化的主要途径有哪几种?第Ⅰ相生物转化的主要目的是增加药物的极性,使之容易排泄。
第Ⅰ相生物转化有如下几种途径①葡萄糖醛酸结合;②硫酸结合;③氨基酸结合;④谷胱甘肽或疏基尿酸结合;⑤甲基化反应;⑥乙酰化反应。
7、简述吗啡及合成镇痛药的立体结构特征。
(1)分子中具有一个平坦的芳环结构,与受体的平坦区通过范德华力结合;(2)有一个叔氮原子的碱性中心,在生理pH条件下,大部分电离为阳离子正电中心,与受体表面的阴离子部位缔合;(3)联结它们两者之间的烃链部分突出于平面的前方,正好与受体的凹槽相适应。
青霉素类药物的结构改造.1

苯唑西林
Cl
O HH
N
S
Cl
H
N O
CH3 O
N
H COOH
双氯西林
N O
O HH
N H CH3 O
S N
H COOH
3.广谱青霉素
将青霉素6位侧链α-碳原子上引入亲水性基团,可扩大 抗菌谱,得到广谱抗生素
如:
氨苄西林
O
HO
HH
N NH2 H
O
S N
H COOH
阿莫西林
O HH
N NH2 H
O
二、青霉素类结构改造
1.耐酸青霉素
在青霉素6位侧链α碳上引入吸电子基团,阻碍了青霉素在酸 性条件下的电子重排,增加了对酸的稳定性。
如:
非奈西林
O
O
HH
N
SБайду номын сангаас
CH3 H N
O
H COOH
阿度西林
O
H N
H
S
N3 H N
O
H COOH
2.耐酶青霉素
酰胺侧链引入较大的取代基,具有较大的空间位阻,阻
止了β-内酰胺酶的进攻。
青霉素类药物的结构改造
一、青霉素类药物概述
• 1.青霉素结构特征:由β-内酰胺环、氢化噻唑环及
酰基侧链构成
酰胺侧链
O R
HH N H
N O
S COOH
6-氨基青霉烷酸 四氢噻唑环
β-内酰胺环
2. 天然青霉素存在的不足:
不耐酸,只能注射给药 易产生耐药性 抗菌谱窄,仅对革兰阳性菌有效 有严重的过敏反应
S N
H COOH
青霉素类药物的结构改造.

O NH2
H N H
O
H S
N H COOH
知识回顾 Knowledge Review
祝您成功!
二、青霉素类结构改造
1.耐酸青霉素
在青霉素6位侧链α碳上引入吸电子基团,阻碍了青霉素在酸 性条件下的电子重排,增加了对酸的稳定性。
如:
非奈西林
O O
CH3
H N H
O
H S
N H COOH
阿度西林
O
NH H S
N3 H
N
O
H COOH
2.耐酶青霉素
酰胺侧链引入较大的取代基,具有较大的空间位阻,阻
止了β-内酰胺酶的进攻。
如苯Leabharlann 西林ClO HHN
S
Cl
N O
H CH3 O
N H COOH
双氯西林
N O
O HH
N
S
H CH3 O
N H COOH
3.广谱青霉素
将青霉素6位侧链α-碳原子上引入亲水性基团,可扩大 抗菌谱,得到广谱抗生素
如:
氨苄西林
O
HO
HH
N NH2 H
O
S N
H COOH
阿莫西林
青霉素类药物的结构改造
一、青霉素类药物概述
• 1.青霉素结构特征:由β-内酰胺环、氢化噻唑环及
酰基侧链构成
酰胺侧链
O R
H N H
O
H S
N COOH
6-氨基青霉烷酸 四氢噻唑环
β-内酰胺环
2. 天然青霉素存在的不足:
不耐酸,只能注射给药 易产生耐药性 抗菌谱窄,仅对革兰阳性菌有效 有严重的过敏反应
中国药科大学2009,2010年《药物化学》试题以及答案
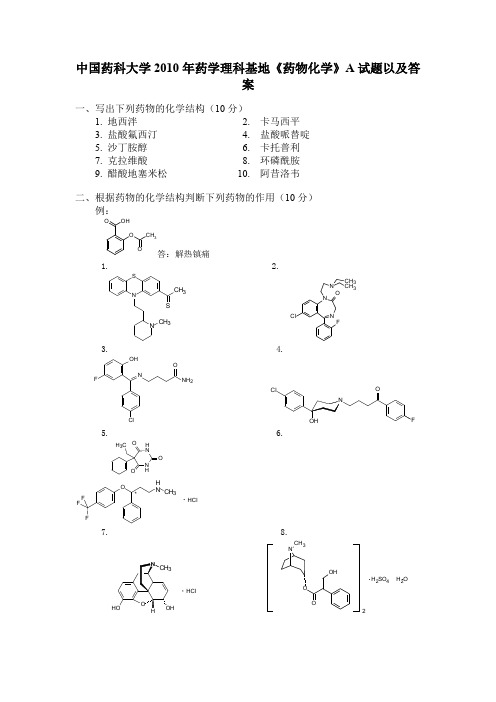
中国药科大学2010年药学理科基地《药物化学》A 试题以及答案一、写出下列药物的化学结构(10分)1. 地西泮2. 卡马西平3. 盐酸氟西汀4. 盐酸哌替啶5. 沙丁胺醇6. 卡托普利7. 克拉维酸8. 环磷酰胺9. 醋酸地塞米松 10. 阿昔洛韦二、根据药物的化学结构判断下列药物的作用(10分)例:答:解热镇痛1. 2.3. 4.5. 6.7. 8.OH OO CH 3O NS CH 3S N CH 3NN OClFN CH 3CH 3OHNClNH 2OFH NN HOOOH 3C OHN CH 3F FF HCl*HClH 2SO 4 H2OCH 329. . 10.11. 12.13. 14.15. 16.17. 18.19. 20.21 22. .3H 332Cl5H 2OH 2N ClF 3CNHOHCH 3CH 3CH 3.N CH 3ClCH 3NOOOH OH HCl 1 H 2O12N CH32HClOOOHClNNCH 3MeO MeOOSNH N H CH 3N CH 3CH 3NO2CH 3H N O CH 3N CH 3N CH 3H 3C CH 3ON H CH 3CH 3OH 3COOHNHNSCH 3CH 3OOMe MeO3O 33·H Cl23. 24.25. 26.27. 28.29. 30.31. 32.33. 34ON CH 3S N HO O NH 2H 3CNN N H OOO CH 2CH 3HNS ON H OCH 3COOH2OOH NOH 3CNO OOH H NO OH 3C N NO O OHNOOHNNN CH 3CH 3CH33H 2H3H 3OOHO H 3C CH 3H 3CCH 3N OCH 3N N OOF OO33O N O CH 3Cl FN NCH 3FFFS H 2NO O35. 36.37.38. 38.39. 40三、填空(20分)1.奥沙西泮是-----------的代谢产物。
青霉素类抗生素

抗菌谱
• 抗菌谱广,抗菌作用强。对G+菌的作用与阿莫西林相 似,对G-菌作用强。 • 对金葡菌的抗菌活性为酰脲基青霉素中最强者,对肠 球菌的作用与氨苄西林相当,对肺炎球菌、化脓性链 球菌具有高度抗菌活性。 • 本品最突出的优点是抗铜绿假单胞菌的作用强,并可 抑制洋葱假单胞菌、嗜麦芽假单胞菌、和荧光假单胞 菌,其活性为同类青霉素中最强。 • 对肠杆菌科细菌较阿洛西林和美洛西林强,但不及氨 基苷类。对部分脆弱类杆菌的作用较阿洛西林和美洛 西林强。 • 与氨基苷类联用,对铜绿假单胞菌、沙雷菌、克雷伯 菌、其他肠杆菌属和葡萄球菌的敏感菌株有协同抗菌 作用。
分类
天然青霉素
半合成青霉素
不耐酸
耐酸青霉素类Βιβλιοθήκη 不耐酶易形成耐药性窄谱
耐酶青霉素类
广谱青霉素类
易过敏
代表药青霉素G
抗铜绿假单孢菌青霉素类
抗革兰氏阴性菌青霉素类
LOREM
02
天然青霉素类
天然青霉素
由青霉菌培养液提取获得,含有5种(X、 F、G、K、双氢F),其中以青霉素G性 质较稳定,作用最强,低毒价廉,可用 于治疗敏感菌所致的各种感染。
首选用于敏感的G+球菌、G-球菌、螺旋体所致的感染,
但须病人对青霉素不过敏
1、G+球菌
2、G+杆菌
草绿色链球菌、肺炎球菌、溶血性链球菌
3、G-球菌感染:脑膜炎球菌引起的流行性脑脊髓膜炎首 选, 不产酶淋球菌引起的淋病首选 4、钩端螺旋体感染:梅毒、钩端螺旋体感染首选
由于天然青霉素存在有抗菌谱窄、不耐胃 酸口服无效及不耐酶易被水解、抗菌谱窄、 过敏性反应大等缺点,因此,通过改变天 然青霉素G的侧链可获得耐酸、耐酶、广谱、 抗铜绿假单胞菌及主要作用于G-菌等等一 系列不同品种的半合成青霉素。 但半合成青霉素的抗菌活性均不及天然 青霉素G
药化大题

1.天然青霉素G有哪些缺点?试述半合成青霉素的结构改造方法。
答:天然青霉素G的缺点为对酸不稳定,不能口服,只能注射给药;抗菌谱比较狭窄,仅对革兰阳性菌的效果好;细菌易对其产生耐药性;有严重的过敏性反应。
在青霉素的侧链上引入吸电子基团,阻止侧链羰基电子向β一内酰胺环的转移,增加了对酸的稳定性,得到一系列耐酸青霉素。
在青霉素的侧链上引入较大体积的基团,阻止了化合物与酶活性中心的结合。
又由于空间阻碍限制酰胺侧链R与羧基间的单键旋转,从而降低了青霉素分子与酶活性中心作用的适应性,因此药物对酶的稳定性增加。
在青霉素的侧链上引入亲水性的基团(如氨基,羧基或磺酸基等),扩大了抗菌谱,不仅对革兰阳性菌有效,对多数革兰阴性菌也有效。
2.简述现代新药开发与研究的内容。
3.巴比妥类药物的一般合成方法中,用卤烃取代丙二酸二乙酯的氢时,当两个取代基大小不同时,一般应先引入大基团,还是小基团?为什么?答:当引入的两个烃基不同时,一般先引入较大的烃基到次甲基上。
经分馏纯化后,再引入小基团。
这是因为,当引入一个大基团后,因空间位阻较大,不易再接连上第二个基团,成为反应副产物。
同时当引入一个大基团后,原料、一取代产物和二取代副产物的理化性质差异较大,也便于分离纯化。
4.以captopril为例,简要说明ACEI类抗高血压药的作用机制及为克服captopril的缺点及对其进行结构改造的方法。
答:血管紧张素转化酶抑制剂(ACEI)类抗高血压药主要是通过抑制血管紧张素转化酶(ACE)的活性、,使血管紧张素I(AngI)不能转化为血管紧张素Ⅱ(AngⅡ),导致血浆中AngⅡ数量下降,无法发挥其收缩血管的作用及促进醛固酮分泌作用,ACEI还能抑制缓激肽的降解,上述这些作用结果均使血压下降。
卡托普利(Captopril)是根据ACE的结构设计出来的第一个上市的ACEI,为脯氨酸的衍生物,脯氨酸氮原子上连一个有甲基和巯基取代的丙酰基侧链,使Captopril具有良好的抗高血压作用,但用药后易产生皮疹、干咳、嗜酸性粒细胞增高、味觉丧失和蛋白尿的副作用.,味觉丧失可能与结构中的巯基有关,考虑到脯氨酸的吡咯环及环上的羧基阴离子对结合酶部位起到重要的作用,故在尽可能保留该部分结构特点的同时,用α一羧基苯丙胺代替巯基如依那普利(Enalapril),或用含次膦酸基的苯丁基代替巯基福辛普利(Fosinpril),再将羧基或次膦酸基成酯,则可得到一类长效的ACEI,上述不良反应也减少。
试述半合成青霉素的结构改造方法

半合成青霉素的结构改造方法引言半合成青霉素是一种重要的抗生素,由于其广谱抗菌活性和良好的耐受性,被广泛应用于临床医学领域。
然而,青霉素的天然产物含量较低,大规模生产困难,因此,寻找半合成方法来改造青霉素的结构以提高其产量成为一个重要的研究方向。
本文将详细探讨半合成青霉素的结构改造方法。
I. 青霉素的结构与合成方法回顾1. 青霉素的结构青霉素是一种含有β-内酰胺环的混合酸氨酯类抗生素,其结构由苯丙侧链、β-内酰胺环和五元内酯环组成。
2. 青霉素的天然合成路径青霉素的天然合成路径包括青霉素酸的生物合成和β-内酰胺环、五元内酯环的合成。
青霉素酸由L-α-酸乳酸经过环化反应形成β-内酰胺环,然后与半胱氨酸形成五元内酯环。
3. 青霉素的全合成方法青霉素的全合成方法较为复杂,包括多步反应和复杂条件,因此不适合用于大规模生产。
为了解决这一问题,研究人员提出了半合成方法来改造青霉素的结构以提高产量。
II. 基于半合成的青霉素结构改造方法1. 引入新的化学修饰基团通过在青霉素分子中引入新的化学修饰基团,可以改变其物理化学性质和抗菌活性。
例如,引入氯原子可以增强青霉素的抗菌活性。
这种方法需要合成新的衍生物,并通过活性筛选来评估其抗菌活性。
2. 修改侧链结构通过修改苯丙侧链结构,可以改变青霉素的溶解性、稳定性和生物利用度。
例如,引入疏水基团可以提高青霉素在脂肪组织中的分配,从而增强其抗菌活性。
3. 调节β-内酰胺环和五元内酯环的形成通过调节β-内酰胺环和五元内酯环的形成,可以改变青霉素的稳定性和抗菌活性。
例如,通过改变环化反应的反应条件和催化剂,可以调节β-内酰胺环的形成速度和稳定性。
4. 制备半合成前体物制备半合成前体物是半合成青霉素的关键步骤。
通过合成不同的前体物,可以改变青霉素的结构和物理化学性质。
例如,通过合成β-内酰胺环和五元内酯环的前体物,可以改变青霉素的稳定性和活性。
III. 半合成青霉素的应用与展望半合成青霉素在临床医学领域具有广阔的应用前景。
药物化学简答题题库

1].巴比妥类药物的一般合成方法中,用卤烃取代丙二酸二乙酯的氢时,当两个取代基大小不同时,应先引入大基团,还是小基团?为什么?当引入的两个烃基不同时,一般先引入较大的烃基到次甲基上。
经分馏纯化后,再引入小基团。
这是因为,当引入一个大基团后,因空间位阻较大,不易再接连上第二个基团,成为反应副产物。
同时当引入一个大基团后,原料、一取代产物和二取代副产物的理化性质差异较大,也便于分离纯化。
2].试说明异戊巴比妥的化学命名。
异戊巴比妥的化学命名采用芳杂环嘧啶作母体。
按照命名规则,应把最能表明结构性质的官能团酮基放在母体上。
为了表示酮基(=O)的结构,在环上碳2,4,6均应有连接两个键的位置,故采用添加氢(Added Hydrogen)的表示方法。
所谓添加氢,实际上是在原母核上增加一对氢(即减少一个双键),表示方法是在结构特征位置的邻位用带括号的H表示。
本例的结构特征为酮基,因有三个,即表示为2,4,6-(1H,3H,5H)嘧啶三酮。
2,4,6是三个酮基的位置,1,3,5是酮基的邻位。
该环的编号依杂环的编号,使杂原子最小,则第五位为两个取代基的位置,取代基从小排到大,故命名为5-乙基-5(3-甲基丁基)-2,4,6(1H,3H,5H)嘧啶三酮。
3].巴比妥药物具有哪些共同的化学性质?1)呈弱酸性,巴比妥类药物因能形成内酰亚氨醇一内酰胺互变异构,故呈弱酸性。
2)水解性,巴比妥类药物因含环酰脲结构,其钠盐水溶液,不够稳定,甚至在吸湿情况下,也能水解。
3)与银盐的反应,这类药物的碳酸钠的碱性溶液中与硝酸银溶液作用,先生成可溶性的一银盐,继而则生成不溶性的二银盐白色沉淀。
4)与铜吡啶试液的反应,这类药物分子中含有-CONHCONHCO-的结构,能与重金属形成不溶性的络合物,可供鉴别。
11) 为什么巴比妥C5次甲基上的两个氢原子必须全被取代才有疗效?其原因是:一般来说,未解离的巴比妥类药物分子较其离子易于透过细胞膜而发挥作用。
生产青霉素的利用原理

生产青霉素的利用原理青霉素是一种广谱抗生素,广泛应用于临床医疗和养殖业。
它能有效地杀死多种细菌,使其无法生存和繁殖,从而对抗感染疾病。
青霉素的生产利用了青霉属真菌(Penicillium)的特殊代谢能力,通过在适当的发酵条件下生产和提取青霉素。
青霉素的利用原理主要包括以下几个方面:1. 青霉属真菌的培养和筛选:青霉属真菌尤其是青霉菌(Penicillium chrysogenum)是生产青霉素的主要菌种,需要在适宜的培养基中进行培养。
培养基通常包含碳源、氮源、微量元素和适宜的pH。
通过培养基的优化,可以提高青霉素的产量和质量。
2. 青霉素的生物合成:青霉素的生物合成过程是一系列复杂的酶催化反应。
首先,真菌通过特殊的酶合成青霉素的骨架结构,即β-内酰胺环。
随后,通过非酶催化的氧化和还原反应,在骨架结构上引入各种功能基团,最终形成成熟的青霉素分子。
由于青霉属菌种的遗传多样性,不同的菌株在青霉素生物合成途径中可能存在差异。
3. 青霉素的产量调控:青霉素的产量受到多种因素的调控,包括培养基成分、培养条件、发酵过程等。
一般来说,生产青霉素的青霉属真菌会在培养基中营养不足时产生青霉素。
因此,通过调节培养基成分和培养条件,可以增加青霉素的产量。
同时,通过对菌株的基因工程改造,也可以提高青霉素的产量和稳定性。
4. 青霉素的提取和纯化:在青霉属真菌发酵过程结束后,需要对发酵液进行提取和纯化,以获取纯净的青霉素产品。
常用的提取方法包括萃取、溶剂萃取和离子交换等。
提取后,还需要通过过滤、浓缩、结晶和干燥等工艺步骤进行纯化。
纯净的青霉素产品可以作为药物或饲料添加剂使用。
总之,生产青霉素的利用原理是通过培养和筛选青霉属真菌,利用其特殊的生物合成能力合成青霉素,然后对发酵液进行提取和纯化,最终得到纯净的青霉素产品。
青霉素的生产过程需要严格控制培养条件和工艺参数,以及通过基因工程技术提高产量和品质。
青霉素的产业化生产对于保障人类和动物健康有着重要意义,也为抗生素的合理应用提供了重要支持。
青霉素类药物的缺点及其结构修饰原理及方法

青霉素类药物的缺点及其结构修饰原理及方法
缺点:青霉素的价格是比较便宜,但是疗效是不错的。
现在很多的新药都是从青霉素衍生而来。
现在医院里面使用的量较少,是因为很多头孢类药物的使用,导致了细菌对青霉素耐药或者疗效打了折扣。
所以医生使用青霉素的时候,如果效果一般,就会及时更换药物。
还是青霉素是可以进入大脑,治疗脑膜炎的一种,目前依然在使用的一种有效抗生素。
但是青霉素的缺点也是有很多的,例如最严重的过敏反应,轻者可以引起水肿、皮肤红等,重的话可以引起休克、或者是死亡的。
所以使用之前一定要皮试的,阳性者不能用。
然后就是容易使细菌产生耐药性,这是现在普遍存在的问题的了。
结构作用方法:青霉素之所以具有强大的抗菌作用是由于青霉素与细菌细胞壁可以发生作用。
黏肽是细菌细胞壁的主要成分,也是细菌细胞壁中最坚硬的一层。
它的存在可以维持细菌细胞的外形,保持其细胞壁的通透性。
青霉素的结构同黏肽的末端结构丙氨酰丙氨酸相似,其可以取代丙氨酰丙氨酸与酶的活性中心结合,从而使组成黏肽的多肽不能交联形成网状的黏肽,导致细菌细胞壁不能形成,从而使细菌被溶解而死亡。
而人类的细胞没有细胞壁,只有细胞膜,所以人类细胞受青霉素的影响很小。
青霉素的合成方法与工艺优化策略

青霉素的合成方法与工艺优化策略青霉素是一种广泛应用于临床治疗的抗生素,被誉为“抗生素之王”。
它是由青霉菌属真菌产生的一类天然产物,具有广谱的抗菌活性,对革兰阳性细菌尤为有效。
本文将探讨青霉素的合成方法以及工艺优化策略,以期为医学人员提供更多的科学依据和实践指导。
一、青霉素的合成方法青霉素的合成方法主要分为天然合成和半合成两种。
1. 天然合成:青霉素的天然合成是通过青霉菌属真菌自身的代谢途径合成的。
青霉素的合成过程包括青霉素酸的合成、侧链的合成以及酸酐的合成等。
其中,青霉素酸是青霉素的前体,通过一系列酶的作用,最终合成出青霉素。
2. 半合成:半合成是在天然合成的基础上,通过化学手段对青霉素的结构进行改造,以获得更多种类和更高效的青霉素类似物。
半合成青霉素的合成方法主要包括侧链改造、半合成酸酐和半合成青霉素的合成等。
二、青霉素的工艺优化策略青霉素的工艺优化策略主要包括改进合成方法、提高产量和纯度、减少污染物产生等方面。
1. 改进合成方法:通过改进合成方法,可以提高青霉素的产量和纯度,并减少副产物的生成。
例如,引入新的催化剂、优化反应条件、改变反应顺序等,可以提高合成效率和产物纯度。
2. 提高产量和纯度:青霉素的产量和纯度是评价合成工艺的重要指标。
通过优化培养条件、改进发酵工艺、提高菌株的发酵能力等手段,可以提高青霉素的产量和纯度。
3. 减少污染物产生:在青霉素的合成过程中,会产生一些副产物和污染物,对产品质量和纯度产生不利影响。
通过优化反应条件、改进分离纯化工艺、加强废水处理等措施,可以减少污染物的产生,提高产品质量。
4. 提高抗菌活性:除了改进合成方法和工艺优化,还可以通过改变青霉素的结构,提高其抗菌活性。
例如,通过半合成的方法,可以引入新的官能团或改变侧链结构,以增强青霉素的抗菌活性。
总结:青霉素的合成方法和工艺优化策略是医学领域的重要研究方向。
通过不断改进合成方法、提高产量和纯度、减少污染物产生等措施,可以获得更高效、更纯净的青霉素产品,为临床治疗提供更好的药物选择。
青霉素的分类与特点解析

青霉素的分类与特点解析青霉素是一类广泛应用于临床的抗生素,它具有独特的分类和特点。
本文将对青霉素的分类和特点进行深入解析,以帮助读者更好地了解和应用这一药物。
一、青霉素的分类青霉素是指由青霉菌属(Penicillium)产生的一类天然抗生素。
根据其化学结构和作用机制的不同,青霉素可以分为以下几类:1. 天然青霉素:天然青霉素是指从青霉菌属真菌中提取的原始抗生素,如青霉素G(Penicillin G)。
天然青霉素具有广谱抗菌活性,对革兰氏阳性菌和一些革兰氏阴性菌均有较好的杀菌作用。
2. 半合成青霉素:半合成青霉素是在天然青霉素的基础上进行改造,通过化学合成或半合成方法制得的抗生素。
半合成青霉素包括青霉素V(Penicillin V)和青霉素G的衍生物,如氨苄青霉素(Ampicillin)和苄西林(Benzylpenicillin)。
半合成青霉素具有更广泛的抗菌谱,对某些革兰氏阴性菌和耐青霉素酶产生菌有较好的疗效。
3. 后期半合成青霉素:后期半合成青霉素是在半合成青霉素的基础上进一步改进的抗生素,如青霉素类似物(Penicillinase-Resistant Penicillins)和氨基苷青霉素(Aminopenicillins)。
后期半合成青霉素具有更强的抗菌活性和更好的耐药性。
4. 抗假单胞菌青霉素:抗假单胞菌青霉素是专门针对假单胞菌属细菌的抗生素,如甲氧西林(Methicillin)和万古霉素(Vancomycin)。
抗假单胞菌青霉素通常用于治疗假单胞菌感染和耐药性较强的细菌感染。
二、青霉素的特点青霉素作为一类重要的抗生素,具有以下几个特点:1. 强效抗菌作用:青霉素对革兰氏阳性菌和一些革兰氏阴性菌具有广谱的杀菌作用。
它通过抑制细菌细胞壁的合成,破坏细菌细胞壁的完整性,导致细菌死亡。
2. 低毒性:青霉素在治疗剂量下对人体的毒副作用较小。
它主要通过抑制细菌细胞壁的合成来发挥抗菌作用,而人体细胞没有细菌细胞壁,因此对人体细胞的影响较小。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
H2N NH O H O H S H Nlin acylase
H O H N
OH
Penicillin G
6-APA
16
6-APA与相应的侧链酸缩合
17
谢
谢!
甲氧西林
OCH3
OCH3 O
NH
S H O
H O H
N OH
第一个用于临床的耐酶青霉素。
10
3.3 耐酶青霉素
在青霉素 6 位侧链酰胺基上引入较大空间位阻的基团,阻 止药物与酶的活性中心作用,保护药物分子中的β-内酰胺酶。
苯 基 ( 空 间 位 阻 ,吸 电 子 )
O N O 异噁唑基 甲基 H N O
3.4 广谱青霉素
在青霉素酰基α位引入极性亲水性基团-NH2、COOH、-SO3H等,发展了广谱的半合成青霉素。
阿莫西林
阿莫西林(Amoxicillin)
OH NH2
对羟基苯甘 氨酸
H
有一个手性C 临床用右旋体, 构型为R-构型
O
NH
S H O
H O H
N OH
广谱的半合成青霉素
14
4、半合成青霉素衍生物的化学合成方法
青霉素的结构改造
药物化学教研室: 田海英
1
1、青霉素结构特点和作用机制
6-氨基青霉烷酸 酰胺侧链
Acyl side chain
6-Aminapenicillanic Acind 6-APA
H H H N S O C N O H O ONa
β-内酰胺环
β-Lactam Ring
四氢噻唑环
Thiazolidine Ring
3.2 耐酸青霉素
青霉素V
设计合成了在酰胺基α位引入吸电子基团的化合物, 如非奈西林、丙匹西林和阿度西林,口服吸收良 好。
非奈西林
phenethillin
丙匹西林
propicillin
阿度西林
azidocillin
3.3 耐酶青霉素
最早发现三苯甲基青霉素可耐酶,由于三苯甲基的 空间位阻,阻止了化合物与酶活性中心的结合。
1、青霉素结构特点和作用机制
β-内酰胺环的作用:
四元环张力较大,其化学性质不稳定,易发生
开环导致失活; β-内酰胺环开环与细菌发生酰化作用,抑制细 菌的生长。
2、青霉素使用缺点及不良反应
对酸不稳定
只能注射给药,不能口服;
抗菌谱窄
对G+的活性比较高
产生耐药性 体内作用时间短 ,每天至少注射两次;肌 注疼痛。 对某些病人引起过敏反应,严重时会死亡
O
定性更好,抗菌谱更广,
耐酸、耐酶
6
3.1 延长作用时间的方法
与分子较大的胺制成难溶性盐,作用时间延长。– 普鲁卡因青霉素、苄星西林;
普鲁卡因青霉素 procaine benzylpenicillin
苄星青霉素 benzathine benzylpenicillin
羧基酯化,延长作用时间–醋甲西林。
以 Penicillin G 为原料,经青霉素酰化 酶(Penicillin acylase)进行酶解,生成 6- 氨基青霉烷酸 (6-APA) ,是半合成青 霉素的主要中间体。 得到6-APA后,再与相应的侧链酸进行 缩合
(1)酰氯法
(2)酸酐法
(3)DCC法
Penicillin G经酶解生成6-APA
H H S N . H2O COONa
苯唑西林:第一个耐酶、耐酸的青霉素,可口服、注射,引入苯
甲异噁唑环是重大进展。
3.4 广谱青霉素
从头孢霉菌发酵液中分离出的青霉素 N 对 G+ 菌作 用比青霉素弱,但是对G-菌作用强于青霉素; 其6位有D-α-氨基己二酸单酰胺侧链,侧链上的氨 基是产生对G-菌活性的重要基团。
4
3、半合成抗生素
通过结构改造
增加稳定性 降低毒副作用 扩大抗菌谱
减少耐药性
改善生物利用度 提高治疗效力
改变用药途径
5
天然青霉素:发酵制得 H H 4 H2N 半合成青霉素 S
以6-氨基青霉烷酸(6-
6
APA)为基本母核,引入
适当的侧链而获得的稳
C N 1 O H 2 OH