英文 原料药批生产记录 模板
中英文对照FDA原料药GMP指南

Q7a(中英文对照)FDA原料药GMP指南Table of Contents 目录1. INTRODUCTION 1. 简介1.1 Objective 1.1目的1.2 Regulatory Applicability 1.2法规的适用性1.3 Scope 1.3范围2. QUALITY MANAGEMENT 2.质量管理2.1 Principles 2.1总则2.2 Responsibilities of the Quality Unit(s) 2.2质量部门的责任2.3 Responsibility for Production Activities 2.3生产作业的职责2.4 Internal Audits (Self Inspection) 2.4内部审计(自检)2.5 Product Quality Review 2.5产品质量审核3. PERSONNEL 3. 人员3.1 Personnel Qualifications 3.人员的资质3.2 Personnel Hygiene 3.2 人员卫生3.3 Consultants 3.3 顾问4. BUILDINGS AND FACILITIES 4. 建筑和设施4.1 Design and Construction 4.1 设计和结构4.2 Utilities 4.2 公用设施4.3 Water 4.3 水4.4 Containment 4.4 限制4.5 Lighting 4.5 照明4.6 Sewage and Refuse 4.6 排污和垃圾4.7 Sanitation and Maintenance 4.7 卫生和保养5. PROCESS EQUIPMENT 5. 工艺设备5.1 Design and Construction 5.1 设计和结构5.2 Equipment Maintenance and Cleaning 5.2 设备保养和清洁5.3 Calibration 5.3 校验5.4 Computerized Systems 5.4 计算机控制系统6. DOCUMENTATION AND RECORDS 6. 文件和记录6.1 Documentation System andSpecifications6.1 文件系统和质量标准6.2 Equipment cleaning and Use Record 6.2 设备的清洁和使用记录6.3 Records of Raw Materials, Intermediates, API Labeling and Packaging Materials 6.3 原料、中间体、原料药的标签和包装材料的记录6.4 Master Production Instructions (MasterProduction and Control Records)6.4 生产工艺规程(主生产和控制记录)6.5 Batch Production Records (BatchProduction and Control Records)6.5 批生产记录(批生产和控制记录)6.6 Laboratory Control Records 6.6 实验室控制记录6.7 Batch Production Record Review 6.7批生产记录审核7. MATERIALS MANAGEMENT 7. 物料管理7.1 General Controls 7.1 控制通则7.2 Receipt and Quarantine 7.2接收和待验7.3 Sampling and Testing of IncomingProduction Materials7.3 进厂物料的取样与测试7.4 Storage 7.4储存7.5 Re-evaluation 7.5复验8. PRODUCTION AND IN-PROCESSCONTROLS8. 生产和过程控制8.1 Production Operations 8.1 生产操作8.2 Time Limits 8.2 时限8.3 In-process Sampling and Controls 8.3 工序取样和控制8.4 Blending Batches of Intermediates orAPIs8.4 中间体或原料药的混批8.5 Contamination Control 8.5 污染控制9. PACKAGING AND IDENTIFICATIONLABELING OF APIs ANDINTERMEDIATES9. 原料药和中间体的包装和贴签9.1 General 9.1 总则9.2 Packaging Materials 9.2 包装材料9.3 Label Issuance and Control 9.3 标签发放与控制9.4 Packaging and Labeling Operations 9.4 包装和贴签操作10. STORAGE AND DISTRIBUTION 10.储存和分发10.1 Warehousing Procedures 10.1 入库程序10.2 Distribution Procedures 10.2 分发程序11. LABORATORY CONTROLS 11.实验室控制11.1 General Controls 11.1 控制通则11.2 Testing of Intermediates and APIs 11.2 中间体和原料药的测试11.3 Validation of Analytical Procedures 11.3 分析方法的验证11.4 Certificates of Analysis 11.4 分析报告单11.5 Stability Monitoring of APIs 11.5 原料药的稳定性监测11.6 Expiry and Retest Dating 11.6 有效期和复验期11.7 Reserve/Retention Samples 11.7 留样12. V ALIDATION 12.验证12.1 Validation Policy 12.1 验证方针12.2 Validation Documentation 12.2 验证文件12.3 Qualification 12.3 确认12.4 Approaches to Process Validation 12.4 工艺验证的方法12.5 Process Validation Program 12.5 工艺验证的程序12.6 Periodic Review of Validated Systems 12.6验证系统的定期审核12.7 Cleaning Validation 12.7 清洗验证12.8 Validation of Analytical Methods 12.8 分析方法的验证13. CHANGE CONTROL 13.变更的控制14. REJECTION AND RE-USE OFMATERIALS14.拒收和物料的再利用14.1 Rejection 14.1 拒收14.2 Reprocessing 14.2 返工14.3 Reworking 14.3 重新加工14.4 Recovery of Materials and Solvents 14.4 物料与溶剂的回收14.5 Returns 14.5 退货15. COMPLAINTS AND RECALLS 15.投诉与召回16. CONTRACT MANUFACTURERS(INCLUDING LABORATORIES)16.协议生产商(包括实验室)17. AGENTS, BROKERS, TRADERS, DISTRIBUTORS, REPACKERS, AND RELABELLERS 17.代理商、经纪人、贸易商、经销商、重新包装者和重新贴签者17.1 Applicability 17.1适用性17.2 Traceability of Distributed APIs andIntermediates17.2已分发的原料药和中间体的可追溯性17.3 Quality Management 17.3质量管理17.4 Repackaging, Relabeling, and Holding of APIs and Intermediates 17.4原料药和中间体的重新包装、重新贴签和待检17.5 Stability 17.5稳定性17.6 Transfer of Information 17.6 信息的传达17.7 Handling of Complaints and Recalls 17.7 投诉和召回的处理17.8 Handling of Returns 17.8 退货的处理18. Specific Guidance for APIs Manufactured by Cell Culture/Fermentation 18. 用细胞繁殖/发酵生产的原料药的特殊指南18.1 General 18.1 总则18.2 Cell Bank Maintenance and RecordKeeping18.2细胞库的维护和记录的保存18.3 Cell Culture/Fermentation 18.3细胞繁殖/发酵18.4 Harvesting, Isolation and Purification 18.4收取、分离和精制18.5 Viral Removal/Inactivation steps 18.5 病毒的去除/灭活步骤19.APIs for Use in Clinical Trials 19.用于临床研究的原料药19.1 General 19.1 总则19.2 Quality 19.2 质量19.3 Equipment and Facilities 19.3 设备和设施19.4 Control of Raw Materials 19.4 原料的控制19.5 Production 19.5 生产19.6 Validation 19.6 验证19.7 Changes 19.7 变更19.8 Laboratory Controls 19.8 实验室控制19.9 Documentation 19.9 文件20. Glossary 20. 术语Q7a GMP Guidance for APIs Q7a原料药的GMP指南1. INTRODUCTION 1. 简介1.1 Objective 1.1目的This document is intended to provide guidance regarding good manufacturing practice (GMP) for the manufacturing of active pharmaceutical ingredients (APIs) under an appropriate system for managing quality. It is also intended to help ensure that APIs meet the quality and purity characteristics that they purport, or are represented, to possess. 本文件旨在为在合适的质量管理体系下制造活性药用成分(以下称原料药)提供有关优良药品生产管理规范(GMP)提供指南。
原料药GMP规范指南中英文对照.doc

Q7a(中英文对照)FDA原料药GMP指南Table of Contents 目录1. INTRODUCTION 1. 简介1.1 Objective 1.1目的1.2 Regulatory Applicability 1.2法规的适用性1.3 Scope 1.3范围2. QUALITY MANAGEMENT 2.质量管理2.1 Principles 2.1总则2.2 Responsibilities of the Quality Unit(s) 2.2质量部门的责任2.3 Responsibility for Production Activities 2.3生产作业的职责2.4 Internal Audits (Self Inspection) 2.4内部审计(自检)2.5 Product Quality Review 2.5产品质量审核3. PERSONNEL 3. 人员3.1 Personnel Qualifications 3.人员的资质3.2 Personnel Hygiene 3.2 人员卫生3.3 Consultants 3.3 顾问4. BUILDINGS AND FACILITIES 4. 建筑和设施4.1 Design and Construction 4.1 设计和结构4.2 Utilities 4.2 公用设施4.3 Water 4.3 水4.4 Containment 4.4 限制4.5 Lighting 4.5 照明4.6 Sewage and Refuse 4.6 排污和垃圾4.7 Sanitation and Maintenance 4.7 卫生和保养5. PROCESS EQUIPMENT 5. 工艺设备5.1 Design and Construction 5.1 设计和结构5.2 Equipment Maintenance and Cleaning 5.2 设备保养和清洁5.3 Calibration 5.3 校验5.4 Computerized Systems 5.4 计算机控制系统6. DOCUMENTATION AND RECORDS 6. 文件和记录6.1 Documentation System andSpecifications6.1 文件系统和质量标准6.2 Equipment cleaning and Use Record 6.2 设备的清洁和使用记录6.3 Records of Raw Materials, Intermediates, API Labeling and Packaging Materials 6.3 原料、中间体、原料药的标签和包装材料的记录6.4 Master Production Instructions (MasterProduction and Control Records)6.4 生产工艺规程(主生产和控制记录)6.5 Batch Production Records (BatchProduction and Control Records)6.5 批生产记录(批生产和控制记录)6.6 Laboratory Control Records 6.6 实验室控制记录6.7 Batch Production Record Review 6.7批生产记录审核7. MATERIALS MANAGEMENT 7. 物料管理7.1 General Controls 7.1 控制通则7.2 Receipt and Quarantine 7.2接收和待验7.3 Sampling and Testing of IncomingProduction Materials7.3 进厂物料的取样与测试7.4 Storage 7.4储存7.5 Re-evaluation 7.5复验8. PRODUCTION AND IN-PROCESSCONTROLS8. 生产和过程控制8.1 Production Operations 8.1 生产操作8.2 Time Limits 8.2 时限8.3 In-process Sampling and Controls 8.3 工序取样和控制8.4 Blending Batches of Intermediates orAPIs8.4 中间体或原料药的混批8.5 Contamination Control 8.5 污染控制9. PACKAGING AND IDENTIFICATIONLABELING OF APIs ANDINTERMEDIATES9. 原料药和中间体的包装和贴签9.1 General 9.1 总则9.2 Packaging Materials 9.2 包装材料9.3 Label Issuance and Control 9.3 标签发放与控制9.4 Packaging and Labeling Operations 9.4 包装和贴签操作10. STORAGE AND DISTRIBUTION 10.储存和分发10.1 Warehousing Procedures 10.1 入库程序10.2 Distribution Procedures 10.2 分发程序11. LABORATORY CONTROLS 11.实验室控制11.1 General Controls 11.1 控制通则11.2 Testing of Intermediates and APIs 11.2 中间体和原料药的测试11.3 Validation of Analytical Procedures 11.3 分析方法的验证11.4 Certificates of Analysis 11.4 分析报告单11.5 Stability Monitoring of APIs 11.5 原料药的稳定性监测11.6 Expiry and Retest Dating 11.6 有效期和复验期11.7 Reserve/Retention Samples 11.7 留样12. V ALIDATION 12.验证12.1 Validation Policy 12.1 验证方针12.2 Validation Documentation 12.2 验证文件12.3 Qualification 12.3 确认12.4 Approaches to Process Validation 12.4 工艺验证的方法12.5 Process Validation Program 12.5 工艺验证的程序12.6 Periodic Review of Validated Systems 12.6验证系统的定期审核12.7 Cleaning Validation 12.7 清洗验证12.8 Validation of Analytical Methods 12.8 分析方法的验证13. CHANGE CONTROL 13.变更的控制14. REJECTION AND RE-USE OFMATERIALS14.拒收和物料的再利用14.1 Rejection 14.1 拒收14.2 Reprocessing 14.2 返工14.3 Reworking 14.3 重新加工14.4 Recovery of Materials and Solvents 14.4 物料与溶剂的回收14.5 Returns 14.5 退货15. COMPLAINTS AND RECALLS 15.投诉与召回16. CONTRACT MANUFACTURERS(INCLUDING LABORATORIES)16.协议生产商(包括实验室)17. AGENTS, BROKERS, TRADERS, DISTRIBUTORS, REPACKERS, AND RELABELLERS 17.代理商、经纪人、贸易商、经销商、重新包装者和重新贴签者17.1 Applicability 17.1适用性17.2 Traceability of Distributed APIs andIntermediates17.2已分发的原料药和中间体的可追溯性17.3 Quality Management 17.3质量管理17.4 Repackaging, Relabeling, and Holding of APIs and Intermediates 17.4原料药和中间体的重新包装、重新贴签和待检17.5 Stability 17.5稳定性17.6 Transfer of Information 17.6 信息的传达17.7 Handling of Complaints and Recalls 17.7 投诉和召回的处理17.8 Handling of Returns 17.8 退货的处理18. Specific Guidance for APIs Manufactured by Cell Culture/Fermentation 18. 用细胞繁殖/发酵生产的原料药的特殊指南18.1 General 18.1 总则18.2 Cell Bank Maintenance and RecordKeeping18.2细胞库的维护和记录的保存18.3 Cell Culture/Fermentation 18.3细胞繁殖/发酵18.4 Harvesting, Isolation and Purification 18.4收取、分离和精制18.5 Viral Removal/Inactivation steps 18.5 病毒的去除/灭活步骤19.APIs for Use in Clinical Trials 19.用于临床研究的原料药19.1 General 19.1 总则19.2 Quality 19.2 质量19.3 Equipment and Facilities 19.3 设备和设施19.4 Control of Raw Materials 19.4 原料的控制19.5 Production 19.5 生产19.6 Validation 19.6 验证19.7 Changes 19.7 变更19.8 Laboratory Controls 19.8 实验室控制19.9 Documentation 19.9 文件20. Glossary 20. 术语Q7a GMP Guidance for APIs Q7a原料药的GMP指南1. INTRODUCTION 1. 简介1.1 Objective 1.1目的This document is intended to provide guidance regarding good manufacturing practice (GMP) for the manufacturing of active pharmaceutical ingredients (APIs) under an appropriate system for managing quality. It is also intended to help ensure that APIs meet the quality and purity characteristics that they purport, or are represented, to possess. 本文件旨在为在合适的质量管理体系下制造活性药用成分(以下称原料药)提供有关优良药品生产管理规范(GMP)提供指南。
原料药GMP指南(中英文对照)

Q7a(中英文对照)FDA原料药GMP指南Table ofContents目录1、INTRODUCTION 1、简介1、1 Objective1、1目得1、2Regulatory Applicability 1、2法规得适用性1、3 Scope 1、3范围2、QUALITY MANAGEMENT2、质量管理2、1 Principles 2、1总则2、2质量部门得责任2、2Responsibilities of the Quality Unit(s)2、3生产作业得职责2、3 Responsibility for ProductionActivities2、4内部审计(自检)2、4 InternalAudits (Self Inspection)2、5 Product Quality Review2、5产品质量审核3、PERSONNEL3、人员3、1 PersonnelQualifications 3、人员得资质3、2 Personnel Hygiene 3、2 人员卫生3、3 Consultants 3、3 顾问4、建筑与设施4、BUILDINGS ANDFACILITIES4、1Designand Construction4、1 设计与结构4、2 Utilities4、2 公用设施4、3 Water4、3 水4、4 Containment4、4 限制4、5 Lighting 4、5 照明4、6Sewage and Refuse 4、6 排污与垃圾4、7Sanitation andMaintenance 4、7卫生与保养5、PROCESS EQUIPMENT5、工艺设备5、1DesignandConstruction 5、1 设计与结构5、2Equipment Maintenance and5、2设备保养与清洁Cleaning5、3Calibration 5、3 校验5、4 puterized Systems5、4 计算机控制系统6、DOCUMENTATION AND RECORDS6、文件与记录6、1 Documentation System andSpecifications6、1 文件系统与质量标准6、2EquipmentcleaningandUseRecord6、2 设备得清洁与使用记录6、3 Recordsof Raw Materials,Intermediates,APILabeling andPackaging Materials 6、3 原料、中间体、原料药得标签与包装材料得记录6、4Master Production Instructions (Master Production and ControlRecords)6、4 生产工艺规程(主生产与控制记录)6、5 BatchProduction Records(Batch Production andControlRecords)6、5 批生产记录(批生产与控制记录)6、6Laboratory ControlRecords 6、6 实验室控制记录6、7 Batch Production RecordReview6、7批生产记录审核7、MATERIALSMANAGEMENT7、物料管理7、1GeneralControls 7、1 控制通则7、2Receiptand Quarantine 7、2接收与待验7、3Sampling andTesting of IningProduction Materials7、3 进厂物料得取样与测试7、4 Storage 7、4储存7、5Re-evaluation 7、5复验8、PRODUCTION ANDIN—PROCESS CONTROLS8、生产与过程控制8、1ProductionOperations 8、1 生产操作8、2 Time Limits 8、2 时限8、3 In-process Sampling and Controls8、3 工序取样与控制8、4 BlendingBatches ofIntermediatesor APIs8、4 中间体或原料药得混批8、5 Contamination Control 8、5 污染控制9、PACKAGING AND IDENTIFICATION LABELING OF APIs AND INTERMEDIATES9、原料药与中间体得包装与贴签9、1General 9、1 总则9、2Packaging Materials 9、2 包装材料9、3Label Issuance andControl 9、3标签发放与控制9、4Packaging and LabelingOp9、4包装与贴签操作erations10、储存与分发10、STORAGE AND DISTRIBUTION10、1 Warehousing Procedures 10、1 入库程序10、2DistributionProcedures 10、2 分发程序11、LABORATORY CONTROLS 11、实验室控制11、1 General Controls 11、1控制通则11、2中间体与原料药得测试11、2 Testing ofIntermediatesandAPIs11、3 Validation of Analytical11、3 分析方法得验证Procedures11、4CertificatesofAnalysis11、4 分析报告单11、5 原料药得稳定性监测11、5 Stability Monitoringof APIs11、6 Expiryand RetestDating11、6 有效期与复验期11、7Reserve/Retention Samples 11、7 留样12、VALIDATION 12、验证12、1 Validation Policy 12、1 验证方针12、2 Validation Documentation12、2 验证文件12、3Qualification 12、3 确认12、4 工艺验证得方法12、4Approachesto ProcessValidation12、5 Process Validation Program12、5 工艺验证得程序12、6PeriodicReviewof12、6验证系统得定期审核Validated Systems12、7 CleaningValidation 12、7 清洗验证12、8 分析方法得验证12、8 Validation of Analytical Methods13、CHANGECONTROL 13、变更得控制14、REJECTIONANDRE-USEO14、拒收与物料得再利用FMATERIALS14、1 Rejection 14、1 拒收14、2Reprocessing14、2 返工14、3Reworking 14、3 重新加工14、4Recovery of Materialsand14、4 物料与溶剂得回收Solvents14、5Returns14、5 退货15、PLAINTS AND RECALLS 15、投诉与召回16、CONTRACTMANUFACTURERS (INCLUDING LABORATORIES)16、协议生产商(包括实验室)17、AGENTS,BROKERS, TRADERS,DISTRIBUTORS,REPACKERS,AND RELABELLERS17、代理商、经纪人、贸易商、经销商、重新包装者与重新贴签者17、1Applicability 17、1适用性17、2Traceabilityof DistributedAPIs and Intermediates17、2已分发得原料药与中间体得可追溯性17、3QualityManagement 17、3质量管理17、4Repackaging, Relabeling,and Holding of APIsandInterm ediates17、4原料药与中间体得重新包装、重新贴签与待检17、5Stability 17、5稳定性17、6 TransferofInformation 17、6 信息得传达17、7Handling ofplaints andRecalls17、7投诉与召回得处理17、8Handlingof Returns 17、8 退货得处理18、Specific Guidance for APIs Manufactured byCell Culture/Fermentation18、用细胞繁殖/发酵生产得原料药得特殊指南18、1 General 18、1总则18、2Cell Bank Maintenanceand Record Keeping18、2细胞库得维护与记录得保存18、3 CellCulture/Fermentation18、3细胞繁殖/发酵18、4 Harvesting, IsolationandPurification18、4收取、分离与精制18、5Viral Removal/Inactivation steps18、5 病毒得去除/灭活步骤19、APIsfor Use in Clinical Trials19、用于临床研究得原料药19、1General 19、1 总则19、2 Quality 19、2 质量19、3Equipment and Facilities 19、3设备与设施19、4 ControlofRaw Materials 19、4原料得控制19、5Production 19、5 生产19、6Validation 19、6 验证19、7 Changes19、7变更19、8 Laboratory Controls 19、8实验室控制19、9 Documentation 19、9 文件20、Glossary 20、术语Q7a GMP Guidance forAPIs Q7a原料药得GMP指南1、INTRODUCTION1、简介1、1 Objective1、1目得Thisdocument isintendedto provide guidance regarding good manufacturingpractice (GMP) for the manufacturing of active pharmaceuticalingredients (APIs)underan app ropriate systemfor managing qualit y、Itis also intended tohelp ensure that APIsmeet the qualityand puritycharacteristicstha ttheypurport,or arerepresented,to possess、本文件旨在为在合适得质量管理体系下制造活性药用成分(以下称原料药)提供有关优良药品生产管理规范(GMP)提供指南。
原料药批生产记录模板

********有限公司****批生产记录
品名:
规格:
批号:
理论产量:
成品数:
成品率:
生产日期:
生产部审阅:
质管部审阅:
目录
批生产指令单
开工前现场检查表
检查日期:年月日
生产管理员:QA检查员:
***** 岗位生产记录(三)
***** 岗位生产记录(四)
原料药一般生产区岗位清场记录
生产管理员:QA检查:
***** 岗位生产记录(一)
***** 岗位生产记录(二)
***** 岗位生产记录(三)
原料药一般生产区岗位清场记录
生产管理员:QA检查:
***** 岗位生产记录(一)
原料药一般生产区岗位清场记录
生产管理员:QA检查:
***** 干燥岗位生产记录
原料药洁净区岗位清场记录
生产管理员:QA检查:
批包装指令单
可编辑
***** 粉碎过筛岗位生产记录
原料药洁净区岗位清场记录
生产管理员:QA检查:
可编辑
***** 混合包装岗位生产记录(一)
***** 混合包装岗位生产记录(二)
原料药洁净区岗位清场记录
生产管理员:QA检查:
原料药包装岗位清场记录
生产管理员:QA检查:
***** 批生产汇总表
生产管理员:QA检查员:
产品生产检验报告单记录表
日期:年月日
生产管理员:QA检查员:
***** 关键岗位工艺查证记录(一)
生产管理员:QA检查员:
生产管理员:QA检查员:
批生产记录部门审核表
日期:年月日
可编辑
精品。
原料药批生产记录簿

******** 有限公司****批生产记录品名:___________________________ 规格: ___________________________ 批号:___________________________ 理论产量:________________________ 成品数:__________________________ 成品率:__________________________ 生产日期:________________________ 生产部审阅:______________________ 质管部审阅:______________________实用标准文案目录批生产指令单开工前现场检查表检查日期:年月曰生产管理员:QA检查员:实用标准文案***** 岗位生产记录(二)生产管理员:检查员:生产管理员: QA检查员:生产管理员:检查员:原料药一般生产区岗位清场记录生产管理员:QA检查:生产管理员: QA检查员:实用标准文案***** 岗位生产记录(三)生产管理员:检查员:原料药一般生产区岗位清场记录生产管理员:QA检查:实用标准文案***** 岗位生产记录(三)生产管理员:检查员:实用标准文案***** 岗位生产记录(五)原料药一般生产区岗位清场记录生产管理员:QA检查:***** 干燥岗位生产记录原料药洁净区岗位清场记录品名批号岗位清场日期有效期生产管理员:QA检查:批包装指令单原料药洁净区岗位清场记录品名批号岗位清场日期有效期生产管理员:QA检查:原料药洁净区岗位清场记录品名批号岗位清场日期有效期生产管理员:QA检查:原料药包装岗位清场记录生产管理员:QA检查:***** 批生产汇总表QA检查员:生产管理员: 精彩文档产品生产检验报告单记录表日QA检查员:***** 关键岗位工艺查证记录(一)生产管理员:QA检查员:生产管理员: QA检查员:批生产记录部门审核表。
原料药GMP规范指南中英文对照.doc

Q7a(中英文对照)FDA原料药GMP指南Table of Contents 目录1. INTRODUCTION 1. 简介1.1 Objective 1.1目的1.2 Regulatory Applicability 1.2法规的适用性1.3 Scope 1.3范围2. QUALITY MANAGEMENT 2.质量管理2.1 Principles 2.1总则2.2 Responsibilities of the Quality Unit(s) 2.2质量部门的责任2.3 Responsibility for Production Activities 2.3生产作业的职责2.4 Internal Audits (Self Inspection) 2.4内部审计(自检)2.5 Product Quality Review 2.5产品质量审核3. PERSONNEL 3. 人员3.1 Personnel Qualifications 3.人员的资质3.2 Personnel Hygiene 3.2 人员卫生3.3 Consultants 3.3 顾问4. BUILDINGS AND FACILITIES 4. 建筑和设施4.1 Design and Construction 4.1 设计和结构4.2 Utilities 4.2 公用设施4.3 Water 4.3 水4.4 Containment 4.4 限制4.5 Lighting 4.5 照明4.6 Sewage and Refuse 4.6 排污和垃圾4.7 Sanitation and Maintenance 4.7 卫生和保养5. PROCESS EQUIPMENT 5. 工艺设备5.1 Design and Construction 5.1 设计和结构5.2 Equipment Maintenance and Cleaning 5.2 设备保养和清洁5.3 Calibration 5.3 校验5.4 Computerized Systems 5.4 计算机控制系统6. DOCUMENTATION AND RECORDS 6. 文件和记录6.1 Documentation System andSpecifications6.1 文件系统和质量标准6.2 Equipment cleaning and Use Record 6.2 设备的清洁和使用记录6.3 Records of Raw Materials, Intermediates, API Labeling and Packaging Materials 6.3 原料、中间体、原料药的标签和包装材料的记录6.4 Master Production Instructions (MasterProduction and Control Records)6.4 生产工艺规程(主生产和控制记录)6.5 Batch Production Records (BatchProduction and Control Records)6.5 批生产记录(批生产和控制记录)6.6 Laboratory Control Records 6.6 实验室控制记录6.7 Batch Production Record Review 6.7批生产记录审核7. MATERIALS MANAGEMENT 7. 物料管理7.1 General Controls 7.1 控制通则7.2 Receipt and Quarantine 7.2接收和待验7.3 Sampling and Testing of IncomingProduction Materials7.3 进厂物料的取样与测试7.4 Storage 7.4储存7.5 Re-evaluation 7.5复验8. PRODUCTION AND IN-PROCESSCONTROLS8. 生产和过程控制8.1 Production Operations 8.1 生产操作8.2 Time Limits 8.2 时限8.3 In-process Sampling and Controls 8.3 工序取样和控制8.4 Blending Batches of Intermediates orAPIs8.4 中间体或原料药的混批8.5 Contamination Control 8.5 污染控制9. PACKAGING AND IDENTIFICATIONLABELING OF APIs ANDINTERMEDIATES9. 原料药和中间体的包装和贴签9.1 General 9.1 总则9.2 Packaging Materials 9.2 包装材料9.3 Label Issuance and Control 9.3 标签发放与控制9.4 Packaging and Labeling Operations 9.4 包装和贴签操作10. STORAGE AND DISTRIBUTION 10.储存和分发10.1 Warehousing Procedures 10.1 入库程序10.2 Distribution Procedures 10.2 分发程序11. LABORATORY CONTROLS 11.实验室控制11.1 General Controls 11.1 控制通则11.2 Testing of Intermediates and APIs 11.2 中间体和原料药的测试11.3 Validation of Analytical Procedures 11.3 分析方法的验证11.4 Certificates of Analysis 11.4 分析报告单11.5 Stability Monitoring of APIs 11.5 原料药的稳定性监测11.6 Expiry and Retest Dating 11.6 有效期和复验期11.7 Reserve/Retention Samples 11.7 留样12. V ALIDATION 12.验证12.1 Validation Policy 12.1 验证方针12.2 Validation Documentation 12.2 验证文件12.3 Qualification 12.3 确认12.4 Approaches to Process Validation 12.4 工艺验证的方法12.5 Process Validation Program 12.5 工艺验证的程序12.6 Periodic Review of Validated Systems 12.6验证系统的定期审核12.7 Cleaning Validation 12.7 清洗验证12.8 Validation of Analytical Methods 12.8 分析方法的验证13. CHANGE CONTROL 13.变更的控制14. REJECTION AND RE-USE OFMATERIALS14.拒收和物料的再利用14.1 Rejection 14.1 拒收14.2 Reprocessing 14.2 返工14.3 Reworking 14.3 重新加工14.4 Recovery of Materials and Solvents 14.4 物料与溶剂的回收14.5 Returns 14.5 退货15. COMPLAINTS AND RECALLS 15.投诉与召回16. CONTRACT MANUFACTURERS(INCLUDING LABORATORIES)16.协议生产商(包括实验室)17. AGENTS, BROKERS, TRADERS, DISTRIBUTORS, REPACKERS, AND RELABELLERS 17.代理商、经纪人、贸易商、经销商、重新包装者和重新贴签者17.1 Applicability 17.1适用性17.2 Traceability of Distributed APIs andIntermediates17.2已分发的原料药和中间体的可追溯性17.3 Quality Management 17.3质量管理17.4 Repackaging, Relabeling, and Holding of APIs and Intermediates 17.4原料药和中间体的重新包装、重新贴签和待检17.5 Stability 17.5稳定性17.6 Transfer of Information 17.6 信息的传达17.7 Handling of Complaints and Recalls 17.7 投诉和召回的处理17.8 Handling of Returns 17.8 退货的处理18. Specific Guidance for APIs Manufactured by Cell Culture/Fermentation 18. 用细胞繁殖/发酵生产的原料药的特殊指南18.1 General 18.1 总则18.2 Cell Bank Maintenance and RecordKeeping18.2细胞库的维护和记录的保存18.3 Cell Culture/Fermentation 18.3细胞繁殖/发酵18.4 Harvesting, Isolation and Purification 18.4收取、分离和精制18.5 Viral Removal/Inactivation steps 18.5 病毒的去除/灭活步骤19.APIs for Use in Clinical Trials 19.用于临床研究的原料药19.1 General 19.1 总则19.2 Quality 19.2 质量19.3 Equipment and Facilities 19.3 设备和设施19.4 Control of Raw Materials 19.4 原料的控制19.5 Production 19.5 生产19.6 Validation 19.6 验证19.7 Changes 19.7 变更19.8 Laboratory Controls 19.8 实验室控制19.9 Documentation 19.9 文件20. Glossary 20. 术语Q7a GMP Guidance for APIs Q7a原料药的GMP指南1. INTRODUCTION 1. 简介1.1 Objective 1.1目的This document is intended to provide guidance regarding good manufacturing practice (GMP) for the manufacturing of active pharmaceutical ingredients (APIs) under an appropriate system for managing quality. It is also intended to help ensure that APIs meet the quality and purity characteristics that they purport, or are represented, to possess. 本文件旨在为在合适的质量管理体系下制造活性药用成分(以下称原料药)提供有关优良药品生产管理规范(GMP)提供指南。
BM双语(英汉)批生产记录模板,中美双报

XXX制药有限公司XXX Pharmaceuticals Co., Ltd.批制作记录BPR For Manufacture产品分步收率1.Tableting Yield压片工序收率Tableting Yield limits for FetteP2020: 98.5%~100.5%; Tableting Yield limits for GEA Courtoy Modul TM D: 95.0%~100.5%; Reconciliation: 99.0%~100.3%;FetteP2020压片收率限度98.5%~100.5%;GEA Courtoy Modul TM D压片机压片收率限度95.0%~100.5%;物料平衡限度99.0%~100.3%Limits限度:99.7%~100.5%)2.Coating Yield包衣工序收率(Coating Material Dispensing配料工序1.Preparation & inspection before dispensing配料前准备和检查:2.Balance information配料操作所用电子秤3.Record the temperature, humidity, differential pressure at the starting time.操作前记录操作间的温湿度、压差,符合规定才能进行生产。
rmation of PE bag所使用的PE塑料袋信息5.Material confirmation: confirm material information on the label is consistent with the following table before dispensing.物料确认:确认待配物料的收料标签卡上的物料名称、代号、入厂号与下表中的一致,然后进行称量操作。
检查产品名称,批号等无误后将配料单粘于第四页。
片剂批生产记录表格模板

片剂批生产记录
产品名称
批号
成品量
生产周期:车间审核意见
规格
理论量
包装规格
********有限公司
月日至年月
审核人审核日期
日期
领料人
领料单
保管人
批生产指令单
起草人审核人接收人
称量配料岗位生产记录
年月曰
物料核对记录年月曰
清场记录
年月曰
原产品名称: 批 号: 调换产品名称: 批 号: 清场人: 质监员:
清场日期:年月日时 有效期至:年月日时
备注
清场人:
质监员:
清场合格证(正本)
M-J
>
岗
位:
称量配料
粉碎岗位生产记录
产品名称生产日期规格
清场记录
年月日
混合制粒干燥岗位生产记录
年月曰产品名称规格
清场记录
年月曰
整理总混岗位生产记录
年月曰
清场记录
年月曰
清场人:质监员:
填充装囊岗位生产记录
年月日
清场记录
年月日
领料单
保管人:
退料单
日期:年月日
退料人:
塑瓶内包装岗位生产记录
年月日
清场记录
年月日。
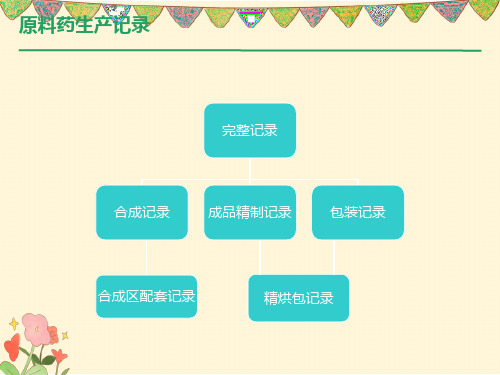
原料药生产记录
填写生产中遇到一些生产或设备运转异常情况及处理方法,并要有操作人签字,没有异常就划“/” (有异常写偏差)
清场检查记录 6.特殊问题或异常 事件记录
合成、成品精制记录
生产操作记录
生产操作记录中的内容是根据工艺规程按步骤编定的,操作记录针对操作内容主要从时间、温度、数量、检测结果、收率等方面全面、准确地反映生产情况 。 例如( 1) 投料时:操作记录需填写投料时间、净重; 升温(降温)至保温反应nhr:要填写开始升温(降温)时间,此时的温度;中间保温反应较长的,根据工艺要求每间隔一段时间记录时间、温度;
合成、成品精制记录
2.称量记录 需有原料名称、批号、日期、毛重、余重、净重、操作者、复核者、 QA签字审核,备注,称量时首先检查秤是否调零,按投料的时间先后顺序填写,试剂级(或固体)可以一起称量并记录;液体需每桶称量并记录,以kg为单位保留小数点后一位,以g为单位保留到g,物料均按“先进先出”的顺序使用。记录中填写内容与上项相同时应重复抄写,不得用“…”或“同上”表示;不得留有空格,如无内容填写要用 “/”划掉。
具体如下:
合成、成品精制记录组成
合成、成品精制记录
封面(包含公司名称)
品名:写工艺上统一的名称,不得随意简写,若是成品应按现行标准写全称。
产品批号:原料药的合成和成品批号均由年-月-年流水号6位数组成,合成(中间体)批号中“月”按投料当月月份定,成品批号中“月”按收料当月月份定。成品返工批号在原批号后加2,合成返工批号在原批号后加-1、-02…..
精烘包记录
设备日志(精制釜、微粉机、离心机、烘箱、粉碎机) 每次生产使用上述这些设备时填写,主要操作内容是指XX产品精制或微粉等。
精烘包记录
批生产记录--模板

主要设备
胶塞清洗机(设备编号)、电子秤(设备编号)
计划产量
瓶
执行SOP
操作指令
记录
操作人
复核人
胶塞洗涤灭菌标准操作程序
全自动胶塞清洗机标准操作程序
准备(班组质检员执行):
1)确认岗位有效期内的《清场合格证》,并附批记录。
2)确认设备内、生产线、生产区内无上次生产遗留物品,无与生产无关的物品。
3)确认操作间相对A/B级区负压>10pa、
7)“自动上料”时将待清洗胶塞(<46000个)加入料斗。漂洗后QA人员取样检查洗涤胶塞的注射用水的可见异物及细菌内毒素应合格。
8)灭菌结束时,记录灭菌结束时间。
9)打印灭菌曲线,确认灭菌温度、时间应符合要求。
10)灭菌曲线签字后附在本记录后面。
11)通知灌装岗位灭菌结束时间。
12)允许卸料后QA人员取样检查胶塞外观应合格。
3)确认操作间相对A/B级区负压>10pa、
温度18~35℃、
相对湿度45~75%。
4)确认设备、器具在清洁有效期内。
5)确认注射用水、纯化水、电、压缩空气供应正常。
6)设备试运转正常。
7)洗瓶机及隧道烘箱挂“运行中”状态标志。
8)岗位挂“正在生产”状态标志。
:
□已进行
□已进行
Pa
℃
%
□确认
□确认
□正常
13)清洗100支供QA人员检查合格。
14)确认烘箱温度升到设定温度后,关闭网带。
15)开始连续清洗,西林瓶进入隧道烘箱进行干燥、灭菌和除热原。
16)每小时检查记录一次注射用水压力、循环水的温度和压力、压缩空气压力、洗灌间压差。
□确认
ICH-Q7(中英文对照)

Q7a(中英文对照)FDA原料药GMP指南Table of Contents 目录1. INTRODUCTION 1. 简介1.1 Objective 1.1目的1.2 Regulatory Applicability 1.2法规的适用性1.3 Scope 1.3范围2. QUALITY MANAGEMENT 2.质量管理2.1 Principles 2.1总则2.2 Responsibilities of the Quality Unit(s) 2.2质量部门的责任2.3 Responsibility for Production Activities 2.3生产作业的职责2.4 Internal Audits (Self Inspection) 2.4内部审计(自检)2.5 Product Quality Review 2.5产品质量审核3. PERSONNEL 3. 人员3.1 Personnel Qualifications 3.人员的资质3.2 Personnel Hygiene 3.2 人员卫生3.3 Consultants 3.3 顾问4. BUILDINGS AND FACILITIES 4. 建筑和设施4.1 Design and Construction 4.1 设计和结构4.2 Utilities 4.2 公用设施4.3 Water 4.3 水4.4 Containment 4.4 限制4.5 Lighting 4.5 照明4.6 Sewage and Refuse 4.6 排污和垃圾4.7 Sanitation and Maintenance 4.7 卫生和保养5. PROCESS EQUIPMENT 5. 工艺设备5.1 Design and Construction 5.1 设计和结构5.2 Equipment Maintenance and Cleaning 5.2 设备保养和清洁5.3 Calibration 5.3 校验5.4 Computerized Systems 5.4 计算机控制系统6. DOCUMENTATION AND RECORDS 6. 文件和记录6.1 Documentation System andSpecifications6.1 文件系统和质量标准6.2 Equipment cleaning and Use Record 6.2 设备的清洁和使用记录6.3 Records of Raw Materials, Intermediates, API Labeling and Packaging Materials 6.3 原料、中间体、原料药的标签和包装材料的记录6.4 Master Production Instructions (MasterProduction and Control Records)6.4 生产工艺规程(主生产和控制记录)6.5 Batch Production Records (BatchProduction and Control Records)6.5 批生产记录(批生产和控制记录)6.6 Laboratory Control Records 6.6 实验室控制记录6.7 Batch Production Record Review 6.7批生产记录审核7. MATERIALS MANAGEMENT 7. 物料管理7.1 General Controls 7.1 控制通则7.2 Receipt and Quarantine 7.2接收和待验7.3 Sampling and Testing of IncomingProduction Materials7.3 进厂物料的取样与测试7.4 Storage 7.4储存7.5 Re-evaluation 7.5复验8. PRODUCTION AND IN-PROCESSCONTROLS8. 生产和过程控制8.1 Production Operations 8.1 生产操作8.2 Time Limits 8.2 时限8.3 In-process Sampling and Controls 8.3 工序取样和控制8.4 Blending Batches of Intermediates orAPIs8.4 中间体或原料药的混批8.5 Contamination Control 8.5 污染控制9. PACKAGING AND IDENTIFICATIONLABELING OF APIs ANDINTERMEDIATES9. 原料药和中间体的包装和贴签9.1 General 9.1 总则9.2 Packaging Materials 9.2 包装材料9.3 Label Issuance and Control 9.3 标签发放与控制9.4 Packaging and Labeling Operations 9.4 包装和贴签操作10. STORAGE AND DISTRIBUTION 10.储存和分发10.1 Warehousing Procedures 10.1 入库程序10.2 Distribution Procedures 10.2 分发程序11. LABORATORY CONTROLS 11.实验室控制11.1 General Controls 11.1 控制通则11.2 Testing of Intermediates and APIs 11.2 中间体和原料药的测试11.3 Validation of Analytical Procedures 11.3 分析方法的验证11.4 Certificates of Analysis 11.4 分析报告单11.5 Stability Monitoring of APIs 11.5 原料药的稳定性监测11.6 Expiry and Retest Dating 11.6 有效期和复验期11.7 Reserve/Retention Samples 11.7 留样12. V ALIDATION 12.验证12.1 Validation Policy 12.1 验证方针12.2 Validation Documentation 12.2 验证文件12.3 Qualification 12.3 确认12.4 Approaches to Process Validation 12.4 工艺验证的方法12.5 Process Validation Program 12.5 工艺验证的程序12.6 Periodic Review of Validated Systems 12.6验证系统的定期审核12.7 Cleaning Validation 12.7 清洗验证12.8 Validation of Analytical Methods 12.8 分析方法的验证13. CHANGE CONTROL 13.变更的控制14. REJECTION AND RE-USE OFMATERIALS14.拒收和物料的再利用14.1 Rejection 14.1 拒收14.2 Reprocessing 14.2 返工14.3 Reworking 14.3 重新加工14.4 Recovery of Materials and Solvents 14.4 物料与溶剂的回收14.5 Returns 14.5 退货15. COMPLAINTS AND RECALLS 15.投诉与召回16. CONTRACT MANUFACTURERS(INCLUDING LABORATORIES)16.协议生产商(包括实验室)17. AGENTS, BROKERS, TRADERS, DISTRIBUTORS, REPACKERS, AND RELABELLERS 17.代理商、经纪人、贸易商、经销商、重新包装者和重新贴签者17.1 Applicability 17.1适用性17.2 Traceability of Distributed APIs andIntermediates17.2已分发的原料药和中间体的可追溯性17.3 Quality Management 17.3质量管理17.4 Repackaging, Relabeling, and Holding of APIs and Intermediates 17.4原料药和中间体的重新包装、重新贴签和待检17.5 Stability 17.5稳定性17.6 Transfer of Information 17.6 信息的传达17.7 Handling of Complaints and Recalls 17.7 投诉和召回的处理17.8 Handling of Returns 17.8 退货的处理18. Specific Guidance for APIs Manufactured by Cell Culture/Fermentation 18. 用细胞繁殖/发酵生产的原料药的特殊指南18.1 General 18.1 总则18.2 Cell Bank Maintenance and RecordKeeping18.2细胞库的维护和记录的保存18.3 Cell Culture/Fermentation 18.3细胞繁殖/发酵18.4 Harvesting, Isolation and Purification 18.4收取、分离和精制18.5 Viral Removal/Inactivation steps 18.5 病毒的去除/灭活步骤19.APIs for Use in Clinical Trials 19.用于临床研究的原料药19.1 General 19.1 总则19.2 Quality 19.2 质量19.3 Equipment and Facilities 19.3 设备和设施19.4 Control of Raw Materials 19.4 原料的控制19.5 Production 19.5 生产19.6 Validation 19.6 验证19.7 Changes 19.7 变更19.8 Laboratory Controls 19.8 实验室控制19.9 Documentation 19.9 文件20. Glossary 20. 术语Q7a GMP Guidance for APIs Q7a原料药的GMP指南1. INTRODUCTION 1. 简介1.1 Objective 1.1目的This document is intended to provide guidance regarding good manufacturing practice (GMP) for the manufacturing of active pharmaceutical ingredients (APIs) under an appropriate system for managing quality. It is also intended to help ensure that APIs meet the quality and purity characteristics that they purport, or are represented, to possess. 本文件旨在为在合适的质量管理体系下制造活性药用成分(以下称原料药)提供有关优良药品生产管理规范(GMP)提供指南。
ICH-Q7(中英文对照)【范本模板】

Q7a(中英文对照)FDA原料药GMP指南Table of Contents 目录1。
INTRODUCTION 1。
简介1.1 Objective 1.1目的1.2 Regulatory Applicability 1。
2法规的适用性1.3 Scope 1。
3范围2。
QUALITY MANAGEMENT 2.质量管理2。
1 Principles 2。
1总则2。
2 Responsibilities of the Quality Unit(s) 2。
2质量部门的责任2。
3 Responsibility for Production Activities 2。
3生产作业的职责2。
4内部审计(自检)29589 7395 玕€32268 7E0C 縌21637 5485咅-26995 6973 楳2。
4 Internal Audits (Self Inspection)2.5 Product Quality Review 2。
5产品质量审核3. PERSONNEL 3。
人员3。
1 Personnel Qualifications 3。
人员的资质3.2 Personnel Hygiene 3.2 人员卫生3.3 Consultants 3。
3 顾问4。
BUILDINGS AND FACILITIES 4。
建筑和设施4.1 Design and Construction 4。
1 设计和结构4.2 Utilities 4.2 公用设施4。
3 水ct[?d#37711 934F 鍏4。
3 Water4。
4 Containment 4.4 限制4。
5 Lighting 4.5 照明4。
6 Sewage and Refuse 4。
6 排污和垃圾4。
7 Sanitation and Maintenance 4.7 卫生和保养5. PROCESS EQUIPMENT 5。
工艺设备5.1 Design and Construction 5.1 设计和结构5.2 Equipment Maintenance and Cleaning 5。
原料药研发流程-中英文

Schedule regular meetings (such as once a month) to discuss any related issuesand make any plans
定期(例如每月一次)讨论任何相关问题并制订实施计划;
Monitorany progress of on-goingproject.
The step 6-8 can be conducted in parallel with above development work
6-8步骤可以与上述的研发工作同步开展
6
a)Purchase USP reference standard (if it is commercially available in USP)
相同批次物料也可以作为公司自己的标准品和或成为内部工作标准物。
Note备注:
All spectroscopic data should be interpreted and presented in tabularformat.
所有实验分析数据均应以表格的形式记录和说明。
1)Qualification protocoland related SOPs forreferencestandard and/or in-houseworking standard should beprepared.
监管正在进行的项目的进展情况
System based CGMPauditingshould beconducted to identify any deficiencies.
执行基于六大系统的CGMP审计以寻找任何GMP缺陷或不足。
Training should be scheduled toprovideCGMP related subjects based on the deficiencies found.
药品生产技术《原料药批生产记录》

XXXXXX
X X X X X批生产记录
品名:
规格:
批号:
理论产量:
成品数:
成品率:
生产日期:
生产部审阅:
质管部审阅:
目录
生产管理员:QA检查员:XXXXXXXX岗位生产记录〔三〕
生产管理员:QA检查员:
原料药洁净区岗位清场记录
生产管理员:QA检查:
批包装指令单
日期:年月日
XXXXX粉碎过筛岗位生产记录
生产管理员:QA检查员:
原料药洁净区岗位清场记录
生产管理员:QA检查:
生产管理员:QA检查员:
生产管理员:QA检查员:
原料药洁净区岗位清场记录
生产管理员:QA检查:
原料药包装岗位清场记录
生产管理员:QA检查:
XXXXX批生产汇总表
生产管理员:
QA检查员:
产品生产检验报告单记录表
日期:年月
日
生产管理员:QA检查员:
XXXXX关键岗位工艺查证记录〔一〕
生产管理员:QA检查员:
XXXXX关键岗位工艺查证记录〔二〕
生产管理员:QA检查员:
生产管理员:QA检查员:
批生产记录部门审核表
日期:年月日
成品检验报告单
检验单号:
成品审核记录
XXXXXXXXXXXXXX RQA027-00
第
一
联
‥
生
产
部
第
二
联
‥
仓
库。
常用制药GMP英文词汇

常用制药GMP英文词汇————————————————————————————————作者: ————————————————————————————————日期:ﻩ国际组织ISO(InternationalOrganization for Standardization):国际标准化组织日常办事机构是中央秘书处,设在瑞士日内瓦WHO(World Health Organization):世界卫生组织是联合国属下的专门机构,国际最大的公共卫生组织,总部设于瑞士日内瓦PIC/S(Pharmaceutical Inspection Convention/Pharmaceutical Inspec tion Cooperation Scheme):国际医药品稽查协约组织由欧洲自由贸易区(EFTA)组建ICH(International Conferenceon Harmonizationof TechnicalRequirementsforRegistrationof PharmaceuticalsforHuman Use):人用药物注册技术要求国际协调会由欧盟(EU)、欧洲制药工业协会联合会(EFPIA)、日本厚生省(MHW)、日本制药工业协会(JPMA)、美国FDA、美国药物研究生产联合会(PRMA)等机构组成WHO、EFTA、加拿大卫生保健局(CHPB)为观察员ISPE(International Societyfor PharmaceuticalEngineering):国际制药工程协会是致力于培训制药领域专家并提升制药行业水准的世界最大的非盈利性组织之一,在美国坦帕州设有全球总部,在布鲁塞尔设有欧洲总部,亚洲总部在新加坡HHS(United StatesDepartment of HealthandHuman Services):美国卫生及公共服务部(美国卫生部)FDA(Food and DrugAdministration):美国食品药品监督管理局(HHS下属机构)PDA(Parenteral DrugAssociation):美国注射剂协会EPA(Environmental Protection Agency):美国国家环境保护局CDER(Center for Drug Evaluationand Research):FDA药物评价与研究中心EMEA(The European Agency for the Evaluationof MedicinalProducts):欧洲药物评审组织MHW(Ministry of Health and Welfare):日本厚生省,现改为厚生劳动省MHLW(Mini stryofHealth,Laborand Welfare),负责医疗卫生和社会保障的主要部门D&B(Dun&Bradstreet):邓白氏公司DUNS(Data UniversalNumbering System):邓白氏公司提供的唯一的公司代号,用于信用评级等在SMF文件中会用到ATCC(AmericanType Culture Collection):美国模式培养物集存库ASTM(AmericanSocietyfor Testing Materials):美国材料与试验协会法规GMP(Good Manufacturing Practice):药品良好生产规范cGMP(Current Good Manufacture Practices):动态药品生产管理规范,即现行的GLP(Good Laboratory Practice):药物非临床研究质量管理规范,及优良实验室规范GSP(GoodSupplyingPractice):药品经营质量管理规范,及良好的药品供应规范GAP(Good Agricultural Practice for Chinese Crude Drugs):中药材生产质量管理规范GDP(GoodDocumentationPractice):良好文件管理GEP(GoodEngineeringPractice):工程设计规范GAMP(GoodAutomated Manufacturing Practice):优良自动化生产规范USP(united states pharmacopeia):美国药典EP(EuropeanPharmacopeia):欧洲药典JP(Japanese Pharmacopoeia):日本药典CFR(Code ofFederal Regulations):美国联邦法律CFR21 Part 11(Code of Federal RegistryPart11):联邦法规法律标题21第11部分CEP/COS(Certificateo f S uitability to themonographsofE uropeanPharmacopoeia):欧洲药典适应性认证证书CEP认证,COS证书CTD(Common Technical Document):国际注册用常规技术文件CTD文件是国际公认的文件编写格式,用来制作一个向药品注册机构递交的结构完善的注册申请文件EHS(Environment、Health、Safety):环境-健康-安全管理体系HACCP(HazardAnalysisand Critical Control Point):(保健食品)危害分析和关键控制点REACH(REGULATIONconcerning the Registration, Evaluation, Authori zationandRestriction of Chemicals):欧盟规章《化学品注册、评估、许可和限制》,欧盟建立的,并于2007年6月1日起实施的化学品监管体系ICH法规ICH-Q1A:新原料药和制剂的稳定性试验ICH-Q1B:稳定性试验:新原料药和制剂的光稳定性试验ICH-Q1C:稳定性试验:新剂型的要求ICH-Q1D:新原料药和制剂的稳定性试验的括号法和矩阵法设计ICH-Q1E:稳定性数据的评价ICH-Q1F:气候带Ⅲ和Ⅳ注册申请的稳定性数据ICH-Q2A:分析步骤验证:正文ICH-Q2B:分析步骤验证:方法学ICH-Q3A:原料药中的杂质ICH-Q3B:新制剂中的杂质ICH-Q3C:杂质;残留溶剂的指导原则ICH-Q4:药典ICH-Q4A:药典的同一化ICH-Q4B:各地区使用的药典正文评估和建议ICH-Q5A:来源于人或动物细胞系的生物技术产品的病毒安全性评价ICH-Q5B:生物技术产品的质量:rDNA衍生蛋白质产品生产细胞的表达构建体分析ICH-Q5C:生物技术产品的质量:生物制品/生物技术产品的稳定性试验ICH-Q5D:用于生物技术产品及生物制品生产的细胞基质的来源和鉴定ICH-Q5E:生物技术产品/生物制品在工艺变更时的可比性ICH-Q6A:质量标准新原料药和制剂的检测以及可接受标准:化学物质ICH-Q6B:质量标准:生物技术产品及生物制品的检测方法和可接受标准ICH-Q7:原料药良好制造规范(ICH-Q7A的新版)ICH-Q7A:原料药的GMP规范ICH-Q8:药物研发指南ICH-Q9:质量风险管理ICH-Q10(PQS):药物质量体系ICH-Q11:原料药研发与生产常见术语QA(Quality Assurance):质量保证QC(Quality Control):质量控制CQA(CriticalQualityAttribute):关键质量属性QRM(QualityRisk Management):质量风险管理IPC(InproceicsQuality Control):制程品质控制/中控OOS(Out of Specification):检验结果超标OOT(Out of Trend):超趋势结果OOL(Out of Limit):超出极限的结果,如温湿度等OOE(Out of Expectation):超期望结果SOP(Standard OperationProcedure):标准操作规程DMF(Drug MasterFile):药品主文件SMF(Site MasterFile):工厂主文件URS(User Requirement Specification):用户需求标准FAT(Factory AcceptanceTest):工厂验收测试SAT(SiteAcceptance Test):现场验收测试FS(Functional Specification):功能标准DS(Design Specification):设计标准DQ(Design Qualification):设计确认IQ(Installation Qualification):安装确认OQ(Operational Qualification):运行确认PQ(Performance Qualification):性能确认RQ(Requalification):再确认CAPA(Corrective Action& Preventive Action):纠正预防系统,Q10的四大要素之一QbD(Quality by Design):质量源于设计PMC(ProductMaterial Control):生产物料控制PC生产控制;MC物料控制CMC(Chemistry and manufacture control):生产和化学控制APR(AnnualProducts Review):年度质量回顾CNC(Controlled Non-Classified Area):受控非洁净区应用技术APS(Aseptic Processing Simulation):培养基模拟灌装CIP(Cleaning in Place):原位清洗(全自动,如针剂配制系统)WIP(Washing inPlace):在线清洁(半自动,需要手动的拆卸,如流化床)SIP(Sterilization in Place):在线灭菌BFS(BlowingFilling and Sealing):吹-灌-封PAT(Process AnalyticalTechnology):过程分析技术PLC(Programmable Logic Controller):可编程逻辑控制EDI(Electrodeionization):一种制备纯化水的离子交换技术MAC(Minimum Acceptable Cycle):最低可接受程序SAM(Steam-Air Mixture):蒸汽空气混合气体灭菌程序WIT(WaterIntrusion Test):水侵入测试(东富龙疏水性滤器的在线进行完整性测试的方法)BP(BubblePoint Test):起跑点试验FF(ForwardFlow/Diffusive Flow):前进流、扩散流试验HPLC(High Performance Liquid Chromatography):高效液相色谱GC(Gas Chromatography):气相色谱FTIR(Fourier TransformInfrared spectroscopy):傅氏转换红外线光谱分析仪MS(Mass Spectroscopy):质谱LC/MS:液质联用GC/MS:气质联用TOC(Total Organic Carbon):总有机碳NVR(Nonvolatile Residue):不挥发残留物RFS(Ready for Sterilization):免洗胶塞RFU(Ready for Use):即用胶塞物品名称SVP(Small Volume Parenteral):小容量注射剂LVP(LargeVolume Parenteral):大容量注射剂APA(Aseptic ProcessingArea):无菌区P&ID(Piping and Instrument Diagram):工艺管道仪表流程图PFD(Process Flow Diagram):工艺流程图UFD(UtilityFlow Diagram):公用工程流程图HVAC(Heating Ventilation Air Conditioning):供热空气调节净化系统HEPA(HighEfficiencyParticulate Air Filter):高效过滤器FFU(Fan Filter Units):风机滤器单元AHU(Air Handling Unit):空气处理单元COA(Certificate ofAnalysis):分析证书/检验报告书/检验报告单BPR(Batch Production Record):批生产记录API(Active Pharmaceutical Ingredients):药物活性成分,通常指的原料药WFI(Waterfor Injection):注射用水DOP:为邻苯二甲酸二辛酯,HEPA检漏用的气溶胶PAO:聚-α-烯烃,HEPA检漏用的气溶胶IBC(IntermediateBulk Container):中型散装容器FBD(Fluid BedDryer):流化床IRTD(Intelligent ResistanceTemperature Detector):智能热电阻温度探头,标准温度探头SV(Solenoid Valve):电磁阀FV:气动阀P/HG(Porous/Hard Goods Loads):多孔/坚硬装载,包括过滤器、胶塞、软管、拖把、工作服、塞子、清洁器具或设备的更换部件。
中英文对照FDA原料药GMP指南

Q7a(中英文对照)FDA原料药GMP指南Table of Contents 目录1. INTRODUCTION 1. 简介1.1 Objective 1.1目的1.2 Regulatory Applicability 1.2法规的适用性1.3 Scope 1.3范围2. QUALITY MANAGEMENT 2.质量管理2.1 Principles 2.1总则2.2 Responsibilities of the Quality Unit(s) 2.2质量部门的责任2.3 Responsibility for Production Activities 2.3生产作业的职责2.4 Internal Audits (Self Inspection) 2.4内部审计(自检)2.5 Product Quality Review 2.5产品质量审核3. PERSONNEL 3. 人员3.1 Personnel Qualifications 3.人员的资质3.2 Personnel Hygiene 3.2 人员卫生3.3 Consultants 3.3 顾问4. BUILDINGS AND FACILITIES 4. 建筑和设施4.1 Design and Construction 4.1 设计和结构4.2 Utilities 4.2 公用设施4.3 Water 4.3 水4.4 Containment 4.4 限制4.5 Lighting 4.5 照明4.6 Sewage and Refuse 4.6 排污和垃圾4.7 Sanitation and Maintenance 4.7 卫生和保养5. PROCESS EQUIPMENT 5. 工艺设备5.1 Design and Construction 5.1 设计和结构5.2 Equipment Maintenance and Cleaning 5.2 设备保养和清洁5.3 Calibration 5.3 校验5.4 Computerized Systems 5.4 计算机控制系统6. DOCUMENTATION AND RECORDS 6. 文件和记录6.1 Documentation System andSpecifications6.1 文件系统和质量标准6.2 Equipment cleaning and Use Record 6.2 设备的清洁和使用记录6.3 Records of Raw Materials, Intermediates, API Labeling and Packaging Materials 6.3 原料、中间体、原料药的标签和包装材料的记录6.4 Master Production Instructions (MasterProduction and Control Records)6.4 生产工艺规程(主生产和控制记录)6.5 Batch Production Records (BatchProduction and Control Records)6.5 批生产记录(批生产和控制记录)6.6 Laboratory Control Records 6.6 实验室控制记录6.7 Batch Production Record Review 6.7批生产记录审核7. MATERIALS MANAGEMENT 7. 物料管理7.1 General Controls 7.1 控制通则7.2 Receipt and Quarantine 7.2接收和待验7.3 Sampling and Testing of IncomingProduction Materials7.3 进厂物料的取样与测试7.4 Storage 7.4储存7.5 Re-evaluation 7.5复验8. PRODUCTION AND IN-PROCESSCONTROLS8. 生产和过程控制8.1 Production Operations 8.1 生产操作8.2 Time Limits 8.2 时限8.3 In-process Sampling and Controls 8.3 工序取样和控制8.4 Blending Batches of Intermediates orAPIs8.4 中间体或原料药的混批8.5 Contamination Control 8.5 污染控制9. PACKAGING AND IDENTIFICATIONLABELING OF APIs ANDINTERMEDIATES9. 原料药和中间体的包装和贴签9.1 General 9.1 总则9.2 Packaging Materials 9.2 包装材料9.3 Label Issuance and Control 9.3 标签发放与控制9.4 Packaging and Labeling Operations 9.4 包装和贴签操作10. STORAGE AND DISTRIBUTION 10.储存和分发10.1 Warehousing Procedures 10.1 入库程序10.2 Distribution Procedures 10.2 分发程序11. LABORATORY CONTROLS 11.实验室控制11.1 General Controls 11.1 控制通则11.2 Testing of Intermediates and APIs 11.2 中间体和原料药的测试11.3 Validation of Analytical Procedures 11.3 分析方法的验证11.4 Certificates of Analysis 11.4 分析报告单11.5 Stability Monitoring of APIs 11.5 原料药的稳定性监测11.6 Expiry and Retest Dating 11.6 有效期和复验期11.7 Reserve/Retention Samples 11.7 留样12. V ALIDATION 12.验证12.1 Validation Policy 12.1 验证方针12.2 Validation Documentation 12.2 验证文件12.3 Qualification 12.3 确认12.4 Approaches to Process Validation 12.4 工艺验证的方法12.5 Process Validation Program 12.5 工艺验证的程序12.6 Periodic Review of Validated Systems 12.6验证系统的定期审核12.7 Cleaning Validation 12.7 清洗验证12.8 Validation of Analytical Methods 12.8 分析方法的验证13. CHANGE CONTROL 13.变更的控制14. REJECTION AND RE-USE OFMATERIALS14.拒收和物料的再利用14.1 Rejection 14.1 拒收14.2 Reprocessing 14.2 返工14.3 Reworking 14.3 重新加工14.4 Recovery of Materials and Solvents 14.4 物料与溶剂的回收14.5 Returns 14.5 退货15. COMPLAINTS AND RECALLS 15.投诉与召回16. CONTRACT MANUFACTURERS(INCLUDING LABORATORIES)16.协议生产商(包括实验室)17. AGENTS, BROKERS, TRADERS, DISTRIBUTORS, REPACKERS, AND RELABELLERS 17.代理商、经纪人、贸易商、经销商、重新包装者和重新贴签者17.1 Applicability 17.1适用性17.2 Traceability of Distributed APIs andIntermediates17.2已分发的原料药和中间体的可追溯性17.3 Quality Management 17.3质量管理17.4 Repackaging, Relabeling, and Holding of APIs and Intermediates 17.4原料药和中间体的重新包装、重新贴签和待检17.5 Stability 17.5稳定性17.6 Transfer of Information 17.6 信息的传达17.7 Handling of Complaints and Recalls 17.7 投诉和召回的处理17.8 Handling of Returns 17.8 退货的处理18. Specific Guidance for APIs Manufactured by Cell Culture/Fermentation 18. 用细胞繁殖/发酵生产的原料药的特殊指南18.1 General 18.1 总则18.2 Cell Bank Maintenance and RecordKeeping18.2细胞库的维护和记录的保存18.3 Cell Culture/Fermentation 18.3细胞繁殖/发酵18.4 Harvesting, Isolation and Purification 18.4收取、分离和精制18.5 Viral Removal/Inactivation steps 18.5 病毒的去除/灭活步骤19.APIs for Use in Clinical Trials 19.用于临床研究的原料药19.1 General 19.1 总则19.2 Quality 19.2 质量19.3 Equipment and Facilities 19.3 设备和设施19.4 Control of Raw Materials 19.4 原料的控制19.5 Production 19.5 生产19.6 Validation 19.6 验证19.7 Changes 19.7 变更19.8 Laboratory Controls 19.8 实验室控制19.9 Documentation 19.9 文件20. Glossary 20. 术语Q7a GMP Guidance for APIs Q7a原料药的GMP指南1. INTRODUCTION 1. 简介1.1 Objective 1.1目的This document is intended to provide guidance regarding good manufacturing practice (GMP) for the manufacturing of active pharmaceutical ingredients (APIs) under an appropriate system for managing quality. It is also intended to help ensure that APIs meet the quality and purity characteristics that they purport, or are represented, to possess. 本文件旨在为在合适的质量管理体系下制造活性药用成分(以下称原料药)提供有关优良药品生产管理规范(GMP)提供指南。
药品化妆品批生产记录模板

审核结论
经对:
1、物料、批生产记录、检验监控各审核内容的审核;
2、产品的注册批准的法规性、质量标准、检验标准的审核;
3、12项审查内容的审核;
本批产品:
□符合规定□不符合规定
批产品:(同意/不同意)放行。
质检部门负责人:日期:年月日
静置时间
操作人
检查人
质检员
紫外灯使用记录
开启时间
关闭时间
使用时间
操作人
检查人
质检员
生产后清场清洁记录
清场清洁内容
清场清洁标准
清场清洁结果
操作人
检查人
质检员
按规程清洁静置间及设备
静置间及设备、容器具清洁消毒与下次生产无关的物料、废弃物清除
是□否□
清场后,开启室内紫外线消毒,悬挂清场合格证。
开启室内紫外线消毒30分钟,悬挂清场合格证
清场清洁内容
清场清洁标准
清场清洁结果
操作人
检查人
质检员
按规程清洁操作间及设备
操作间及设备、容器具、管道清洁消毒与下次生产无关的物料、废弃物清除
是□否□
清场后,开启室内紫外线消毒,悬挂清场合格证。
开启室内紫外线消毒30分钟,悬挂清场合格证
是□否□
备注:
取样检验合格后出料并称好重量,贴好桶卡,注明品名、重量,出料完毕计算收率,明确标识后,移至静置间。
个/每包装单位
包材根据实际情况可以高于理论用量领取
说明书
张
张/每包装单位
塑膜
Kg
Kg/每包装单位
箱
个
个/每包装单位
标注标示要求
瓶包装:
批号:生产日期:
盒包装:
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
BATCH PRODUCTION RECORD
The following code has been given to the finished product.
Revision level:
Reasons for Revision:
INPUT DETAILS
Product Name: Flunixin meglumine Doc No:
Batch No: Stage: Ⅰ
PRODUCT OUTPUT DETAILS
Out put weight/ Output Molecule mass Actual yield ratio calculation: Yield ratio = -------------------------------------------------- Input weight / Input Molecule mass Calculated by Verified by
Product Name: Flunixin meglumine Doc No: Batch No: Stage: Ⅰ
Op. No Operation Date Time (hrs) Remarks Done
by
Checked
by
From To Duration
01 Make sure that R2201 is clean and
dry
02 Charge 98kg of purified water , 53kg
of MA , 38.7kg of CA , and 3.67kg
of TS into R2201
03 Start agitation and keep for
15~20minutes
04 Heat to reflux and maintain the reflux
for 2 hours at 10±5℃
05 Prepare 36% potassium hydroxide
solution by adding 25.2kg of
potassium hydroxide and
44.8kg of purified water
06 Add 30.5kg of hydrochloric acid and
9.5kg of purified water to prepare
another 23% hydrochloric acid
solution
07 Then charge excess 4 kg of purified
water to R2201
08 Adjust the reaction mass at pH=9
by dropping 36% potassium
hydroxide solution
09 Cool the temperature below 10℃
and recheck whether pH is still at
9~10, if not , add 36% potassium
hydroxide further to correct it
10 Make sure that ST2202 is dry and
clean
11 Centrifuge the mass from R2201 and
wash the wet cake and R2201 with
100kg of purified water (clean the
R2201 as per procedure specified in
Page No of)
12 Spin dry for 40~50minutes and
introduce mother liquid back R2201
Note: Shift in–charge should cross check the S. No:and shall sign in ‘checked by’ column.
BATCH PRODUCTION RECORD
Product Name: Flunixin meglumine Doc No: Batch No: Stage: Ⅰ
ANALYTICAL DATA RECORD
、
BATCH PRODUCTION RECORD
Product Name: Flunixin meglumine Doc No: Batch No: Stage: Ⅰ
Deviation Record
1) Deviations in Operation :
2) Deviations in Yield :
Remarks:
Production
BATCH PRODUCTION RECORD
Product Name:Flunixin meglumine Doc No: Batch No: Stage: Ⅰ
Temperature Sheet
BATCH PRODUCTION RECORD
Product Name:Flunixin meglumine Doc No: Batch No: Stage: Ⅰ
WEIGHING RECORD
INPUTS
EQUIPMENT BATCH TO BATCH CLEANING PROCEDURE
Product Name: Flunixin meglumine Doc No:
Batch No: Stage: Ⅰ
Ref. No :
Operation No. :EQ. ID No.: RE402
Operation no: (visual observation) should be done by shift in charge.。