厦门大学 物理化学 (上) 各章 知识点 总结
物理化学各章概念总结、公式总结电子版1 表面化学与胶体

第8章 表面化学与胶体8.1 重要概念和规律1.比表面能与表面张力物质的表面是指约几个分子厚度的一层。
由于表面两侧分子作用力不同,所以在表面上存在一个不对称力场,即处在表面上的分子都受到一个指向体相内部的合力,从而使表面分子具有比内部分子更多的能量。
单位表面上的分子比同样数量的内部分子多出的能量称为比表面能(也称比表面Gibbs函数)。
表面张力是在表面上的相邻两部分之间单位长度上的相互牵引力,它总是作用在表面上,并且促使表面积缩小。
表面张力与比表面能都是表面上不对称力场的宏观表现,即二者是相通的,它们都是表面不对称力场的度量。
它们是两个物理意义不同,单位不同,但数值相同,量纲相同的物理量。
2.具有巨大界面积的系统是热力学不稳定系统物质表面所多余出的能量γA称表面能(亦叫表面Gibbs函数),它是系统Gibbs函数的一部分,表面积A越大,系统的G值越高。
所以在热力学上这种系统是不稳定的。
根据热力学第二定律,在一定温度和压力下,为了使G值减少,系统总是自发地通过以下两种(或其中的一种)方式降低表面能γA:①在一定条件下使表面积最小。
例如液滴呈球形,液面呈平面;②降低表面张力。
例如溶液自发地将其中能使表面张力降低的物质相对浓集到表面上(即溶液的表面吸附),而固体表面则从其外部把气体或溶质的分子吸附到表面上,从而改变表面结构,致使表面张力降低。
3.润湿与铺展的区别润湿和铺展是两种与固—液界面有关的界面过程。
两者虽有联系,但意义不同。
润湿是液体表面与固体表面相互接触的过程1因此所发生的变化是由固—液界面取代了原来的液体表面和固体表面。
润湿程度通常用接触角表示,它反映液、固两个表面的亲密程度。
当θ值最小(θ=0o)时,润湿程度最大,称完全润湿。
铺展是指将液体滴洒在固体表面上时,液滴自动在表面上展开并形成一层液膜的过程,因此所发生的变化是由固—液界面和液体表面取代原来的固体表面。
铺展的判据是上述过程的∆G:若∆G<0,则能发生铺展;若∆G≥0,则不能铺展。
物理化学各章知识点总结-86页

经此过程,所有状态函数的变量均为零。
4. 功和热
注意:
W pambdV
• 不论是膨胀还是压缩,体积功都用- pambdV
计算,公式中的压力都用外压
• 公式中的pamb不能随便写到积分号外,只有 常数才行
• 只有- pambdV这个量才是体积功,pambV或 Vdpamb都不是体积功。
不同过程功的计算分析:W p amb V
W pamb dV
(1)环境为真空态(自由膨胀):
pamb= 0,W = 0
(2)恒外压过程(pamb恒定): W = -pambΔV = -pamb(V2-V1)
(3)恒压过程(pamb=p=定值):
W=-p△V
(4)恒容过程(△V =0):
W=0
例如:在298.15 K时
1 2
H2
(g,
p
)
1 2
Cl2
(g,
p
)
HCl(
g,
p
)
反应焓变为:
r
H
m
(298
.15
K
)
92.31kJ.mol 1
这就是HCl(g)的标准摩尔生成焓:
f Hm (HCl, g,298.15K) 92.31kJ.mol1
10.可逆过程与可逆体积功
可逆过程:系统内部及系统与环境之间在无限接近平 衡条件下进行的过程,称为可逆过程,否则称为不可 逆过程。
生成物来表示反应进行的程度,所得的值都是相
同的,即:
d dnD dnE dnF dnG D E F G
当反应按所给反应式的系数比进行了一个单位的化 学反应时,即 nB / mol B,这时反应进度就是1mol。
《大学物理化学》知识点总结

第一章 理想气体1、理想气体:在任何温度、压力下都遵循PV=nRT 状态方程的气体。
2、分压力:混合气体中某一组分的压力。
在混合气体中,各种组分的气体分子分别占有相同的体积(即容器的总空间)和具有相同的温度。
混合气体的总压力是各种分子对器壁产生撞击的共同作用的结果。
每一种组分所产生的压力叫分压力,它可看作在该温度下各组分分子单独存在于容器中时所产生的压力B P 。
P y P B B =,其中∑=BBB B n n y 。
分压定律:∑=BB P P道尔顿定律:混合气体的总压力等于与混合气体温度、体积相同条件下各组分单独存在时所产生的压力的总和。
∑=BB V RT n P )/(3、压缩因子ZZ=)(/)(理实m m V V 4、范德华状态方程 RT b V V ap m m=-+))((2 nRT nb V Van p =-+))((225、临界状态(临界状态任何物质的表面张力都等于0)临界点C ——蒸气与液体两者合二为一,不可区分,气液界面消失; 临界参数:(1)临界温度c T ——气体能够液化的最高温度。
高于这个温度,无论如何加压 气体都不可能液化;(2)临界压力c p ——气体在临界温度下液化的最低压力; (3)临界体积c V ——临界温度和临界压力下的摩尔体积。
6、饱和蒸气压:一定条件下,能与液体平衡共存的它的蒸气的压力。
取决于状态,主要取决于温度,温度越高,饱和蒸气压越高。
7、沸点:蒸气压等于外压时的温度。
8、对应状态原理——处在相同对比状态的气体具有相似的物理性质。
对比参数:表示不同气体离开各自临界状态的倍数 (1)对比温度c r T T T /= (2)对比摩尔体积c r V V V /= (3)对比压力c r p p p /= 9、rr r c r r r c c c T Vp Z T V p RT V p Z =⋅=10、压缩因子图:先查出临界参数,再求出对比参数r T 和r p ,从图中找出对应的Z 。
厦门大学物理化学近年真题考点归纳
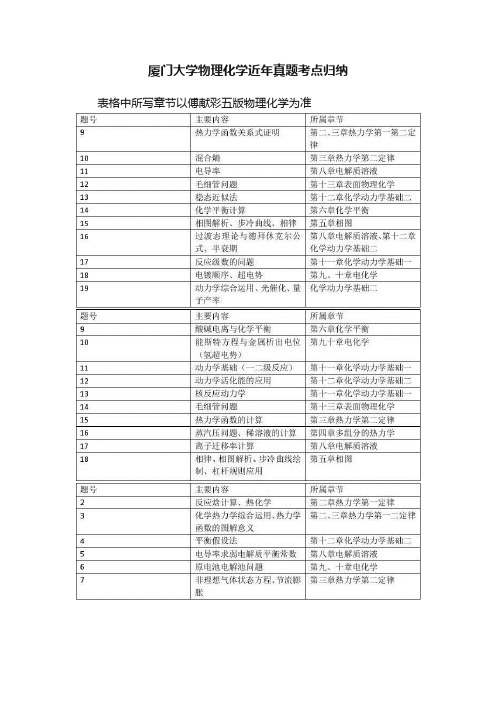
厦门大学物理化学近年真题考点归纳表格中所写章节以傅献彩五版物理化学为准
以上列了近几年厦门大学物理化学考试科目大题的主要考点。
厦门大学的物理化学不同于其他学校,他考察的题型比较单一。
一般12~16分的选择题,然后剩下的大概十道左右的大题。
首先,复习过程中一般使用傅献彩的物理化学课本,据悉厦大本校上课也是使用这本教材。
第一章气体的不用看,统计热力学一般就考一个选择,也可舍弃(明确说明只考概念),第十四章胶体近年来也只考选择,也可考虑舍弃。
厦大的物化热力学考察并非重点,但热力学函数的关系、麦克斯韦关系要会熟练推导,并要求熟悉各个函数的意义,今年来有向热化学、能源方面考察的趋势。
相图每年必考,且分值较大,考察的相图也较为常规,多进行几个典型相图的练习总结规律就行,步冷曲线也一般会要求绘制,杠杆规则的应用,并注意这部分可以和第四章结合考察。
化学平衡也几乎年年考,这部分相对简单。
电解质这一章本身就比较简单,一般是求电导率以及弱电解质平衡常数。
第九十章电化学每年必考大题,能斯特方程要熟练运用,注意超电势的问题以及电解过程中离子浓度的改变。
第十一十二章动
力学每年考察的比重比较大,常用的反应级数求解、稳态近似平衡假设的使用及其使用条件、过渡态理论中热力学函数与活化能的关系、重要的关系式的推导。
第十三章也年年考大题,开尔文公式、毛细现象,都很简单,但要注意浸润与不浸润时方程中R的正负(14年考的汞和玻璃,非常遗憾做错了)
最后,厦大物化最重要的参考书是孙世刚编写的物理化学的学习指导以及物理化学题库,历年真题很多出自上面。
要将上面的习题反复练习。
物理化学知识点总结

第一章 热力学第一定律一、基本概念系统与环境,状态与状态函数,广度性质与强度性质,过程与途径,热与功,内能与焓。
二、基本定律热力学第一定律:ΔU =Q +W 。
焦耳实验:ΔU =f (T ) ; ΔH =f (T ) 三、基本关系式1、体积功的计算 δW = -p e d V恒外压过程:W = -p e ΔV可逆过程:1221ln ln p p nRT V V nRT W ==2、热效应、焓等容热:Q V =ΔU (封闭系统不作其他功) 等压热:Q p =ΔH (封闭系统不作其他功) 焓的定义:H =U +pV ; d H =d U +d(pV )焓与温度的关系:ΔH =⎰21d p T T T C3、等压热容与等容热容热容定义:V V )(T U C ∂∂=;p p )(T H C ∂∂=定压热容与定容热容的关系:nR C C =-V p 热容与温度的关系:C p =a +bT +c’T 2 四、第一定律的应用1、理想气体状态变化等温过程:ΔU =0 ; ΔH =0 ; W =-Q =⎰-p e d V 等容过程:W =0 ; Q =ΔU =⎰T C d V ; ΔH =⎰T C d p 等压过程:W =-p e ΔV ; Q =ΔH =⎰T C d p ; ΔU =⎰T C d V 可逆绝热过程:Q =0 ; 利用p 1V 1γ=p 2V 2γ求出T 2,W =ΔU =⎰T C d V ;ΔH =⎰T C d p不可逆绝热过程:Q =0 ; 利用C V (T 2-T 1)=-p e (V 2-V 1)求出T 2,W =ΔU =⎰T C d V ;ΔH =⎰T C d p2、相变化可逆相变化:ΔH =Q =n Δ_H ;W=-p (V 2-V 1)=-pV g =-nRT ; ΔU =Q +W3、热化学物质的标准态;热化学方程式;盖斯定律;标准摩尔生成焓。
摩尔反应热的求算:)298,()298(B H H m f B m r θθν∆=∆∑反应热与温度的关系—基尔霍夫定律:)(])([,p B C T H m p BB m r ∑=∂∆∂ν。
物理化学基础知识总结上册

物理化学基础知识总结上册第一章热力学第一定律及其应用1.体系与环境:我们用观察,实验等方法进行科学研究时,必须先确定所要研究的对象,把一部分物质与其余的分开(可以是实际的,也可以是想像的)。
这种被划定的研究对象,就称为体系或系统,而在体系以外与体系密切相关,影响所能及的部分,则称为环境。
根据体系和环境之间的关系,可以把体系分为三种:体系完全不受环境的影响,和环境之间没有物质或能量的交换者,称为隔离体系或孤立体系;体系与环境之间没有物质的交换,但可以发生能量的交换者,称为封闭体系;体系不受上述限制,即体系与环境之间可以有能量以及物质交换者,称为敞开体系。
明确所研究的体系属于何种体系是至关重要的。
由于处理问题的对象不同,描述他们的变量不同,所适用的热力学公式也有所不同。
描述体系宏观性质的热力学变量可分为两类:广度性质(容量性质)和强度性质。
广度性质的数值与体系的数量成正比。
此种性质具有加和性,即整个性质的某种广度性质是体系中各部分该种性质的总和。
广度性质在数学上是一次齐函数。
强度性质此种性质不具有加和性,其数值取决于体系自身的特性,与体系的数量无关。
强度性质在数学上是零次齐函数。
体系的某种广度性质除以总质量或物质的量(或者把体系的两个容量性质相除)之后就成为强度性质。
若体系中所含物质的量是单位量,即一摩尔,则广度性质就成为强度性质。
2.热力学平衡态和状态函数:热平衡,力学平衡,相平衡,化学平衡。
当体系处于一定的状态时,其广度性质和强度性质都具有一定的数值。
但是体系的这些性质彼此之间是相互关联的,通常只需要指定其中的几个,其余的也就随之而定了。
也就是说,在这些性质之中只有部分是独立的。
体系的某些性质的改变只与始态和终态有关,而与变化时所经历的途径无关。
在热力学中,把具有这种特性的物理量叫做状态函数。
热和功与其改变的途径有关,是过程函数,从微观角度来说,功是大量质点以有序运动而传递的能量,热量是大量质点以无序运动方式而传递的能量。
物理化学课本重点全方位总结

第一章热力学第一定律1、热力学三大系统:(1)敞开系统:有物质和能量交换;(2)密闭系统:无物质交换,有能量交换;(3)隔绝系统(孤立系统):无物质和能量交换。
2、状态性质(状态函数):(1)容量性质(广度性质):如体积,质量,热容量。
数值与物质的量成正比;具有加和性。
(2)强度性质:如压力,温度,粘度,密度。
数值与物质的量无关;不具有加和性,整个系统的强度性质的数值与各部分的相同。
特征:往往两个容量性质之比成为系统的强度性质。
3、热力学四大平衡:(1)热平衡:没有热隔壁,系统各部分没有温度差。
(2)机械平衡:没有刚壁,系统各部分没有不平衡的力存在,即压力相同(3)化学平衡:没有化学变化的阻力因素存在,系统组成不随时间而变化。
(4)相平衡:在系统中各个相(包括气、液、固)的数量和组成不随时间而变化。
4、热力学第一定律的数学表达式:∆U = Q + W Q为吸收的热(+),W为得到的功(+)。
12、在通常温度下,对理想气体来说,定容摩尔热容为:单原子分子系统,V m C =32R 双原子分子(或线型分子)系统 ,V m C =52R 多原子分子(非线型)系统 ,V mC 632R R == 定压摩尔热容:单原子分子系统 ,52p m C R =双原子分子(或线型分子)系统 ,,p m V m C C R -=,72p m C R =多原子分子(非线型)系统 ,4p m C R =可以看出:,,p m V m C C R -=13、,p m C 的两种经验公式:,2p m C a bT cT =++ (T 是热力学温度,a,b,c,c ’ 是经,2'p m c C a bT T=++ 验常数,与物质和温度范围有关)14、在发生一绝热过程时,由于0Q δ=,于是dU Wδ=理想气体的绝热可逆过程,有:,V m nC dT pdV =- ⇒ 22,11lnln V m T V C R T V =- 21,12ln ,ln V m p V C Cp m p V ⇒= ,,p mV mC pV C γγ=常数 =>1. 15、-焦耳汤姆逊系数:J T T =()H pμ∂∂- J T μ->0 经节流膨胀后,气体温度降低;J T μ-<0 经节流膨胀后,气体温度升高; J T μ-=0 经节流膨胀后,气体温度不变。
物化上册知识点总结

物化上册知识点总结一、物理化学基本概念和原理1. 物理化学的范围和任务物理化学是研究物质的物理性质与化学性质之间的关系的一门科学。
其任务是探讨物质的结构和变化规律,揭示物质变化的机理。
2. 物理化学基本概念物理化学的基本概念包括物质、物理量、物态、物质的结构等。
其中,物质是构成世界一切事物的基本成分,具有质量和体积;物理量是用来描述物质的特性或者物理过程的量;物态是物质的存在状态,包括固态、液态和气态等;物质的结构是指物质内部组织和排列的方式。
3. 物理化学的基本原理物理化学的基本原理包括热力学、动力学、统计力学等。
热力学是研究能量转化和能量传递的规律以及物质变化过程的规律;动力学是研究物质变化速率和变化规律的科学;统计力学是研究大量微粒系统的宏观性质与微观结构之间的关系的一门学科。
二、热力学1. 热力学基本概念热力学的基本概念包括热力学系统、状态参量和热力学定律。
热力学系统是指能够发生能量交换的物理系统;状态参量是用来描述系统状态的参量,包括内能、焓、熵等;热力学定律包括热力学第一定律、热力学第二定律和热力学第三定律。
2. 热力学状态函数热力学状态函数是用来描述系统状态的函数,包括内能、焓、熵等。
这些状态函数在对恒定温度和压力下的过程中不随着时间的改变而改变。
3. 理想气体状态方程理想气体状态方程描述了理想气体的状态与压力、体积、温度之间的关系。
它可以用来描述气体在不同条件下的状态。
4. 热力学第一定律热力学第一定律描述了能量守恒的原理,即系统的内能增加等于系统所吸收的热量与所做的功之和。
5. 热力学第二定律热力学第二定律描述了系统熵的增加原理,即在热平衡状态下,熵增不可能减少,熵在不可逆过程中总是增加。
6. 热力学第三定律热力学第三定律描述了熵的极限原理,即在零温度下,系统的熵为零。
7. 热力学循环热力学循环是指在热机中热能和机械能相互转化的过程。
热力学循环包括卡诺循环、斯特林循环等。
三、溶液1. 溶解过程溶解过程是指溶质与溶剂之间相互作用并形成溶液的过程。
物理化学各章小结

第一章 气体本章小结1.理想气体状态方程 pV =nRT pV m =RT pV =(m /M ) RT气体的密度 ρ =m /V =pM /(RT ) 2. 道尔顿分压定律 B p p =∑B B n RTp V =BB p x p= B B p x p = 3. 实际气体的液化和临界点实际气体在临界温度以下通过加压可以被液化。
理想气体则不能。
临界温度T c 是实际气体能被液化的最高温度,在临界温度时使气体液化所需要的最小压力叫临界压力p c 。
在描述实际气体液化的p -V 图上,临界温度和临界压力所对应的点称为临界点。
0cT p V ∂⎛⎫= ⎪∂⎝⎭ 220cT p V ⎛⎫∂= ⎪∂⎝⎭ 临界温度和临界压力时所对应的体积称为临界摩尔体积V m,c 。
临界温度、临界压力和临界摩尔体积统称为临界参数,各种实际气体的临界参数可以在各类物理化学数据手册中查得。
4. 实际气体的范德华方程范德华研究了实际气体与理想气体产生偏差的两个因素-分子本身占有体积和分子间存在作用力,由此引入两个校正项,得适用于1mol 气体的范德华方程为()2m m a p V b RT V ⎛⎫+-= ⎪⎝⎭适用于n mol 气体的范德华方程为()22an p V nb nRT V ⎛⎫+-= ⎪⎝⎭公式中的a 和b 称为范德华常数,可以通过气体的临界参数计算2227,648c c ccR T RT a b p p == 符合范德华方程的气体称为范德华气体,范德华气体的玻意尔温度为,00B m B T p pV a T p Rb →⎛⎫∂=⇒=⎪∂⎝⎭5. 压缩因子与压缩因子图m pV pV Z RT nRT ==Z 称为压缩因子,Z >1,气体较难压缩,Z <1,气体较易压缩,Z =1,还原为理想气体。
Z 值可由对比温度(/c T T τ=)和对比压力(/c p p π=)通过压缩因子图查得。
查得Z 值后可用上述方程求算实际气体的p -V -T 。
厦门大学 物理化学 (上) 各章 知识点 总结

第1章第零定律与物态方程一、基本要点公式及其适用条件1.系统的状态和状态函数及其性质系统的状态—就是系统物理性质和化学性质的综合表现,它采用系统的宏观性质来描述系统的状态,系统的宏观性质,也称为系统的"状态函数"。
系统的宏观性质(状态函数)—就是由大量(摩尔级)的分子、原子、离子等微观粒子组成的宏观集合体所表现出的集团行为,简称"热力学性质"或“热力学函数”如p、V、T、U、H、S、A、G 等。
Z=f(x,y)表示一定量、组成不变的均相系统,其任意宏观性质(Z)是另两个独立宏观性质(x,y)的函数。
状态函数Z具有五个数学特征:(1),状态函数改变量只决定于始终态,与变化过程途径无关。
(2),状态函数循环积分为零,这是判断Z是否状态函数的准则之一。
(3),系Z的全微分表达式(4),系Z的Euler 规则,即微分次序不影响微分结果。
(5),系Z、x、y满足循环式,亦称循环规则。
2.热力学第零定律即热平衡定律:当两个物态A和B分别与第三个物体C处于热平衡,则A和B之间也必定彼此处于热平衡。
T =t+273.15,T是理想气体绝对温标,以"K"为单位。
t是理想气体摄氏温标,以"℃"为单位。
绝对温标与摄氏温标在每一度大小是一样的,只是绝对温标的零度取在摄氏温标的-273.15℃处,可以看出,有了绝对温标的概念后,只需确定一个固定参考点(pV)0p=0,依国际计量大会决定,这个参考点选取在纯水三相点,并人为规定其温度正好等于273.16K。
3.理想气态方程及其衍生式为:;式中p、V、T、n单位分别为Pa、m3、K、mol;R=8.314J〃mol-1〃K-1,V m为气体摩尔体积,单位为m3〃mol-1,ρ 为密度单位kg〃m-3,M 为分子量。
此式适用于理想气或近似地适用于低压气。
4.理想混合气基本公式(1)平均摩尔质量;式中M B和y B分别为混合气中任一组份B 的摩尔质量与摩尔分数。
物理化学各章概念总结、公式总结电子版1___电化学

7.1 重要概念、方法及注意事项
1.关于物质的量的基本单元 物质B的物质的量nB正比于B的特定单元的数目NB,即nB=(1/L)NB,其中L为阿伏加德 罗(Avogadro)常数。这种特定单元叫做基本单元,它可以是分子、原子、离子、原子团、电 子、光子及其他粒子或这些粒子的任意特定组合。在讨论电解质溶液导电性质时,为了讨论
问题方便,使公式表示形式简单,人们常以一个元电荷为基础指定物质的基本单元,这样,
相同物质的量的不同物质便具有相同的电关系。例如,1 mo1 的Na+,Cl-,
1 2
Ca
2+
,
1 3
PO43−
等,它们都带有 96500C 的电量。若某强电解质依下式电离
Mν + Aν − → ν + M z+ +ν − Az−
和Λm来计算离子的λB。应用这些公式时,均应以一个元电荷为基础来指定电解质和离子的 基本单元。
13.
α = Λm Λ∞m
式中。是弱电解质的电离度,Λm和Λm∞分别为弱电解质溶液的实际摩尔电导率和极限摩尔电
导率。此式是用电导法测定弱电解质电离常数的依据。它表明,可通过测定弱电解质溶液的
t+ = u+
t− u−
在多电解质溶液中,任意两种离子 i 和 j 的迁移数,不仅取决于它们的电迁移率,而且与它
们的浓度有关,
ti = uici
t j ujcj
其中ci和cj分别代表i和j的浓度,它们的基本单元应以一个元电荷为基础来指定。
4.
n(电解质↓)=n(离子迁出)
或
n(电解质↑)=n(离子迁入)
指定电解质和离子的基本单元。
物理化学各章节总结

物理化学每章总结第1章 热力学第一定律及应用1.系统、环境及性质热力学中把研究的对象(物质和空间)称为系统,与系统密切相关的其余物质和空间称为环境。
根据系统与环境之间是否有能量交换和物质交换系统分为三类:孤立系统、封闭系统和敞开系统。
性质⎩⎨⎧容量性质强度性质2.热力学平衡态系统的各种宏观性质不随时间而变化,则称该系统处于热力学平衡态。
必须同时包括四个平衡:力平衡、热平衡、相平衡、化学平衡。
3.热与功 (1) 热与功的定义热的定义:由于系统与环境间温度差的存在而引起的能量传递形式。
以Q 表示,0>Q 表示环境向系统传热。
功的定义:由于系统与环境之间压力差的存在或其它机、电的存在引起的能量传递形式。
以W 表示。
0>W 表示环境对系统做功。
(2) 体积功与非体积功功有多种形式,通常涉及到是体积功,是系统体积变化时的功,其定义为:V p W d δe -=式中e p 表示环境的压力。
对于等外压过程 )(12e V V p W --= 对于可逆过程,因e p p =,p 为系统的压力,则有V p W V V d 21⎰-=体积功以外的其它功,如电功、表面功等叫非体积功,以W ′表示。
4.热力学能热力学能以符号U 表示,是系统的状态函数。
若系统由状态1变化到状态2,则过程的热力学增量为 12U U U -=∆对于一定量的系统,热力学能是任意两个独立变量的状态函数,即 ),(V T f U = 则其全微分为V V U T T U U TVd d d ⎪⎭⎫⎝⎛∂∂+⎪⎭⎫ ⎝⎛∂∂=对一定量的理想气体,则有0=⎪⎭⎫⎝⎛∂∂TV U 或 U =f (T ) 即一定量纯态理想气体的热力学能只是温度的单值函数。
5.热力学第一定律及数学表达式 (1) 热力学第一定律的经典描述① 能量可以从一种形式转变为另一种形式,但在转化和传递过程中数量不变。
② “不供给能量而可连续不断做功的机器称为第一类永动机,第一类永动机是不可能存在的。
物理化学上册知识点总结

第一章:气体1、掌握理想气体的状态方程( )及分压力、分体积等概念,会进行简单计算2、理解真实气体与理想气体的偏差及原因,了解压缩因子Z 的定义及数值大小的意义,熟悉范德华方程(理想气体基础上引入压力、体积修正项)第二章:热力学第二定律;第三章:热力学第三定律(2个计算题)1、系统性质(广度、强度性质)2、状态函数特性(如:异途同归,值变相等;周而复始,数值还原及在数学上具有全微分的性质等)3、热力学第一定律:ΔU =Q+W (Q 、W 取号的规定及各种过程对应计算)4、恒容热、恒压热及之间的关系式,能进行简单计算,掌握焓的定义式,会应用赫斯定律5、掌握各种不同过程的热力学函数计算(单纯PVT 变化时自由膨胀、等温、等压、等容及绝热可逆或不可逆等过程的U 、H 、A 、G 、S 等函变以及正常、非正常相变过程U 、H 、A 、G 、S 等函变计算(状态函数法)6、理解理想气体的一些性质(如U 、H 仅为温度函数、Cp 与Cv 的差值及单原子、双原子理想气体的C V ,m 和绝热可逆过程过程方程式等)、实际气体—节流膨胀过程(等焓过程,了解焦-汤系数等)7、反应进度8、如何由标准摩尔生成焓、燃烧焓计算标准摩尔反应焓变以及相关规定9、反应焓变与温度的关系(基尔霍夫定律)10、自发过程及其共同特征;热力学第二定律文字描述11、卡诺循环、卡诺定理、热机效率;熵的定义式及克劳修斯不等式12、判断过程可逆性及自发变化方向的各种判据13、了解热力学第三定律,掌握根据规定熵、标准摩尔生成焓、标准摩尔生成吉布斯函变计算化学变化过程中对应函数的变化值14、热力学函数间的关系及麦克斯韦关系式的应用(应用于各函数间的相互计算以及一些证明),了解各函数特征变量15、了解Clapeyron 方程,掌握Clausius-Clapeyron 方程各种形式第四章:多组分系统热力学及其在溶液中的应用(1个计算题)1、熟悉偏摩尔量、化学势表示,了解偏摩尔量加和公式和吉布斯-杜亥姆公式,掌握相平衡、化学平衡条件2、了解各种不同情况化学势的表达式,假想标准态等概念3、掌握稀溶液中两个经验定律:拉乌尔、亨利定律表达式及简单计算4、掌握理想液态混合物的通性5、了解依数性的一些结论第五章:化学平衡(1个计算题)1、会表示任意化学反应的标准平衡常数、其它各种平衡常数,并能相互换算2、熟悉化学反应等温方程,并能应用其判断反应方向3、掌握范特霍夫方程各种形式并进行相关计算4、了解温度、压力等各种因素对化学平衡影响的相关结论5、掌握使用标准平衡常数定义式以及热力学相关公式进算平衡组成的计算。
物理化学各章总结

XB
X nB
T, p,
nC
B
GB
G nB
T, p,nC
多 组 分 系 统
化学式 的表达 式
X=f(T,p,n) 偏摩尔量
μB(g)
μ B(g)
RT
ln
pB p
p
1、NH4HS(s)放入真空容器中,并与其分解产物NH3(g)和H2S(g)达到平衡, 则该系统中组分数C= ;相数P= ;自由度F= 。
2、A及B二组分组成的凝聚体系能生成三种稳定的化合物,则 于常压下在液相开始冷却的过程中,最多有 2 种固相同时 析出? 3、单组分系统相图中,点、线、面的自由度数分别 为0 、1 、2 。
2、通常情况下,对于二组分物系能平衡共存的最多相为 D , 最大自由度是 C 。
A1 B2 C3 D4
3、体系中含有H2O、H2SO4·4H2O、H2SO4·2H2O、H2SO4·H2O 、H2SO4 ,其组分数C为 B 。
A 1 B2 C3 D 4
2、1 mol蔗糖(C12H22O11)溶于3 mol水中,蔗糖溶液的蒸汽压 是水的蒸气压的(C)。 A.1/3 B.1/4 C.3/4 D.不知道
学 表 达
U = Q + W 适用于封闭系统
单纯pVT变化过程 T2 QV U n CV ,mdT
T1
T2
H
nC p ,mdT
T1
相变化过程
T2
Hm T2 Hm T1 Cp,m dT
T1
式
化学反应过程 dξ dnB/νB 标准态
大学课程《物理化学》各章节知识点汇总

A (p1V1)
p
D
A S1(等 温)C S2 (等容) B
S
S1
S2
nR ln
V2 V1
C T2
T1 V
dT T
A S '1(等温) D S '2(等压) B
S S '1 S '2 nR ln
p1 p2
C T2
T1 p
dT T
B (p2V2)
T2
C T1
V
dU TdS pdV
dH TdS Vdp (
dU TdS pdV
U S
V ,ni
T
,
U V
S ,ni
p
G U pV TS
dU dG pdV Vdp TdS SdT
dG SdT Vdp BdnB
B
dU TdS pdV BdnB
B
dU TdS pdV
U
nB
dnB
S ,V ,n j B
U B
某系统经一过程由状态1变为状态2之后,如果采用任何 方法都无法使系统和环境都完全复原,则该过程为不可 逆过程。
准静态压或膨胀过程,如果没有因摩擦而造成能量 损失等情况下就是一可逆过程。
可逆过程的主要特点: 1.可逆过程是以无限小的变化进行,系统始终无限接近 平衡态。 2.系统在可逆过程中作最大功,环境在可逆过程中作最 小功即可逆过程效率最高。
B
dH TdS Vdp BdnB
B
dF SdT pdV BdnB
B
dG SdT Vdp BdnB
B
纯理想气体的化学势
p
T
Gm p
T
Vm
d Vmdp
(T , p) (T ) RT ln p
大学物理化学上册知识点归纳

第一章气体的pvT关系一、理想气体状态方程pV=(m/M)RT=nRT(1.1)或pVm=p(V/n)=RT(1.2)式中p、V、T及n的单位分别为P a 、m3、K及mol。
Vm=V/n称为气体的摩尔体积,其单位为m3·mol。
R=8.314510J·mol-1·K-1称为摩尔气体常数。
此式适用于理想,近似于地适用于低压下的真实气体。
二、理想气体混合物1.理想气体混合物的状态方程(1.3)pV=nRT=(∑BBn)RTpV=mRT/Mmix(1.4)式中Mmix为混合物的摩尔质量,其可表示为Mmix def ∑BBy M B(1.5)Mmix=m/n=∑BBm/∑BBn(1.6)式中MB为混合物中某一种组分B的摩尔质量。
以上两式既适用于各种混合气体,也适用于液态或固态等均匀相混合系统平均摩尔质量的计算。
2.道尔顿定律pB=nBRT/V=yBp(1.7)P=∑BBp(1.8)理想气体混合物中某一种组分B的分压等于该组分单独存在于混合气体的温度T及总体积V的条件下所具有的压力。
而混合气体的总压即等于各组分单独存在于混合气体的温度、体积条件下产生压力的总和。
以上两式适用于理想气体混合系统,也近似适用于低压混合系统。
3.阿马加定律V B *=nBRT/p=yBV(1.9)V=∑VB*(1.10)VB*表示理想气体混合物中物质B 的分体积,等于纯气体B在混合物的温度及总压条件下所占有的体积。
理想气体混合物的体积具有加和性,在相同温度、压力下,混合后的总体积等于混合前各组分的体积之和。
以上两式适用于理想气体混合系统,也近似适用于低压混合系统。
三、临界参数每种液体都存在有一个特殊的温度,在该温度以上,无论加多大压力,都不可能使气体液化,我们把这个温度称为临界温度,以Tc 或tc表示。
我们将临界温度Tc时的饱和蒸气压称为临界压力,以pc表示。
在临界温度和临界压力下,物质的摩尔体积称为临界摩尔体积,以Vm,c 表示。
物理化学总结(上册)ppt课件

•
基尔霍夫定律
• 已知某一温度条件下的反应焓变, 可利用基尔霍夫 定律求任意温度条件下的反应焓变.
• 基尔霍夫定律的微分式:
• ( rH/T)p= rCp • 基尔霍夫定律的不定积分式:
•
rHm(T)=∫rCp,mdT+I
• 基尔霍夫定律的定积分式:
•
rHm(T2)= rHm(T1)+ ∫T1T2rCp,,mdT
• 0: 标准状态(温度为T, 压力为1p0)下理想气体化学势.
.
• 实际气体及其逸度:
• = 0+RT ln(f/p0) • f=p
• f:气体的逸度(fugacity); • :逸度系数(fugacity coefficiant).
• 逸度的计算: • ∫RTdlnf=∫Vmdp • lnf=lnp*+1/RT[pVm-RT-∫p*ppdVm] • 范德华气体的逸度: • lnf=ln(RT/(Vm-b))+b/(Vm-b) -2a/RTVm
rHm(T1)=- H1=∫T1T2 Cp(产物)dT
一般可取反应的初始温度T1为298.15K, 有:
rHm(298.15K)=- H1=-∫298..15KT2 Cp(产物)dT 可解出T2•热力学第二定律
• 热力学第二定律是决定自然界一切过程方向与限 度的基本规律.
• Clauxius表述:
•
力平衡(p相同)
•
热平衡(T相同)
•
相平衡
•
化学平衡
• 状态函数: 只取决于体系平衡态的热力学量.
•
如: T,p,V,U,H,S,F,G,n
• 过程量: 与体系经历的过程有关的量.
•
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
第1章第零定律与物态方程一、基本要点公式及其适用条件1.系统的状态和状态函数及其性质系统的状态—就是系统物理性质和化学性质的综合表现,它采用系统的宏观性质来描述系统的状态,系统的宏观性质,也称为系统的"状态函数"。
系统的宏观性质(状态函数)—就是由大量(摩尔级)的分子、原子、离子等微观粒子组成的宏观集合体所表现出的集团行为,简称"热力学性质"或“热力学函数”如p、V、T、U、H、S、A、G 等。
Z=f(x,y)表示一定量、组成不变的均相系统,其任意宏观性质(Z)是另两个独立宏观性质(x,y)的函数。
状态函数Z具有五个数学特征:(1),状态函数改变量只决定于始终态,与变化过程途径无关。
(2),状态函数循环积分为零,这是判断Z是否状态函数的准则之一。
(3),系Z的全微分表达式(4),系Z的Euler 规则,即微分次序不影响微分结果。
(5),系Z、x、y满足循环式,亦称循环规则。
2.热力学第零定律即热平衡定律:当两个物态A和B分别与第三个物体C处于热平衡,则A和B之间也必定彼此处于热平衡。
T =t+273.15,T是理想气体绝对温标,以"K"为单位。
t是理想气体摄氏温标,以"℃"为单位。
绝对温标与摄氏温标在每一度大小是一样的,只是绝对温标的零度取在摄氏温标的-273.15℃处,可以看出,有了绝对温标的概念后,只需确定一个固定参考点(pV)0p=0,依国际计量大会决定,这个参考点选取在纯水三相点,并人为规定其温度正好等于273.16K。
3.理想气态方程及其衍生式为:;式中p、V、T、n单位分别为Pa、m3、K、mol;R=8.314J·mol-1·K-1,V m为气体摩尔体积,单位为m3·mol-1,ρ 为密度单位kg·m-3,M 为分子量。
此式适用于理想气或近似地适用于低压气。
4.理想混合气基本公式(1)平均摩尔质量;式中M B和y B分别为混合气中任一组份B 的摩尔质量与摩尔分数。
此式既适用于各种混和气,也适用于液态或固态等均相系统的平均摩尔质量计算。
(2)道尔顿定律;这里p B只作为组份B单独存在时产生的压力。
此式适用混合理想气或近似适用于低压混和气。
(3)分压力定义与;作为数学定义可适用各种混和气(4)阿马格定律;适用以混合理想气体或近似适用于低压混和气(5)分体积定义与;可适用于混合理想气或近似适用于低压真实混和气5.范德华方程,范氏常数与临界参数关系,范氏对比态方程(1)范德华方程为:or 式中a和b系与气体种类有关的常数,皆称范德华常数。
a的单位为Pa·m6·mol-2,b的单位为m3·mol-1;该方程适用于几个MPa(几十个atm)的中压范围内实际气体的p、V、n的计算(2),,;式中V cm、P c、T c分别为各种气体的临界摩尔体积、临界压力、临界温度,简称临界参数(3),。
;式中P r、T r、V r分别为对比压力、对比温度、对比体积,简称对比参数,意指物质离开临界点的远近(4);系普遍化范氏对比态方程,其适用范围同范德华方程,并无改善。
6.对应态原理与压缩因子图的应用(1);意指不同气体,若有两个对比状态参数彼此相等,则第三个对比状态参数大体上具有相同的值,并称为处于"对应状态"。
处对应态时,不同物质间的物理性质具有简单关系,此经验规律,即"对应态原理"。
(2);为压缩因子Z的定义式,它表示实际气与理想气的偏差,完全由试验测定,是无量纲的纯数。
Z与气体T、p及性质有关,规定T r可实验绘制Z=f(p r)函数图。
故Z=f(T r、p)称"压缩因子图",不受任何限制,可用于高压下实际气的p、V、T及物质逸度、热容、焓等热r力学函数计算。
7.力学响应函数定义及其应用体积膨胀系数;等温压缩系数;压力系数;α、к、β一般是T、p的函数,均为强度量,但他们彼此关联,且与物态方程可互为转换。
他们是研究物质热性质、晶体结构及相变的重要数据。
第2章热力学第一定律一、本章基本要点公式及其适用条件1.热力学能U及其与热Q、功W相互转换关系(1)U是系统的状态函数,其绝对值仍不可知,对一定量定组成系统,可表为任意俩独立变量x、y的函数关系U =f(x,y),U具备五个数学特征为:o U改变量取决于始终态;o循环积分为零;o可表全微分o符合Euler 规则;o满足循环式(2)第一定律数学表达式。
针对封闭系统:ΔU =Q+W;绝热过程:ΔU =W =W v+W′,;绝功过程:ΔU =Q;等容无其他功:ΔU =Q v(3) Q、W是非状态函数,不仅与始终态有关,更与过程途径相关。
只有在特定限制条件下Q、W与某些状态函数改变量相关联时,仅决定于始终态。
2.可逆过程的本质意义是系统复原时不留下永久性变化的过程,或言之系统与环境之间强度因子相差无穷小时所经历一系列平衡态过程。
可逆过程是平衡态过程,但平衡态过程不一定是可逆过程。
可逆过程是理想化概念,其效率最高,是实际过程的极限。
3.焓H的定义及其与U、Q的关系焓是系统状态函数,定义为H =U+PV,对一定量定组成系统可表为函数关系为H =f(x,y),H同样具有五个数学特征,即焓改变量只决定于始终态,其微变之循环积分为零,可表为全微分,符合Euler 规则,满足循环式。
H与Q在概念上有本质的区别:前者为系统的属性,后者与过程途径有关,唯有等压无其他功时:ΔH =Q p4.等容热容C v与等压热容C p的意义特点及其之间转换关系;;C pm与T关系呈级数展开式,常表为C pm =a+bT +cT2 or C pm =a +bT +c'T -2;热容差为:,体现热响应函数与力学响应函数及物态方程相互间的关联。
5.热力学第一定律在pVT变化、理想气体及相变化中的应用:等容过程:适用于真实气体、液固体及理想气体pVT变化适用反抗恒外压P e的体积功计算等压过程:适用于真实气体、液固体及理想气体pVT变化绝热过程:适用于理想气体且不管其可逆与否等温过程:理想气体绝热可逆过程:PV r=常数,TV r-1=常数,T r P 1-r=常数,热容商相变化过程:Δ相H=Q p适用于等T等P的相变W v =-P(Vβ-Vα)适用于等T等P,由α→β相变的体积功适用于当β相为气相且为理想气体条件6.节流膨胀和焦耳-汤姆生效应作为第一定律在实际气体应用体现1.节流膨胀定义:较高压力下的流体(气或液)经多孔塞(或节流阀)向较低压力方向绝热膨胀过程。
2.节流膨胀过程特点是节流前后焓值相等:H1=H2或ΔH=0。
3.焦-汤系统定义式:,因为dp <0,所以表示流体经节流后(1)温度升高(致热),(2)温度不变,(3)温度降低(致冷)。
值得指出在T=f(p)函数图中的等焓线非节流过程所经历的途径。
7.热力学第一定律在化学变化中的应用0.反应进度ξ 必须针对相应的化学计量方程式而言,其定义为:或,Δξ 或ξ 的单位为mol。
1.物质热力学标准态新规定,标准状态压力为标准压力P=100KP a,右上角“”表示标准态的符号。
气体的标准态:无论是纯气B 或是混合气组份B,均是温度为T、压力为P下并表现理想气体特性的气体纯物质B 的(假想)状态;液体(或固体)的标准态;无论纯液(或固)体B,还是其混合物中的组份B,都是温度T、压力P下液(或固)体纯物质B 的状态;物质的热力学标准态温度T是任意的,未作具体规定,但常用热数据的标准态温度为T =298.15K2.化学反应的标准摩尔热力学能变、标准摩尔焓变以及它们之间相互转换。
3.;(为计量方程式中组份B的计量系数)4.,为T.p下物质B 的摩尔焓绝对值;5.6.热化学方程式:注明具体反应条件(T.P.β,焓变值)的化学反应方程式7.盖斯定律:一个化学反应,不管是一步完成或经数步完全,反应的总标准摩尔焓变是相同的,或称为、“热总值不变定律”。
以此可利用热化学方程式的线性组合,由若干已知反应的标准摩尔焓变,求另一反应的反应标准摩尔焓变。
8.利用各种热数据和克希荷甫方程进行各种过程热的计算0.汽化焓、熔化焓与升华焓三种相变焓之间关系为:1.由标准摩尔生成焓变计算反应焓变为:2.由原子化焓和键焓估算生成焓变和反应焓变:3.4.5.其中n i为反应中单质i的原子个数,n j为化合物中A-B 原子键合类的数目。
6.由标准摩尔燃烧焓变计算反应焓变为:7.由克希荷甫方程计算反应焓变和相变焓为:8.9.9.解题思路与基本方法:先明确系统始终态、历经过程途径及应求的未知量,若求状态函数改变量,则应设计合理的过程途径,勾画出框框图。
继而依据已知物理量数据诸条件,同时应注意给定的特殊系统(如理想气体)、特征过程(如绝热等),以便直接应用其结论,尽量让隐蔽条件浮出水面。
如此参考题目的背景材料,再科学取舍最佳热力学公式并查数据表。
最后运算并检验结果的合理性。
第3章热力学第二定律一、基本要点公式及其应用条件1.热力学第二定律的经典表述与实质说法克劳修斯说法:热不能自动地由低温物体传到高温物体。
开尔文说法:不可能从单一热源吸热使之完全转化为当量的功,而不留下其它变化。
实质说法:一切自发过程(实际过程)都是不可逆的。
2.卡诺定理:,系指工作于两个固定温度热源间的任何热机,其效率都不可能超过可逆卡诺机的。
3.熵的定义。
熵以符号S表示,是系统的状态函数且为广度量,定义为为可逆过程中系统吸收的微量热。
4.热力学第二定律的数学表达式即克劳修斯不等式:式中,对不可逆过程应取用不等号,指系统实际过程热,T指环境温度,对可逆过程应取用等号,指可逆过程热,T为系统温度。
5.熵增原理及熵判据(1)熵增原理:(2)环境(su)熵变计算:;其中环境温度T sy恒定,而Q sy指系统实际过程热。
(3)熵判据6.系统熵变的计算(由熵的定义式计算)(将S视为x,y双变量函数,则由全微分计算)(1)P、V、T变化熵变的计算①液体或固体的P、V、T变化等压变温过程:等容变温过程:等温下的P、V变化过程:②理想气体的P、V、T变化(2)相变化熵变计算①在相平衡温度(T e)压力(P e)下的相变②在非相平衡T、P下的相变,为不可逆相变;应设计可逆途径。
如:即所设计的每一步途径均为可逆且有相应公式及数据,则过程总熵变为各步熵变之和。
7.熵的统计意义波尔兹曼熵定理:S=klnW,其中k为波尔兹曼常数,W为热力学概率或微态数,其蕴意"混乱度,无序度",熵值即"混乱度"的量度。
8.热力学第三定律综合表述及数学表达式。