药物的含量测定方法 PPT
合集下载
维生素C片剂的含量测定及杂质检查方法.ppt

比色法
…
所需试剂
固红Al盐试液
0.25%水溶 液(临用新
配)。
维生素C标准溶 液
取维生素C标 准品适量,用 0.5%的柠檬 酸水溶液配制 成5~ 250g/mL的 维生素C溶液。
维生素C样品液
精密取含维 生素C的样 品适量,用 0.5%的柠檬 酸水溶液配 制成约 20g/mL的 溶液备用。
1
2
3
高效液
相色谱法
…
色谱条件
色 谱 柱 : Inertsil ODS - 3,4.6×250mm( GL Sciences hc )。
流动相:己烷磺酸钠溶液(取0.94g己烷磺酸钠置 1000mL量瓶中,加冰醋酸10mL,加水稀释至刻 度,摇匀)-甲醇 6∶4 ,pH =3.1。
流速: 0.8mL/min
在其中一个比色管中加入一定量的维生素C标准
2
溶液,在另一个比色管中不加维生素C标准溶液;
用二次蒸馏水定容,摇匀。
用水作参比,1cm比色皿于265nm处,分别测
3
定试剂空白的吸光度A1和溶液的吸光度A2,并计
算⊿A=A2-Al。
阻抑光度法
在H2SO4-KBr介 质中,维生素C对碘 酸钾氧化氨基苯磺酸 黄的退色反应有抑制 作用,且抑制作用的 强弱与维生素C的含 量之间呈良好的线性 关系,基于这一点建 立了检测食物中维生 素C的阻抑光度法。
–加新沸过 的冷水目 的是为减 少水中溶 解的氧对 测定的影 响。
–滴定前要 进行必要 的前处理, 片剂溶解 后取续滤 液测定 , 注射液测 定时要加 2mL丙酮 。
0
原理
固红Al盐与维生素C在 酸性介质中反应。在碱性 介质中即生成一种蓝色物 质。该物质在630nm处有 最大吸收。而固红Al盐试 液与维生素C溶液在该范 围内均没有吸收峰。
(优选)药物分析课件第四章药物的含量测定方法

b
2
(二)容量分析法的有关计算
• 3.含量计算方法:直接滴定法与间接滴定法 • 直接滴定法
含量(%)= V T 100% W
• 在实际工作中,滴定液的浓度可能与药典规定的 浓度并不一致,所以应该进行校正。
F
实际摩尔浓度 规定摩尔浓度
实际摩尔浓度=F
规定摩尔浓度
(二)容量分析法的有关计算
• 直接滴定法的含量计算方法
直接读取 药典规定 自己标定
含量(%)= V T F 100% W
取样量
(二)容量分析法的有关计算
(1)生成物滴定法
(1)生成物滴定法
• 示例三 葡萄糖酸锑钠的含量测定
本法先用葡萄糖酸锑钠与碘离子反应生成碘,再用碘与硫代 硫酸钠反应生成碘离子,相当于是用硫代硫酸钠在滴定葡萄 糖锑钠,因此要找出它们两者的量反应关系,也就是a和b。
• 示例四 司可巴比妥钠的含量测定
取本品约0.1g,精密称定,置250mL碘瓶中,加水10mL,振 摇使溶解,精密加溴滴定液(0.05mol/L) 25mL,再加盐酸 5mL,立即密塞,并振摇1min,在暗处放置15min后,注 意微开瓶塞,加碘化钾试液10mL,立即密塞,摇匀后,用 硫代硫酸钠滴定液(0.1mol/L)滴定,至终点时,加淀粉指 示液,继续滴定至蓝色消失,并将滴定得的结果用空白试验 校正。
精密称取本品0.3630g加稀盐酸回流1小时后,放冷,用亚 硝酸钠液(0.1010mol/L)滴定,用去20.00mL。每1mL 亚硝酸钠液(0.1mol/L)相当于17.92mg的C10H13O2N, 请计算非那西丁的含量。
W 被测药物反应的质量 A WB 被测药物的摩尔质量 aM A bM B
滴定液反应的质量 滴定液的摩尔质量
巴比妥类药物—巴比妥类药物含量测定(药物分析课件)

取装量差异项下的内容物,混合均匀,精密称取适量(约相当于硫
喷妥钠0.25g),置500ml量瓶中,加水稀释至刻度,摇匀,量取此溶
液用0.4%氢氧化钠溶液定量稀释制成每1ml中约含5μg的溶液;另取
硫喷妥对照品,加0.4%氢氧化钠溶液溶解并定量稀释制成每1ml中约
含5μg的溶液。照分光光度法,在304nm波长处测定吸收度,根据每
药物分析技术
1. 直接测定的紫外分光光度法 本法是将供试品溶解后,根据溶液的pH,在最大吸收波
长(λmax)处,直接测定对照品溶液和供试品溶液的吸收 度,再计算药物的含量。中国药典对注射用硫喷妥钠的含 量测定采用本法。 Beer-Lambert定律
A = -㏒T =ElC A∝C
药物分析技术
以注射用硫喷妥钠的含量测定为例:
5 1.095
C对
A样 A对
药物分析技术
含量测定结果计算如下:
D% V空 V样 f T 100%
W T cM
n 0.1mol L 260.27g mol
2 13.01mg ml
药物分析技术
注意事项: (1)操作中要防止溴和碘的逸失。 (2)平行条件进行空白试验可以减少溴和碘
逸失带来的误差。
药物分析技术
(三)酸碱滴定法(酸量法)
药物分析技术
例如:异戊巴比妥的含量测定
取本品约0.5g,精密称定,加乙醇20ml溶解后,加麝香草酚 酞指示剂6滴,用氢氧化钠滴定液(0.1mol/L)滴定,并将滴 定 结 果 用 空 白 试 验 校 正 , 即 得 。 每 1ml 氢 氧 化 钠 滴 定 液 (0.1mol/L)相当于22.63mg的C11H18N2O3。
含量测定计算公式:
D% V f T 100% W
药物分析-含量测定结果计算ppt课件

× 100%
分光光度法——(2)对照品法
A E Cl
1 % 1 cm
A A
ቤተ መጻሕፍቲ ባይዱ
对
供
C C
对
供
A C C A
供 供 对
对
标示量%=
_ A供 ×C对 ×D ×W 100% A 对× W × 标示量
盐酸吗啡片的含量测定方法:取本品20片,精密称定,重量 为2.0246g,研细,精密称取0.1108g,置100ml量瓶中,加水 50ml,振摇,使盐酸吗啡溶解,摇匀,滤过,精密量取续滤 液15ml,置50ml量瓶中,加0.2mol/L氢氧化钠溶液25ml, 加水稀释至刻度,摇匀,作为供试品溶液;另精密称取吗啡 对照品适量,用0.1mol/L 氢氧化钠溶液溶液配制成每1ml含 20.18μg的溶液作为对照品溶液。取上述两种溶液照紫外分 光光度法在250nm处测定吸收度。A对=0.453, A供=0.562 已知:标示量为10mg/片 盐酸吗啡(C17H19NO3· HCl· 3H2O)分子量375.85 吗啡(C17H19NO3)分子量285.34 求:供试品中盐酸吗啡(C17H19NO3· HCl· 3H2O)的含量?
0.312 ×1000 ×0.2408/20 ×100 =105.7 标示量%= 323 ×100 ×0.0110 ×0.01
第四节 含量测定结果计算
对乙酰氨基酚片的含量测定: 取 本 品 ( 规 格 为 0.5g/ 片 )10 片 , 精 密 称 定 为 5.2500g ,研细,精密称取 0.0450g ,置 250ml 量 瓶中,加 0.4% 氢氧化钠溶液 50ml 及水 50ml ,振 摇15分钟,加水至刻度,摇匀,用干燥滤纸滤过, 精密量取续滤液 5ml ,置 100ml 量瓶中,加 0.4% 氢氧化钠溶液10ml,加水至刻度,摇匀,照分光 光度法在 257nm 波长处测定吸收度为 0.610 。已 知对乙酰氨基酚的吸收系数 (E1%1cm) 为 715 ,求对 乙酰氨基酚片的标示百分含量。
维生素类药物含量测定2021精选PPT

❖若(A328校正-A328)*100%/A328 所得的数值在±3.0%,则仍不用校正 公式计算吸收度。可直接用A328代入E=A/C.L
❖若(A328校正-A328)*100%/A328 所得的数值 在-15.0%到-3.0%,则应用A328校正代入E=A/C.L
❖若(A328校正-A325%或大于+3% ,则不能用本法测定,应使用第二法。
A 2 = A3 =6/7A 1 1= 325nm, 2=310nm, 3 =334nm Ch.P用于测定维生素A醇
一、维生素A含量测定及方法
3. 杂质的吸收 • 对V.A有影响的杂质主要有以下几种: • (1) V.A2 和V.A3 • (2)维生素A的氧化产物 • (3) 维生素A在光照下产生的无活性的聚合物 • (4)维生素A的异构体等。
② 物质对光的吸收具有加和性。
一、维生素A含量测定及方法
2.波长的选择:最大吸收波长,及两侧各选一波长 ① 第一法(等波长差法) 1 =328nm,
2 =316nm, 3 =340nm,△ =12nm VitA的max(328nm) Ch.P用于测定维生素A醋酸酯
一、维生素A含量测定及方法
② 第二法(等吸收比法) 分别在1的两侧各选一点
一、维生素A含量测定及方法
立体异构体 氧化产物及光照产物 合成中间体 去氢维生素A( VitA2) 去水维生素A( VitA3)
均对测定有干扰 ,故采用三点校 正法
一、维生素A含量测定及方法
1:测定原理,三个波长的吸收度,公式计算校正吸收度, 再计算含量。故得名。该法的依据:
① 杂质的吸收在310~340nm波长范围内呈一条直线,且 随波长的增大吸收度减小;
一、维生素A含量测定及方法
❖若(A328校正-A328)*100%/A328 所得的数值 在-15.0%到-3.0%,则应用A328校正代入E=A/C.L
❖若(A328校正-A325%或大于+3% ,则不能用本法测定,应使用第二法。
A 2 = A3 =6/7A 1 1= 325nm, 2=310nm, 3 =334nm Ch.P用于测定维生素A醇
一、维生素A含量测定及方法
3. 杂质的吸收 • 对V.A有影响的杂质主要有以下几种: • (1) V.A2 和V.A3 • (2)维生素A的氧化产物 • (3) 维生素A在光照下产生的无活性的聚合物 • (4)维生素A的异构体等。
② 物质对光的吸收具有加和性。
一、维生素A含量测定及方法
2.波长的选择:最大吸收波长,及两侧各选一波长 ① 第一法(等波长差法) 1 =328nm,
2 =316nm, 3 =340nm,△ =12nm VitA的max(328nm) Ch.P用于测定维生素A醋酸酯
一、维生素A含量测定及方法
② 第二法(等吸收比法) 分别在1的两侧各选一点
一、维生素A含量测定及方法
立体异构体 氧化产物及光照产物 合成中间体 去氢维生素A( VitA2) 去水维生素A( VitA3)
均对测定有干扰 ,故采用三点校 正法
一、维生素A含量测定及方法
1:测定原理,三个波长的吸收度,公式计算校正吸收度, 再计算含量。故得名。该法的依据:
① 杂质的吸收在310~340nm波长范围内呈一条直线,且 随波长的增大吸收度减小;
一、维生素A含量测定及方法
第七章--药物含量测定-幻灯片

×100%
供试品重量(g)×(1-水分或干燥失重百分数)
制剂含量=
实测的供试品量 供试品的标示量
×100%
标 示 量
含量测定的基本规则
1、所用器具均应校正后使用;所用试液均应按药典规定配 制;
2、称取或量取药品的量应符合规定要求; 3、称量挥发性或吸湿性的物质,必须用密封性好的容器进
行称量操作 4、测定必须排除干扰(专属性); 5、结果起码要平行测定两次,合格,其结果应在允许的相
例:丙酸倍氯米松的百分含量
M样=12.60mg A样=4487 C内标=0.1223mg/ml A内标’=2042
M对=12.50mg A对=4346 C内标=0.1223mg/ml A内标=2027
97.0-103.0%
取样量:0.3003g; 滴定液浓度:0.1005mol/L;
99.1%
用量:9.33ml;
空白试验用量:0.10ml;T:32.09mg/ml
滴定法测含量
❖ 1,直接滴定(水杨酸的含量测定) ❖ 2,需做空白的直接滴定(盐酸异丙嗪的含量测定) ❖ 3,片剂的含量滴定(布洛芬片的含量测定) ❖ 4,注射剂的含量滴定(安乃近注射液的含量测定) ❖ 5,剩余滴定法(氯贝丁酯的含量测定)
引入——阿司匹林的[含量测定]项
标准用的什么方法? 如何计算
内容
1
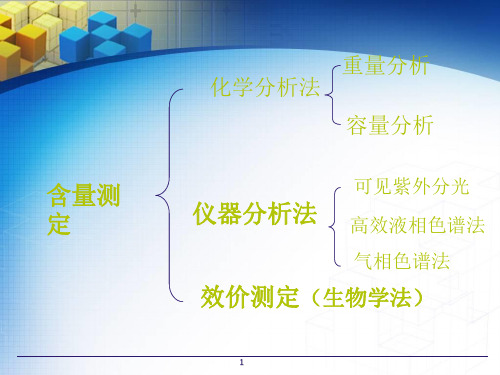
概述
2
容量分析法
3 紫外可见分光光度法
4 高效液相色谱法
概述
解决以下四个问题?
1、含量测定的重要性? 2、常用的含量测定方法? 3、含量的表示方法? 4、含量测定的基本规则?
概述——含量表示方法
中药
含量计算公式
实测的供试品量(g) 原料药含量=
药物的含量测定方法ppt课件
23
(2)测含量(用样品与内标物)
含量(Cx
)
f
Ax AS' / CS'
As’:内标物质峰面积或者峰高 Ax: 供试品的峰面积或者峰高 Cs’: 内标物质浓度 f:校正因子
24
示例一 炔诺酮的含量测定
照高效液相色谱法(附录ⅤD测定)
色谱条件与系统适用性试验 用十八烷基硅烷键 合硅胶为填充剂;以甲醇-水(65:35)为流动相, 检测波长为244nm。理论塔板数按炔诺酮峰计 算不低于1500,炔诺酮峰与内标物质峰的分离 度及主成分峰后一杂质峰与主成分峰的分离度均 应符合要求。 内标溶液的制备 取黄体酮约25mg,精密称定, 置25ml量瓶中,加甲醇溶解并稀释至刻度。
12
二、光谱分析法
红外分光 度法
光谱分析法
药典 收载
火焰光度 紫外可见 荧光分析
法
分光度法
法
原子吸收分 光光度法
13
紫外可 见分光
度法
基于物质对紫外区 (200~400nm)和可见光 区(400~760nm)的单色 光辐射建立起来的光谱
分析法
基 础
朗伯-比尔定律
A lg 1 Ecl T
c(g
W测 0.13263 VitC% = W样 = 0.1332 =99.57%
10
又例 非那西丁含量测定
精密称取本品0.3630g加稀盐酸回流1小时后,放冷, 用亚硝酸钠滴定液(0.1010mol/L)滴定,用去 20.00ml。每1ml亚硝酸钠液(0.1mol/L)相当于 17.92mg的C10H13O2N,请计算非那西丁的含量。
25
测定法 取本品约25mg,精密称定,置25ml量瓶
中,加甲醇溶解并稀释至刻度,摇匀,精密量取
药物分析课件-第四章_药物的含量测定方法[专业内容]
![药物分析课件-第四章_药物的含量测定方法[专业内容]](https://img.taocdn.com/s3/m/d864265ace2f0066f53322ed.png)
T m a M 0.05 1176.13 8.806(mg / mL)
b
1
高等教育
10
(二)容量分析法的有关计算
T (mg / mL) m a M b
• 示例二 异烟肼的滴定度
• 溴酸钾法测定异烟肼含量[M(C6H7N3O)=137.14], C溴酸钾=0.01667mol/L
3C6H7N3O 2KBrO3 3C6H5NO2 3N2 2KBr 3H2O
b
1
含量(%)=(VB0 VBS ) FB TA 100% W
高等教育
22
讨论
• 在剩余量滴定法中,第一滴定液与第二滴 定液的浓度是相当的。
• 如上例中,第一滴定液为溴滴定液 (0.05mol/L),第二滴定液为硫代硫酸 钠滴定液(0.1mol/L)。
• Br2~I2~2Na2S2O3
高等教育
23
• 示例三 葡萄糖酸锑钠的含量测定
本法先用葡萄糖酸锑钠与碘离子反应生成碘,再用碘与硫代 硫酸钠反应生成碘离子,相当于是用硫代硫酸钠在滴定葡萄 糖锑钠,因此要找出它们两者的量反应关系,也就是a和b。
• 0.3g供试品+水100mL+盐酸15mL+碘化钾试液 10mL——密塞,振摇后暗处放置10min——碘量法
分含量较高的样品分析。
• 适用范围:
• 广泛应用于化学原料的测定,较少应用于药物制剂的测定。
高等教育
6
(二)容量分析法的有关计算
• 滴定度:每1mL规定浓度的滴定液所相当 的被测药物质量。《中国药典》用毫克 (mg)表示。
高等教育
7
(二)容量分析法的有关计算
• 滴定度的计算
aA bB cC dD 用B滴定A
药物分析维生素C含量测定PPT课件

与文献数据的比较
文献数据收集
收集相关文献资料,获取不同研究或生产条件下维生素C含量的测 定结果。
数据对比分析
将实验结果与文献数据进行对比分析,了解本实验测定结果的可靠 性、准确性和一致性。
差异原因探讨
探讨实验结果与文献数据存在差异的原因,为后续研究提供改进方 向和思路。
06
结论与展望
结论总结
维生素C含量测定在药物分析 中具有重要意义,能够确保药 品质量和安全。
详细描述
荧光分光光度法利用某些荧光物质与维生素C的特异性反应, 生成荧光产物,通过检测荧光产物的强度来推算维生素C的含 量。该方法具有高灵敏度和选择性,适用于对痕量维生素C的 测定。
微生物学方法
总结词
微生物学方法是利用微生物的生长与维生素C之间的依赖关系来测定维生素C含量的方法。
详细描述
微生物学方法利用某些对维生素C有依赖性的微生物,在含有不同浓度维生素C的培养基中生长,通过测定培养基 中微生物的数量或代谢产物来推算维生素C的含量。该方法具有较好的准确性和可靠性,但操作较为繁琐,需要 一定的实验条件和技术水平。
详细描述
高效液相色谱法利用不同物质在固定相和流动相之间的分配差异进行分离,通 过检测器检测待测物质的信号,从而确定维生素C的含量。该方法具有高分辨率 和准确度,适用于多种药物制剂中维生素C的测定。
荧光分光光度法
总结词
荧光分光光度法是一种灵敏度较高的维生素C含量测定方法, 通过荧光物质与维生素C的特异性反应来检测。
维生素C的来源和摄取
来源
富含维生素C的食物包括柑橘类水 果、草莓、番茄、绿叶蔬菜等。
摄取量
根据膳食指南,成年人每天需要 摄取约100-200毫克维生素C。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
取上述两种溶液各10μl进样,测得对照品和内标峰分别为1350 和1040,供试品峰和内标峰分别为1313和1013。求SD的含量。
解:由于对照品和供试品稀释倍数相同,所以直接按质量代 替浓度计算
As/Cs
As/Cs
=f=
Ar/mr
Ax/mx
1040/0.20
1013/0.20
=f=
1350/50.0
(2)测含量(用样品与内标物)
含量(Cx
)
f
Ax AS' / CS'
As’:内标物质峰面积或者峰高 Ax: 供试品的峰面积或者峰高 Cs’: 内标物质浓度 f:校正因子
示例一 炔诺酮的含量测定
照高效液相色谱法(附录ⅤD测定)
色谱条件与系统适用性试验 用十八烷基硅烷键 合硅胶为填充剂;以甲醇-水(65:35)为流动相, 检测波长为244nm。理论塔板数按炔诺酮峰计算 不低于1500,炔诺酮峰与内标物质峰的分离度及 主成分峰后一杂质峰与主成分峰的分离度均应符 合要求。 内标溶液的制备 取黄体酮约25mg,精密称定, 置25ml量瓶中,加甲醇溶解并稀释至刻度。
银量法
AgNO3 NH4SCN AgSCN NH4NO3
返滴定
2、酸水解法
十一烯酸锌 稀 盐酸十一烯酸+氯化锌
EDTA配位滴定
第二节 定量分析样品的前处理方法
(三)、经还原分解后测定法 适用范围:碘原子直接与芳环连接的含碘有机化合物。
泛影酸
COOH
I
I
+ 11 NaOH + 3 Zn
1
T
=m×
a b
×M=0.05× 1 1
×176.13=8.806(mg/ml)
1
M=176.13
T(mg/ ml) m a M b
示例二 异烟肼的滴定度
溴酸钾法测定异烟肼含量[M(C6H7N3O)=137.14], C溴酸钾=0.01667mol/L
3C6H7N3O 2KBrO3 3C6H5NO2 3N2 2KBr 3H2O
T
=m×
a b
×M=0.01667×
3 2
a
×137.14=3.429(mg/ml)
b
滴定含量计算
实际C
取用 倍数
浓度校 正因数
F=
滴定
规定C
度
W测= N F T V
消耗 体积
浓度校 正因数
例:VitC的含量测定:精密称取本品0.1332g,置锥形
瓶中,加稀醋酸与新沸过的冷蒸馏水的混合液
(1:10)110ml, 加淀粉批示液1ml,用碘滴定液
)
207×1
[例]精密称取VitB12供试品25.0mg,加水溶解并稀释 制成1000mL,于1cm厚的吸收池中,在361nm处测 得吸收度为0.507,已知其百分吸收系数为207,求其 百分含量。
用A算出( E
1%
1cm)供
,再用其与已知(E
1%
1cm )标
比值
解:C= m/V = 25×10-3/1000(g/ml)= 2.5×10- 3(g/100ml)
测定法 取本品约25mg,精密称定,置25ml量瓶中,
加甲醇溶解并稀释至刻度,摇匀,精密量取此溶
液2ml与内标溶液3ml,置10ml量瓶中,加甲醇稀
释至刻度,摇匀,取20μl注入液相色谱仪,记录
色谱仪,记录色谱图;另取炔诺酮对照品适量,
精密称定,同法测定。按内标法以峰面积计算,
即得。
测得量
As / Ar 样品 As / Ar 对照品
专属性差
药物的含量测定
适用于灵敏度 要求较高,样 本量较大的分
析项目
三种 分析 方法
光谱分 析法
特点
简便快速、灵敏度 高,有一定的准确 度,但专属性稍差
色谱分 析法
适用于专 属性和灵 敏度要求 较高的复
杂样品
特点
灵敏度与专属性都高, 有一定的准确度,但结
果需要对照品
第一节 定量分析方法的分类与特点
容量分析法 亦称 滴定法
定 义
将已知浓度的滴定液,由滴定管滴加到被 测药物的溶液中,直至滴定液与被测药物 反应完全,然后根据滴定液消耗的浓度和 被消耗的体积,按化学计量关系式计算出 被测药物的含量。
特点
1.方法简便易行:仪器价廉易得,操作简便快速 2.方法耐用性高:影响测定的试验条件与环境因素少 3.测定结果准确:误差低于0.2% 4.方法专属性差:对相近杂质干扰缺少选择,适用 于主成分含量较高的样品(如原料药)分析
Ax E11c%m 100
含量% =(——E—11%c—m —)供
E (
1% 1cm
)标
药典 记载
[例] 已知VitB12在361nm处的百分吸收系数为 207,现测得其水溶液的吸收度为0.414,吸收
池厚度为1cm,求其浓度。
A
解:C=
E
1% 1cm
L
0.414 = ————
= 0.00200( g单/10位0?ml
CH3COHN
NHCOCH3
I COOH
回流
H2N
+ 33NNaaOI H + 2 CH3COONa +3 Na2ZnO2 + 3 H2O NH2
NaI + AgNO3
AgI + NaNO3
第二节 定量分析样品的前处理方法
三、经有机破坏的分析方法
(一)湿法破坏
含氮、硫、硫柳汞、氯化钠的生物制品、含氮有机合
解:C= m/V = 8.2×10-(4 g/100ml)
(
E
1% 1cm
)
供=
A
/
CL
= 0.580 / 8.2×10-4 = 707.3
VitB12%
(= —E—11—%cm—)供 E ( 1% )标
1cm
= 707.3/715 = 98.93%
(二)荧光分析法
特点: 灵敏度高,可达10-10~10-12g/ml 要求低浓度下测定 须做空白测定 荧光弱的药物可衍生化后测定
(
E
1% 1cm
)
供=
A
/
CL
= 0.507 / (2.5×10-3×1)
= 202.8
VitB12%
(= —E—11—%cm—)供 E ( 1% )标
1cm
= 202.8/207 = 97.97%
[例]用UV法测定对乙酰氨基酚的含量:精密称取本品 0.0410g,置250ml量瓶中,加0.4%NaOH溶液50ml, 溶解后,加水于刻度,摇匀。精密量取5ml,置100ml量瓶 中, 加0.4%NaOH溶液10ml,溶解后,加水至刻度,摇匀。 在257nm处测得吸收度为0.580,按其百分吸收系数 715计算,求其百分含量。
滴定液 浓度
T(mg/ ml) m a M b
药物分 子量
T(mg/ ml) m a M b
示例一 计算VitC的滴定度T
碘量法测定VitC含量M(C6H6O6)=176.13, C(I2)=0.05mol/L
m=0.05 C6H8O6 I2 C6H6O6 2HI
mol/L
= 1×—0.00—.5005—14×8.806×10-3 ×15.02=0.13263(g) VitC的百分含量(%)
W测 0.13263 VitC% = W样 = 0.1332 =99.57%
又例 非那西丁含量测定
精密称取本品0.3630g加稀盐酸回流1小时后,放冷, 用亚硝酸钠滴定液(0.1010mol/L)滴定,用去 20.00ml。每1ml亚硝酸钠液(0.1mol/L)相当于 17.92mg的C10H13O2N,请计算非那西丁的含量。
直接测定 氧化还原滴定法 水解后测定 酸水解
碱水解
经还原分解后测定
经有机 破坏
湿法破坏——凯氏定氮法 干法破坏 高温灼烧法
氧瓶燃烧法
第二节 定量分析样品的前处理方法
(一)、直接测定法 适用范围:金属原子不直接于碳原子相连或某些C-
M键结合不牢固的有机金属药物。 葡萄糖酸钙 EDTA配位滴定 富马酸亚铁 邻二氮菲-硫酸铈滴定 葡萄酸锑钠 间接碘量法
1313/mx
解之,得: mx=49.924(mg)
SD%=49.924/50.5=98.86%
2.外标法
含量(Cx)=CR
×
Ax AR
(二) GC
内标法 校正因子f As / Cs AR / CR
含量(Cx
)
f
Ax AS' / CS'
第二节 定量分析样品的前处理方法
不经有 机破坏
配位滴定法
E1% 1cm
本法紫外区须选用 石英比色皿,比色 皿一般为1cm,吸 光度在0.3~0.7之 间为宜,选用最大 吸收光波长。
紫外可见 分光度法
方法特 点与适 用范围
本法比较少应用于 原料药的含量测定, 可用于制剂的含量 测定,更多的应用 于生物制剂的定量 检查
1.简便易行:本法使用的仪器价格比较低廉,操 作简单,易于普及
消耗 体积
(0.05014mol/L)滴定至蓝色,用去15.02ml,每1ml的
碘液(0.05mol/L)相当于8.806mg C6H8O6,计算本 品是否符合药典规定的含量限度。药典规定含
C6H8O6 不得低于99.0%。
滴定
度
计算步骤
0.1332样品中VitC含量(W测)
W测= N F T V
第一节 定量分析方法
容量分析法有 关计算