变性疾病
变性手术疾病

变性手术疾病变性是一种个体的生物学性别与心理性别发生倒错的心理现象变性手术可以使易性癖者生物学上的性别与其心理性别协调全都他们厌恶自己的身体若被恋爱及工作方面的问题所困扰时心理长期处于应激状态使他们表现出一些负性心情反应如:心情不稳定焦虑与恐惊;悲观与孤独等。
症状精确地表达该疾病的特征,变性不是主观所为,而应当是生物学因素所致;癖则是指后天养成的一种习惯。
众多的病例资料表明,变性是与生俱来的,并非积久成习,像这样把常人畏之如虎、避之不及而又遍体鳞伤的手术当成幸福、当成幸运,这也不行能是癖。
鉴别1.异装癖者对自己的生物学性别持确定态度,并无变性要求,属性行为特别,多见于男性,性定向为异性,对性交有爱好,着异性服饰带有性快感追求成分,甚而消失性兴奋或性感满意。
偶有要求变性者,但在明白变性手术实质后,多远而避之不再求医。
有一异装癖者;取得妻子认同,每月着异装,装扮成女性逛街一次,而家庭性关系均正常,近来告知其尚有裹足癖,观赏女性三寸金莲,经7年多交往观看,其为一典型异装癖患者,而本人在律师工作方面很是精彩。
2.同性恋者对自我性别认同,无变性要求,性定向指向同性,是对他人的感觉,以同性个体作为性爱对象。
同性恋多以男性为多见,他们有自己的生活圈,往往结成团伙活动在公园、公厕、公共浴池,以鸡奸为性爱方式,多数人扮演着主动、被动两种角色,性行为完全是宣泄性欲,固定的同性恋情侣很少。
同性恋者并不愿着异装,即使是扮作异性姿势的一方,仍注意同性气质,更无变性的渴望。
女性同性恋者大多是情感型,她们相爱甚深,有时不易与易性病相区分,还有待于深化讨论与甄别。
并发症易性癖病的手术象其他外科手术一样,也会消失一些并发症。
总体上分为两大类: ⑴手术本身引起的并发症;⑵与病例选择不当或手术效果不佳有关的并发症。
生育问题变性手术的男女严格地说都未从生理上、解剖学上和功能上完全实现女性化或男性化。
男变女或女变男都没有生殖力量,这是变性手术将来要解决的一个难题。
临床医学变性的名词解释

临床医学变性的名词解释临床医学变性是指人体出现某种疾病或病变,导致其生理机能、身体特征或行为方式发生明显改变的过程。
在临床医学中,变性是一个广泛的概念,涵盖了许多不同的疾病和条件。
一、变性的概念和原因变性是指个体的性别认同与其生理性别不一致的情况。
性别认同是指一个人内心深处感知自己是男性、女性还是其他的强烈感受。
当个体的性别认同与其生理性别相矛盾时,就会出现变性的现象。
这可能导致性别认同障碍,使得个体产生身份认同困惑和情绪困扰。
变性的主要原因是多样化的。
在生物学上,染色体异常、荷尔蒙水平异常或性腺发育畸形等因素可能导致个体出现变性。
心理社会因素也可能通过对个体性别认同的影响而引起变性。
无论是生物因素还是心理因素,都可能对变性起到一定的作用。
二、变性的分类根据变性的具体条件,我们可以将变性分为多个分类。
其中,最常见的变性是性别焦虑障碍。
这是指个体对自己的生理性别感到极度不满意,出现严重的身份认同困惑和不适感。
对于这些患者,临床医生可以提供心理咨询、荷尔蒙治疗和手术干预等综合治疗方法。
此外,睾丸移植是另一个常见的变性。
睾丸是男性生殖器官,当个体出现男性外生殖器异常或性别认同困惑时,临床医生可以考虑进行睾丸移植手术,以改变生理性别特征,促进个体的性别认同。
三、变性的治疗方法对于患有变性的患者,临床医生在诊断后可以根据具体情况制定个体化的治疗方案。
一般来说,治疗变性的方法包括心理咨询、荷尔蒙治疗和手术干预等。
心理咨询可以帮助患者理解和接受他们的身份认同困惑,并学会适应和处理相关问题。
荷尔蒙治疗是指通过荷尔蒙补充来改变个体身体特征,以匹配其性别认同。
对于男性变女性的患者,激素治疗可以促进乳房发育和皮肤变软;对于女性变男性的患者,激素治疗可以促进胡须生长和肌肉发育。
手术干预是变性治疗中的最后一步,主要用于改变个体体格和生殖器官,以与其性别认同相一致。
这些手术包括女性变男性的乳房切除术、卵巢和子宫摘除术,以及男性变女性的乳房重建术和性器官整形术等。
神经系统变性病---综述

2021年4月
概述
神经系统变性疾病( neurodegenerative diseases) 是一组原因不明的慢性进行性损害 中枢神经系统和周围神经系统等组织的疾病。
许多变性疾病是神经组织在衍化、发育、成熟、衰老等过程中发生于分子生物学水平 的一系列复杂变化,进而表现为结构和功能等方面的障碍。
目前常用的神经变性疾病的分类是基于临床症候群的分类,然而随着神经病理研究的 进展,人们发现不同的神经系统变性疾病有着相同或相似的病理改变。
如帕金森病和路易体痴呆的主要病理改变均是以a-突触核蛋白为主要成分的路易小体, 因此一起被称为a-突触核蛋白病(a-synucleinopathy)。
神经系统变性疾病的分类系统也日益受到挑战。
近年来,随着分子生物学研究的进展,人们发现同一种神经系统变性疾病可以有不同的分 子生物学改变,而不同的神经系统变性疾病的发病可以基于相同的分子生物学改变。
如行为异常型额颞叶痴呆最常见的分子生物学改变是tau蛋白的异常沉积,然而研究发 现有10%的临床诊断为行为异常型额颞叶痴呆患者的分子致病基础是β-淀粉样蛋白的异常 聚集,这一部分患者又可以归为阿尔茨海默病的额叶变异型。
目前常用的神经变性疾病的分类是基于临床症候群的分类,然而随着神经病理研究的
进展,人们发现不同的神经系统变性疾病有着相同或相似的病理改变。
行为异常型额颞叶痴呆、进行性核上性麻痹和皮层基底节变性的主要病理改变均是tau
蛋白的过度磷酸化和异常沉积。因此一起被称为tau蛋白病( tauopathy)。
此外还有β-淀粉样蛋白病( B-amyloidopathy)、TDP-43蛋白病( TDP-43 proteinopathy)
目前对这一系列的动态变化及其机制尚未完全认识。
神经系统变性疾病16681ppt课件

物的种子可能是引起ALS的一个原因。最近的证据显示,真 正的原因可能是食用一种蝙蝠。这种蝙蝠以苏铁类植物种子 为食,并且很可能使它们体内的毒素累积到有毒水平。当关 岛上的蝙蝠消耗量下降时,ALS的发病率也下降了。
.
免疫因素:尽管MND患者血清曾检出多种抗 体和免疫复合物,如抗甲状腺原抗体、GM1 抗体和L-型钙通道蛋白抗体等,但尚无证据 表明这些抗体可选择性以运动神经元为靶细 胞。目前认为,MND不属于神经系统自身免 疫病。
.
病毒感染:由于MND和急性脊髓灰质炎均侵 犯脊髓前角运动神经元,且少数脊髓灰质炎 患者后来发生MND,故有人推测MND与脊髓 灰质炎或脊髓灰质炎样病毒慢性感染有关。 但ALS患者CSF、血清及神经组织均未发现 病毒或相关抗原及抗体。
.
.
.
.
.
.
.
.
.
.
.
.
韩国电影《我的爱在我身边》
.
“身体慢慢地凝固,走向死亡, 这样的我,
你也可以在身边吗?”
.
.
.
.
.
.
.
.
思考
.
阿尔茨海默病
(Alzheimer’s Disease AD)
罗纳德.里根在1994年 的11月5日宣布他被诊 断为阿尔茨海默病: “我最近被告知我是 美国将患阿尔茨海默 病的人中的一员。目 前,我感觉良好。我 打算在上帝赐予我的 有生之年,一如既往 地做我的事情。我还 将和我的爱妻南希和 全家一起在生命的旅 途上行进。我打算多 享受些野外生活的乐 趣并与我的朋友和支 持者们保持联系!”
有人有人推测与pron、 HIV感染有关
第二章 第九节 脑变性病

肾上腺脑白质营养不良
• 性连锁隐性遗传疾患,男孩最多见。 – 肾上腺皮质功能不全; – 脑白质进行性髓鞘脱失; – 饱和长链脂肪酸在细胞内的病理性堆积。
肾上腺脑白质营养不良
• 病因:酰基辅酶A合成酶缺乏,长链脂肪酸在脑 白质及肾上腺皮质内沉积,造成脑白质脱髓鞘及 肾上腺皮质萎缩。
• 临床表现:3-14岁起病,主要表现有行为异常并 伴有视、听障碍和智力低下。病程呈现进行性恶
化直至死亡,一般不超过9年。
影像学表现
• 多位于侧脑室三角区周围对称性发布 • 病变常为大片长T1长T2信号
• 双侧病变通过胼胝体压部相连,呈现典型“蝶翼”
状
• 由后向前发展 • 桥-延脑皮质脊髓束纤维Wallerian变性,在非
典型病例诊断有特殊意义
• 增强扫描:活动期病灶边缘部分可以出现强化, 非活动期病灶不强化。
1
• “四十四只石狮子”
• 我给您一张纸请您按我说的去做,现在开始:“用右手拿着这张纸,
用两只手将它对折起来,放在您的大腿上。”
3
• 请您念一念这句话,并且按照上面的意思去做。
1
• 您给我写一个完整的句子。(句子必须有主语,动词,有意义)1
• (见背面)这是一张图,请您在同一张纸上照样把它画下来。 1
1H MRS
正常老年人海马区MRS
轻度认知障碍 海马区 MRS
来源:钱丽霞(博士论文)
AD患者,海马体积缩小,MRS MI峰升高 NAA峰降低 来源:钱丽霞(博士论文)
mI增高的原因可能为:
①与脑内淀粉蛋白沉积和神经纤维缠结有关; ②与神经胶质反应性增生有关; ③AD病人大脑皮层和海马等结构有广泛神经元缺失,
• 按起病年龄及临床征象,可分为婴儿型(60%~70% )、 少年型和成年型,
神经系统变性病.ppt

鉴别诊断
①脊髓性颈椎病:有感觉障碍,无脑干受累。EMG多无神 经源性改变,特别是舌肌和胸锁乳突肌。CT、MRI可发现颈 椎管狭窄,脊髓受压。ALS的MRI晚期为脊髓萎缩。
②延髓和脊髓空洞:进展更缓慢,节段性分离性痛温觉 障碍,MRI可显示空洞存在。
③MMN:中青年发病,缓慢进展,不对称性肌无力及肌萎 缩。EMG为周围神经节段性、多灶性传导阻滞。GM1抗体滴度 高,免疫治疗有效。
• 反射异常 • 痴呆 • 感觉异常:躯体感觉和特殊感觉。 • 自主神经功能障碍
神经变性病的实验室检查
• MRI、CT:结构性改变。 • fMRI、MRS、 PET:功能性改变。 • 神经电生理:EEG 、EP、EMG。 • 生化、内分泌、免疫、代谢筛查等 • 分子生物学检查 • 病理检查
主要类型及特征
分型
*肌萎缩侧索硬化 (amyotrophic lateral sclerosis,ALS)
*进行性肌萎缩 (progressive muscular atrophy,PMA)
*进行性延髓麻痹 (progressive bulbar palsy,PBP) *原发性侧索硬化 (primary lateral sclerosis,PLS)
1 最常见,40岁以后多,男多于女,3~5%有家族史,慢 性起病、缓慢进行性过程,平均病程3~5年。
2 多单侧起病,远端→近端,上、下运动神经均受累。 3 运动障碍:远端小肌肉→近端→肢带→躯干→球部及
呼吸肌。活动不灵、无力、僵硬、运动不协调等。 4 肌肉萎缩:多自手肌萎缩开始,呈爪形手,肌张力低。
• 氧化应激:中脑自由基清除系统异常
• 线粒体损害、细胞凋亡、兴奋性氨基酸毒性、钙 超载、免疫异常。
病理
神经病学-神经系统变性疾病

第一节 运动神经元病
临床表现 通常起病隐匿,缓慢进展 临床表现为肌无力与肌萎缩、锥体束征的不同组 合
1、进行性脊肌萎缩 (PMA) 2、进行性延髓麻痹 (PBP) 3、原发性侧索硬化 (PLS) 4、肌萎缩侧索硬化(ALS)
第一节 运动神经元病
临床表现
1. 肌萎缩侧索硬化( ALS)
最多见的类型 ➢ 首发症状:手指活动笨拙、无力,手部小肌肉
第一节 运动神经元病
病因及发病机制 1. 感染和免疫 ALS患者CSF免疫球蛋白升高 血中T细胞数目和功能异常,免疫复合物形成 抗神经节苷脂抗体阳性
2. 金属元素
MND患者有铝接触史,血浆和CSF中铝含量增高
第一节 运动神经元病
病因及发病机制 3. 遗传因素 铜(锌)超氧化物歧化酶基因 TAR DNA结合蛋白基因突变 4. 营养障碍
病理
➢ 肉眼:脊髓萎缩变细 ➢ 光镜:脊髓前角细胞、大脑皮质运动区锥体细胞
变性脱失;脑干运动神经核变性 ➢ 泛素化包涵体:存在于患者的神经元细胞胞质内,
其主要成分为TDP-43 ,➢ 脊神经前根变细,轴索断裂,髓鞘脱失,纤维减少 ➢ 锥体束自远端向近端发展,出现脱髓鞘和轴突变性 ➢ 肌肉呈现失神经支配性萎缩 ➢ 晚期,体内其它组织如心肌、胃肠道平滑肌亦可出
鉴别诊断
1、颈椎病
➢肌萎缩常局限于上肢,多见手肌萎缩,不像 ALS那样广泛 ➢常伴上肢或肩部疼痛 ➢客观检查常有感觉障碍 ➢可有括约肌障碍 ➢无延髓麻痹表现
第一节 运动神经元病
鉴别诊断
2、脊髓空洞症
➢节段性感觉分离现象 ➢MRI检查可鉴别脊髓或延髓空洞症、脑干或 颈髓肿瘤以及颈椎病
第一节 运动神经元病
第一节 运动神经元病
病理-神经变性疾病
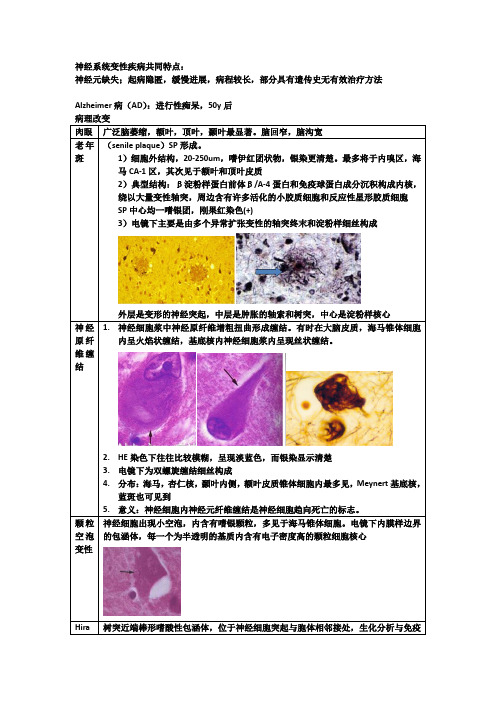
神经系统变性疾病共同特点:神经元缺失;起病隐匿,缓慢进展,病程较长,部分具有遗传史无有效治疗方法Alzheimer病(AD):进行性痴呆,50y后广泛脑萎缩,额叶,顶叶,颞叶最显著。
脑回窄,脑沟宽(senile plaque)SP形成。
1)细胞外结构,20-250um,嗜伊红团状物,银染更清楚。
最多将于内嗅区,海马CA-1区,其次见于额叶和顶叶皮质2)典型结构:β淀粉样蛋白前体β/A-4蛋白和免疫球蛋白成分沉积构成内核,绕以大量变性轴突,周边含有许多活化的小胶质细胞和反应性星形胶质细胞SP中心均一嗜银团,刚果红染色(+)3)电镜下主要是由多个异常扩张变性的轴突终末和淀粉样细丝构成外层是变形的神经突起,中层是肿胀的轴索和树突,中心是淀粉样核心1.神经细胞浆中神经原纤维增粗扭曲形成缠结。
有时在大脑皮质,海马锥体细胞内呈火焰状缠结,基底核内神经细胞浆内呈现丝状缠结。
2.HE染色下往往比较模糊,呈现淡蓝色,而银染显示清楚3.电镜下为双螺旋缠结细丝构成4.分布:海马,杏仁核,颞叶内侧,额叶皮质锥体细胞内最多见,Meynert基底核,蓝斑也可见到5.意义:神经细胞内神经元纤维缠结是神经细胞趋向死亡的标志。
神经细胞出现小空泡,内含有嗜银颗粒,多见于海马锥体细胞。
电镜下内膜样边界的包涵体,每一个为半透明的基质内含有电子密度高的颗粒细胞核心树突近端棒形嗜酸性包涵体,位于神经细胞突起与胞体相邻接处,生化分析与免疫组化证实为肌动蛋白,多见于海马锥体细胞,是一种老化现象的标志广泛的神经下拨脱失和突触减少:黑质细胞变性和脱失;残留神经细胞中Lewy小体形成位于胞浆内,HE染色显示圆形,中心嗜酸性着色,折光性强。
边缘着色浅。
电镜下小体由细丝构成,中心区致密,周围比较松散黑质神经元可见圆形均质,弱嗜酸性包涵体,周围可见空晕。
脑变性疾病

(3)局限性脑萎缩的病因主要是局部脑组织坏 死后软化的负占位效应,如手术后遗症、局部 感染后遗症、出血梗塞后遗症、脑血管畸形周 围脑组织供血不足、外伤后遗症、中毒性脑病 后遗症等,上述病因多数伴软化灶,血管性病 变在MRI上还可见到含铁血黄素沉着。有些病 因如中毒性、重度缺氧、变性等所致的脑萎缩 可无软化灶,如酒精中毒性小脑萎缩、桥小脑 橄榄核萎缩等。
第十二页,共23页。
第十三页,共23页。
4、皮克病
常显遗传,以进行性痴呆为主要症状。影像 以双侧额颞叶底部萎缩,而其他部位正常为主 要特点,MRI多无异常信号。诊断依赖活检。
第十四页,共23页。
5、Huntington舞蹈病
遗传性纹状体(尾状核和壳核)神经元变性, 临床特点为30~50岁成人舞蹈样运动、进行性 痴呆和情感障碍。影像特点为纹状体萎缩,长 T1长T2信号,大脑皮层萎缩。诊断依赖临床。
第十一页,共23页。
CJD的CT检查可能正常或脑萎缩。 MRI对CJD的诊断可见双侧尾状核、壳核于T2加权像呈对
称性均质的高信号,很少波及苍白球,也不呈增强效应,T1可 完全正常,这对CJD的临床诊断有重要意义。双侧尾核、 壳核T2加权像对称性高信号是散发性CJD重要影像学改 变,在特定的临床背景下为散发性CJD临床诊断依据之一。 CJD的主要病理特征为神经细胞脱失、星形胶质细胞增生、 海绵状变性,脑活检发现海绵状变和PrPSC可确诊。
第十五页,共23页。
6、帕金森病:
病理主要特征为黑质多巴胺神经元变性,临床特点为静止 性震颤、肌强直和运动减少三联征,常伴步态不稳和痴呆。 临床表现为此三联征者还有继发于外伤、感染、肿瘤、梗 塞等原因者,称为继发性帕金森病或帕金森综合征。影像 显示正常或弥漫性萎缩、黑质苍白球信号异常等,无特异 性,主要用于特发和继发的鉴别,继发者除发现病因表现 外,还可发现壳核T2低信号。
ald是什么病

ald是什么病ald(自体骨髓移植后遗症)是一种罕见的神经系统变性疾病,它的全称是X连锁性肾上腺性脑白质营养不良症。
ald是由于酯酶A缺乏而引起的脂质代谢障碍,主要表现为神经系统的退化和失调。
在这篇文章中,我们将详细介绍ald的病因、症状、诊断和治疗方法。
ald是一种遗传性疾病,通常通过母系遗传。
这是因为ald与X染色体上的酯酶A基因的突变有关。
男性携带着这个突变的基因,而女性则是携带者。
男性患者通常出现在幼年时期,而女性携带者通常不会出现明显的症状。
这是因为女性拥有两个X染色体,其中一个正常的基因能够弥补突变基因的缺陷。
ald的症状通常在2-10岁之间开始出现。
最常见的症状是运动障碍,包括肌张力障碍、肌肉僵硬和姿势不稳。
此外,患者还可能出现言语和认知能力的退化,以及视力和听力的问题。
ald还可能导致肾上腺功能障碍,引起疲劳、皮肤色素沉着和血压异常。
诊断ald需要进行一系列的测试。
首先,医生会收集患者和家族的病史,并进行体格检查。
然后,通过进行代谢性的血液和尿液检查,可以检测到某些特定的代谢物的异常水平。
最后,进行基因检测可以确认ald的诊断,通过检测酯酶A基因的突变。
目前,没有治愈ald的方法。
然而,早期的干预和治疗可以帮助减缓疾病的进展和症状的严重性。
一种常用的治疗方法是骨髓移植,通过移植健康的骨髓细胞来替代不正常的细胞。
这种治疗方法可以提供正常的酯酶A,从而减缓神经退化的过程。
除了骨髓移植外,对ald患者进行康复治疗也是很重要的。
这种治疗方法可以包括物理治疗、语言治疗和职业治疗等,旨在帮助患者最大限度地保持他们的身体和认知功能。
总而言之,ald是一种罕见的神经系统变性疾病,由X连锁遗传导致。
它主要表现为运动障碍、言语和认知能力的退化,以及可能的肾上腺问题。
通过一系列的测试和基因检测,可以诊断ald。
虽然目前没有治愈这种疾病的方法,但通过骨髓移植和康复治疗可以减缓疾病的进展和改善患者的生活质量。
老年人的神经系统变性疾病

心理支持与康复训练
心理支持:提供心理辅导,帮助老年人调整心态,减轻心理压力 康复训练:进行康复训练,帮助老年人恢复身体机能,提高生活质量 社交活动:鼓励老年人参加社交活动,增强社交能力,提高心理健康水平 健康教育:提供健康教育,提高老年人的健康意识和自我管理能力
添加标题
添加标题
老年人神经系统变性疾病的治疗 方法多样,需要根据患者的具体 情况选择合适的治疗方案
老年人神经系统变性疾病的预防 和康复需要多方面的配合,包括 家庭、社区和医疗机构等
常见的老年神经系
04
统变性疾病
阿兹海默症(老年痴呆症)
病因:神经细胞死亡,导致大脑功能逐渐丧失 症状:记忆力减退,认知功能下降,日常生活能力下降 治疗:目前尚无治愈方法,主要通过药物和康复治疗缓解症状 预防:保持健康的生活方式,加强锻炼,保持良好的心理状态,降低患病风险。
药物选择:根据病情和患者 情况选择合适的药物
药物副作用:注意药物的副 作用,及时调整药物剂量或
更换药物
药物相互作用:注意药物之 间的相互作用,避免药物相
互作用导致的不良反应
非药物治疗
运动疗法:如瑜伽、太极等,有助于改善老年人的神经系统功能 心理疗法:如认知行为疗法、心理辅导等,有助于缓解老年人的心理压力 营养疗法:如补充维生素、矿物质等,有助于改善老年人的神经系统健康
05 预 防 与 治 疗 策 略
06 家 庭 与 社 会 支 持 的 重 要 性
01
添加章节标题
神经系统变性疾病
02
概述
定义与分类
分类:包括阿尔茨海默病、 帕金森病、多发性硬化症、 肌萎缩侧索硬化症等
医学课件神经系统变性性疾病

特点
痴呆必须是获得性而非先天性
智能障碍(高级神经功能紊乱)包括: 记忆、语言、思维、视空间功能不同程度受损 概括、计算、判断、综合、解决问题能力降低 常伴人格异常、行为、情感异常 病人日常生活、社交、工作能力明显减退
认知功能
包括: 1. 注意力 2. 记忆力 3. 定向力 4. 语言能力 5. 视空间能力 6. 执行功能(组织管理能力)
++++
++
+
3y-10y
+
+
++++
治疗
• 对症治疗 • 1.体位性低血压:米多君 • 2.排尿功能障碍:曲司氯铵、奥昔布宁、特 托罗定,解除逼尿肌痉挛 • 3.帕金森综合征:左旋多巴少数有效,帕罗 西汀可能有效
变性相关性痴呆
痴呆(Dementia)为一种临床综 合征,指成年后大脑病变所致的获得性、 持续性智能障碍,即在无意识障碍的情 况下,出现记忆和认知功能障碍阶段, 同时伴有社会、生活活动能力减退。并 且症状和功能损害至少己存在4~6个月。
非变性病:
纹状体--黑质 系( MSA-P)
小脑性 共济失调 (MSA-C)
自主神经功能障碍
胸1-腰3
骶2-4
病因病理
• MSA的病因不明。目前涉及的有脂质过氧 化损伤、酶代谢异常、慢病毒感染、神经 元淍亡、少突胶质细胞胞质内包涵体等。 • 标志是胶质细胞中发现嗜酸性包涵体,神 经细胞变性脱失,胶质细胞增生
概念
多系统萎缩(MSA)是一组原因不明 的散发性、成年起病的进行性神经系统、 多系统变性疾病。
• 包括纹状体黑质系---帕金森综合征(MSAP) 纹状体黑质变性(SND) • 橄榄-脑桥-小脑系--小脑性共济失调(MSAC) 橄榄脑桥小脑萎缩(OPCA) • 脊髓的中间内、外侧细胞柱和Onuf核--自主 神经功能障碍 自主神经功能障碍的ShyDrager综合征(SDS)
变性,变性的症状,变性治疗【专业知识】

变性,变性的症状,变性治疗【专业知识】疾病简介变性是细胞或细胞间质的一系列形态学改变并伴有结构和功能的变化(功能下降),表现为细胞内或细胞间质中出现非生理性物质或生理性物质过度堆集。
但我们通常生活中接触到的变性则多为第三种意思:即易性,通过外科手术方式使人的生殖器官发生改变,切除男性生殖器官移植女性生殖器官或是切除女性生殖器官移植男性生殖器官。
通常想变性(易性)的人群往往患有易性病。
易性病(traexuals)是性身份严重颠倒性疾病,患者通常在3岁时萌发,青春期心理逆变,持续地感受到自身生物学性别与心理性别之间的矛盾或不协调,深信自己是另一性别的人,强烈地要求改变自身的性解剖结构,为此要求作易性手术以达到信念,在易性要求得不到满足时,常因内心冲突而极度痛苦,甚至导致自残、自戕。
疾病病因一、病因易性病发病的原因十分复杂,不少环节还无法解释,至今仍停留在观察和研究阶段,据推测可能与下列因素有关。
1、遗传因素一般认为遗传因素可能与易性病发生有关,但尚无充分资料能加以证明。
曾有2例双胞胎,其中哥哥健康正常,弟弟患有严重易性病,而父母身体健康,其他兄弟精神亦无异常。
2、内分泌因素有学者报告血浆中睾酮水平在易性病中男性患者偏低,女性患者偏高。
亦有人认为男女易性病患者血浆中睾酮均无明显变化。
在笔者接触的病例中,术前术后血浆睾酮水平有改变,但症状并没有随睾丸和卵巢的被切除而消失,可见根源不在于此,性激素水平的差异并非易性病发生的原因。
3、外生殖器大小与形态由于外生殖器与性身份有着密切关系,有人估计外生殖器的大小及形态与易性病的发生有关。
但在笔者目前的资料中,还未发现有男性易性病患者性器官大小、形态异常,以及女性易性病患者的外阴畸形(如阴蒂肥大等)。
4、环境和心理因素人们习惯上认为,产生易性病的原因是父母对幼小儿女按异性打扮或抚养,或患者在异性人群中成长。
女性易性病患者可因为对男性刚强性格和独立生活能力的崇拜而产生对男性性别的向往,这在病例中也确实有所发现。
神经系统变性性疾病PPT课件

6
还有研究显示,有些突变的SOD1蛋白的生理功能增强,可以催化超氧 阴离子和一氧化氮形成过氧亚硝基,而后者可以进一步使络氨酸残基亚 硝基化。而神经丝的低分子量亚单位中富含络氨酸,因此以上反应可以 导致神经丝结构和功能的发生异常改变,进而引起对运动神经元的一系 列病理生理损害。
大量的临床观察和动物实验已经证实在ALS的发病过程中有氧化应
激损伤的参与,包括:蛋白质羰基含量增高、脂质过氧化、DNA氧化损
伤、NOS活性增强等。这些异常变化是由ALS患者体内抗氧化能力减弱
和活性氧中间产物生成增多两方面因素共同导致的。氧化应激损伤可以
在细胞内产生广泛的影响,包括DNA损伤、蛋白质和脂质的修饰等。而
运动神经元由于代谢水平较高、不饱和脂肪酸需要量较大、细胞内钙水
但是这些突变的SOD1的生理功能并未受到影响。所有这些研究表明,仅仅是消
除SOD1的生理功能并不足以导致ALS的发生。在动物和细胞水平的研究都发现,
突变的SOD1有聚集的倾向。而这些异常聚集的SOD1可以损害轴浆运输或者直
接影响运动神经元的存活。因此,现在一般认为突变的SOD1蛋白异常聚集导致
的获得性神经毒性作用在SOD1突变引起的ALS中可能具有重要作用。
2(excitatory amino acid transport 2,EAAT 2)的含量减少。但是EAAT 2
的减少是否可以通过增加兴奋性神经递质的含量导致运动神经元损害还
未得到证实。另有研究显示,谷氨酸可以通过增加细胞内钙离子浓度引
起神经元的损伤。已经有多项实验证实,减少细胞内钙离子水平对运动
外,线粒体功能障碍还可以引起细胞内钙离子浓度升高,并
引起细胞色素C的释放,进而介导程序性细胞死亡的启动。
疾病学变性的名词解释

疾病学变性的名词解释近年来,关于性别认同和性别多样性的讨论不断增加。
在这其中,有一个早已存在的词汇逐渐引起了人们的关注,那就是“疾病学变性”。
在了解这一术语之前,我们需要明确一些相关的概念。
首先,性别认同是指个体对自己的性别意识和认同的心理体验,而不仅仅是指生理上的性别。
对大多数人来说,他们的性别认同与出生时分配给他们的生理性别保持一致,这被称为“性别一致”。
然而,也存在一部分人,他们的性别认同与生理性别不一致。
这些人可能被称为“跨性别者”,他们认为自己的性别身份与其生理性别不匹配。
由于跨性别者在社会中可能面临歧视和隔离,他们有时会寻求专业的支持和介入,以探索更适合他们性别认同的方式。
这就引出了我们要探讨的主题:疾病学变性。
疾病学变性是一个医学术语,用于描述在病理学上对于跨性别者进行性别身份确认和支持的过程。
事实上,这个术语本身也带有一些争议,因为许多跨性别者认为将他们的身份视为“疾病”是对他们的污名化和歧视。
然而,可以确定的是,疾病学变性的目的是为跨性别者提供医学上的支持,而不是试图修复或治疗他们的性别认同。
这种支持可以包括心理咨询、社交转换、激素治疗和/或手术干预等。
在整个过程中,医生和专业人士的角色是为了帮助这些个体获得他们自己的性别认同,而不是尝试改变他们的认同。
对于调查和记录的目的,疾病学变性被列为一种性别身份疾病,但这并不代表跨性别者本身是一种疾病。
相反,越来越多的医学界人士认识到,应将跨性别身份视为自然和正常的发展变异,并为他们提供适当的支持和尊重。
疾病学变性涉及的医学干预方法可以因个体的需求而异。
有些跨性别者可能希望通过社交转换来改变他们的外貌,例如改变服装、发型或化妆。
社交转换还可能包括更改他们在社会中的性别角色,例如用一个新名字或性别代词来标识自己。
对于其他跨性别者来说,他们可能希望通过激素治疗来实现更直接的身体性别改变。
这种治疗可能涉及雌激素或雄激素的使用,以使身体特征更接近他们心目中的性别。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
PD-Dementia complex PD-ALS-Dementia complex ALS-Dementia complex
感觉神经
Leber病 Leber病 遗传性感觉性神经 遗传性感觉性神经 根神经病
感觉+运动
运动神经
•MSA
并发Dementia、PD、ALS Retinal Deg Friedreich ataxia、Hereditary spastic paraplegia、 Olivocerebellar deg、(Holmes Syn)
PD-Dementia complex PD-ALS-Dementia complex ALS-Dementia complex
感觉神经
Leber病 Leber病 遗传性感觉性神经 遗传性感觉性神经 根神经病
感觉+运动
运动神经
ALS、SMA、 ALS、SMA、PLS Progressive Bulbarpaly
腓骨肌萎缩(CMT) 间质性肥大性神经炎 (Dejerine-Sottas Syd) 间质性肥大性神经炎综合征 (Roussy-Cornil Syd)
三、感觉障碍
大脑(痴呆 )
AD、 AD、DLB Pick病和额颞痴呆 Pick病和额颞痴呆 Huntington chorea PD •MSA
并发Dementia、PD、ALS Retinal Deg •Friedreich ataxia、Hereditary spastic paraplegia、 Olivocerebellar deg、(Holmes Syn)
腓骨肌萎缩(CMT) 间质性肥大性神经炎 (Dejerine-Sottas Syd) 间质性肥大性神经炎综合征 (Roussy-Cornil Syd)
Huntington舞蹈病:1872年George Huntington首先报告。常染色体显性遗传, 中年发病、舞蹈、精神障碍→痴呆。
帕金森病:1817年由英国医生Parkinson首
先描述。病理特点:黑质多巴胺神经元变 性脱失和Lewy小体形成。>65岁人群患病率 为2%,我国已>200万人。 Parkinson-Dementia complex Parkinson-Als-Denentia complex Als-Denentia complex
六、脊髓、脑干、小脑、 自主神经系统
腓骨肌萎缩(CMT) 间质性肥大性神经炎 (Dejerine-Sottas Syd) 间质性肥大性神经炎综合征 (Roussy-Cornil Syd)
Leber遗传性视神经病:
•
Leber于1968年首次报道。Nikoskelainen 1984 年发现线粒体DNA突变是本病主要致病因素。
• •
发病年龄多在11~48岁,男:女约为5:1。 : 急性或亚急性原因不明的视力丧失,中心 视觉缺失为特征,早期色觉减退,眼底可 见毛细血管扩张的微血管病,母系阳性家 族史。
球形嗜伊红包涵体,位于黑 质、蓝斑、Meynert核等处的 神经细胞胞浆内,有苍白 “晕环”。
位于大脑皮层细胞者,晕环 欠清。
Lewy’s body,由α-共核蛋白和神经丝异常聚集而成
(三)Pick病和额颞痴呆
Pick病于1892年由Pick首先描述,以额颞 萎缩为病理特征。1911年Alzheimer发现神 经元胞浆中有嗜银包涵体,称Pick小体。 额颞萎缩为特征的痴呆综合征称额颞痴呆, 其中有1/4有Pick小体。
一、痴 呆
中国500万 AD约310万 VD约140万
大脑(痴呆 )
AD、DLB AD、 Pick病和额颞痴呆 Pick病和额颞痴呆 Huntington chorea PD •MSA
并发Dementia、PD、ALS Retinal Deg •Friedreich ataxia、Hereditary spastic paraplegia、 Olivocerebellar deg、(Holmes Syn)
吉林大学第一医院神经内科
饶明俐
定义
慢性、进行性、系统性、以 神经元变性和继发性脱髓鞘为主 要病理特征原因未明的疾病。
病因
是当前研究的前沿课题之一。 1. 遗传因素:有些神经变性病有明确的 家族史,故又可称为遗传变性病。 2. 内源性因素:细胞凋亡?免疫反应? 毒素作用?
分类
大脑(痴呆 )
AD、DLB AD、 Pick病和额颞痴呆 Pick病和额颞痴呆 Huntington chorea PD
大脑(痴呆 )
AD、 AD、DLB Pick病和额颞痴呆 Pick病和额颞痴呆 Huntington chorea PD •MSA
并发Dementia、PD、ALS Retinal Deg •Friedreich ataxia、Hereditary spastic paraplegia、 Olivocerebellar deg、(Holmes Syn)
腓骨肌萎缩(CMT) 间质性肥大性神经炎 (Dejerine-Sottas Syd) 间质性肥大性神经炎综合征 (Roussy-Cornil Syd)
病因
遗传:5%~10%有家族遗传史。日本纪伊半岛、 美国关岛chamorro族发病率高(50~100倍), 15%~25%的FALS存在第21对染色体Cu/ZnSODⅠ型基因突变,致SOD活性丧失,通过H2O2介导 致细胞凋亡? 环境:重金属如铅、汞等(血及CSF中铅↑)、化 学制剂、有机杀虫剂、慢病毒感染与免疫反应、 外伤等?
PD-Dementia complex PD-ALS-Dementia complex ALS-Dementia complex
感觉神经
Leber病 Leber病 遗传性感觉性神经 遗传性感觉性神经 根神经病
感觉+运动
运动神经
ALS、PLS、 ALS、PLS、SMA Progressive Bulbarpaly
PD-Dementia complex PD-ALS-Dementia complex ALS-Dementia complex
感觉神经
Leber病 Leber病 遗传性感觉性神经 遗传性感觉性神经 根神经病
感觉+运动
运动神经
ALS、PLS、 ALS、PLS、SMA Progressive Bulbarpaly
(1869年charcot首次报道)
大脑(痴呆 )
AD、 AD、DLB Pick病和额颞痴呆 Pick病和额颞痴呆 Huntington chorea PD •MSA
并发Dementia、PD、ALS Retinal Deg •Friedreich ataxia、Hereditary spastic paraplegia、 Olivocerebellar deg、(Holmes Syn)
PD-Dementia complex PD-ALS-Dementia complex ALS-Dementia complex
感觉神经
Leber病 Leber病 遗传性感觉性神经 遗传性感觉性神经 根神经病
感觉+运动
运动神经
ALS、SMA、 ALS、SMA、PLS Progressive Bulbarpaly
临床类型
肌萎缩侧索硬化:分散发型(经典型)、 家族型。 美国关岛、日本纪伊半岛的 ALS多合并 PD 和痴呆,称帕金森-痴呆-肌萎缩侧索硬化
(Guam型ALS)
进行性球麻痹:进展快、预后不良。 原发性侧索硬化:少见,慢性缓慢发展,Werdnig-Hoffmann病)(SMAⅠ型):生 后3~6月发病,平均生存7~9月。 慢性婴儿型( SMA Ⅱ型):生后6个月以后发病,肢 体近端无力,多数可活到青少年。 少年型( SMA Ⅲ 型):儿童或青春期缓慢起病,近端 无力,Gowers征,似肌营养不良,50%有肉跳。 成人慢性脊髓萎缩( SMA Ⅳ 型):发病年龄18~60岁, 多以近端为主,可累及球部及面部,发展慢。
斑) 病理改变:老年斑、NFT、颗粒空泡变性、 神经元脱失、血管淀粉样变
阿尔茨海默病理改变
临床症状
早期(1~3年): 近记忆障碍、学习能力 下降、空间定向障碍、淡漠、EEG、 CT/MRI正常 中期(2~10年):智能、人格改变、EEG 节律减慢、脑萎缩 后期(8~12年):智力严重衰退、屈曲体 位、肌强直 、EEG弥漫性慢波、脑萎缩 明显
四、感觉运动性神经病
大脑(痴呆 )
AD、 AD、DLB Pick病和额颞痴呆 Pick病和额颞痴呆 Huntington chorea PD •MSA
并发Dementia、PD、ALS Retinal Deg •Friedreich ataxia、Hereditary spastic paraplegia、 Olivocerebellar deg、(Holmes Syn)
鹤腿、弓形足、槌状趾
五、锥体外系统
大脑(痴呆 )
AD、 AD、DLB Pick病和额颞痴呆 Pick病和额颞痴呆 Huntington chorea PD •MSA
并发Dementia、PD、ALS Retinal Deg •Friedreich ataxia、Hereditary spastic paraplegia、 Olivocerebellar deg、(Holmes Syn)
病程:5~12年
(二)路易体痴呆(DLB)
占老年期痴呆总数的 10%~20%,在变性疾病中 仅次于AD 1912 年 Freidreich Lewy 在 1例PD患者 黑 质中发现 Lewy 体, 1961年Okazaki (日本)等首次在1例 严重痴呆患者的皮质神经元中发现,称DLB 临床特点:进行性痴呆、波动性认知功能障碍 (程度和时间不同,可持续几分钟、几周、几 月),反复发作的视幻觉 (80%), 帕金森综 合征 病程:5~10年