肟醚类化合物作为导向基团在钯催化C-H活化中的应用
过渡金属催化羟基化合成酚类化合物研究进展

·健康科学·163氧化催化合成了2-羟基-2-苯乙醇(图2)。
由于肟类化合物与二价钯的配位能力较低,因此该反应不适合含芳环的酮和醛等化合物。
Sanford研究小组对上述Vavilikolanu研究小组的报道进行了改进,发展了钯催化肟邻位羟基化合成酚类化合物的反应(图3)。
通过对反应条件的考察,发现过醋酸过氧化物是合适的氧化剂,可以减少价格昂贵的碘试剂的使用。
2.铜催化的羟基化反应进展过渡金属催化羟基化合成芳香化合物具有很好的稳定性。
因此,芳香环的修饰存在一定难度。
直到日本Fujiwara研究小组于1990年报道了第一个空气促进,醋酸钯作为催化剂的苯为原料合成苯酚的反应方法。
该反应方法具有里程碑的意义,开辟了应用过渡金属催化合成酚类化合物的先河 (图 1)。
但反应需要在高压和高温下进行,反应条件比较苛刻。
随后,Vavilikolanu研究小组发展了一种钯催化的肟类化合物邻位合成酚类化合物的方法,具体为以醋酸钯作为催化剂,以肟类化合物为原料,经过图1图2图3图4164原料合成酚类化合物的方法(图4)。
该反应体系需要用到化学计算量的催化剂,通过对反应机理进行研究,发现醋酸铜的作用是与羟基配位后能形成保护,防止生成的酚类化合物进一步被氧化。
3.铁催化的羟基化反应进展Matsushita研究小组报道了硫酸铁铵作为催化剂促进合成酚类化合物的方法,即:采用空气或双氧水作为氧化剂,以苯甲醚为原料合成邻位或者间位、对位的酚类化合物的方法(图5)。
其他类型的铁基催化剂,如:Fe/C,Fe/ZSM5沸石和FeSO4/TFA,也分别被报道。
Morooka研究小组报道了以铁配合物作为催化剂合成酚类化合物的方法,通过该方法合成了邻羟基苯甲酸(图6)。
通过对反应机理研究发现,反应过程经历了一个非共价键键合的铁中间体的过程。
4.铱催化的羟基化反应进展Smith Ⅲ研究小组报道了铱催化的一锅法合成3,4,5-三取代酚的方法。
金属有机化学中的钯催化的反应讲解

XXXX大学研究生学位课程论文(2012 ---- 2013 学年第一学期)学院(中心、所):化学化工学院专业名称:应用化学课程名称:高等有机化学论文题目:金属有机化学中的钯催化的反应授课教师(职称)XXXX(教授)研究生姓名:XXXX年级:2012级学号:XXXXXXXXX成绩:评阅日期:XXXX大学研究生学院2012年12 月25 日金属有机化学中的钯催化的反应XXXXXX(XXXX大学化学化工学院,山西,太原,030006)摘要:过渡金属钯在金属有机化学方面具有丰富的反应性,在各类有机化学反应中如氢化、氧化脱氢、偶联、环加成等反应中,钯是优良的催化剂,或是催化剂的重要组分之一。
本文在查阅大量近几年文献资料的基础上,综述了钯催化的反应,同时综述了钯催化反应的机理以及钯催化反应的研究现状。
关键词:钯,催化剂,反应机理,研究进展1钯催化的反应类型及反应机理在现今炼油、石油化工等工业催化反应中,有很多的钯催化反应,尤其是氢化反应中的选择加氢,以及氧化反应中选择氧化生产乙醛、醋酸乙烯、甲基丙烯酸甲酯,均广泛采用和开发钯催化剂。
对石油重整反应,钯也是常选取的催化剂组分之一。
1.1氢化反应钯催化剂具有很大的活性和极优良的选择性,部分氢化选择性高,常用作烯烃选择性氢化催化剂。
1.1.1反应式及反应机理反应底物首先和氢气分子分别被吸附到催化剂上,然后和催化剂的活性中心形成配位键,最后完成氢的转移,氢和反应底物形成σ-键。
1.1.2反应方程式举例1.2氧化反应烯烃和炔烃是十分常见并且重要的有机化合物,选择性地氧化这类不饱和碳氢化合物一直是化学工业和学术界的重要研究目标之一。
1.2.1分子氧参与的钯催化烯烃的氧化反应根据亲核试剂的不同,如氧、氮和碳等亲核试剂,把催化烯烃的氧化反应可以形成C-O、C-N和C-C键。
1.2.1.1反应机理钯催化烯烃的氧化反应都经过三个过程:首先,把插入烯烃形成新的C-Pd键;接着,有机钯中间体进行β-H消除产生Pd(0);最后,Pd(0)被重新氧化为Pd(П)。
“C-H键活化和高效转化的基本科学问题”重大项目指南

“C-H键活化和高效转化的基本科学问题”重大项目指南附件5:“C-H键活化和高效转化的基本科学问题”重大项目指南以C-H键为代表的惰性化学键活化和直接转化是合成化学前沿领域之一。
区别于传统的活泼官能团反应,惰性键的活化和直接转化减少了试剂和原料的预先官能化,体现了高效、原子经济性和环境友好等现代合成理念。
阐明惰性化学键,特别是C-H 键选择性活化转化的内在机制和规律,发展惰性化学键选择性活化的新策略和新模式,是当前有机化学研究的重要科学问题,也是迫切需要解决的关键基础科学问题。
相关研究将为有机化学的发展和创新提供理论基础,推动我国在合成新反应、新试剂、新理论等方面取得更大突破。
本项目以C-H键活化和转化过程中的基本科学问题为研究对象,在充分发挥我国物理有机研究丰富积淀的基础上,利用物理和物理化学的新理论、新技术和新手段,开展重要C-H键活化转化过程的物理有机研究,揭示C-H键活化转化的内在机制和普遍规律,以期进一步提高我国物理有机化学的研究水平。
一、科学目标本项目围绕“C-H键活化和高效转化”中的基本科学问题,集成我国在该领域物理有机研究方面的优势力量,面向科学前沿,在提供C-H活化转化过程中定量的热力学及动力学数据、确定反应活泼中间体或过渡态结构、揭示化学键的断裂及形成的机制和规律等方面做出创新性结果,为理性设计新型C-H活化转化新催化体系、新催化模式和新反应提供理论基础,发展若干具有重要影响的新反应和新试剂。
二、研究内容(一)C-H键能量学。
化学键的核心物理量-断裂能(BDE)对于理性判断化学反应能否发生和如何发生、判断或者预测反应进行的方向和速率、研究反应的机理、认识反应驱动力以及理解反应的热力学本质,都具有十分重要的意义。
本课题将重点围绕C-H等键BDE研究中空缺的能量标度展开,如建立离子溶剂中R-H键能量标度(pKa、BDE 等)以及全系列有机化合物的负氢亲合势等,研究标志性反应的机理,揭示相关有机反应的规律和本质。
钯催化的反应总结

钯催化的反应总结引言钯(Palladium,Pd)是一种常见的过渡金属催化剂,它在有机合成中有着广泛的应用。
由于钯具有良好的催化活性、选择性和功能多样性,钯催化反应已成为有机合成领域备受关注的重要研究方向之一。
本文将对钯催化的一些重要反应进行总结,以便更好地了解和应用这些反应。
催化剂的选择在钯催化反应中,催化剂的选择起着至关重要的作用。
常见的钯催化剂包括[Pd(PPh3)4]、Pd(PPh3)2Cl2、Pd(OAc)2等,这些催化剂具有良好的催化性能和稳定性。
此外,还可以通过对催化剂进行配体修饰来改变其催化性能,如引入膦配体、氨基配体等。
钯催化的碳-碳键形成反应1. Heck反应Heck反应是钯催化的一个重要的碳-碳键形成反应,它通过亲电性或亲核性的烷基化试剂与不饱和化合物间的交叉偶联,在构建碳-碳键的同时保留官能团的特点。
通常情况下,该反应需要碱的存在,并在乙酸盐氛围中进行。
Heck反应适用于合成各类芳香烃、乙烯烃、酮类等化合物。
2. Suzuki-Miyaura偶联反应Suzuki-Miyaura偶联反应是钯催化的另一个重要的碳-碳键形成反应。
该反应利用有机硼酸酯与卤代化合物在碱的存在下进行交叉偶联,生成对应的芳香烃。
Suzuki-Miyaura偶联反应具有底物宽容性和功能团兼容性高的优点,被广泛应用于有机合成中。
钯催化的碳-氮键形成反应1. Buchwald-Hartwig氨基化反应Buchwald-Hartwig氨基化反应是钯催化的一种重要的碳-氮键形成反应,可以将芳香或烯丙基溴化物与氨或胺类化合物发生反应,生成相应的胺化物。
该反应具有反应条件温和、底物宽容性好的特点,被广泛应用于药物合成和天然产物的合成等领域。
2. Sonogashira偶联反应Sonogashira偶联反应是钯催化的一种重要的碳-氮键形成反应,它通过芳香溴化物或卤代烯烃与炔烃发生偶联反应,生成相应的炔烃衍生物。
Sonogashira偶联反应具有底物宽容性好、反应条件温和的特点,被广泛应用于有机合成中。
药物合成中的CH键活化研究

药物合成中的CH键活化研究1. 引言CH键活化是有机合成领域中的一个重要研究方向。
传统的有机合成方法中,CH键通常是非活化的,需要通过复杂的转化步骤才能实现功能基团的引入。
而CH键活化研究的发展,为有机合成提供了一种高效、环境友好的策略。
本文将重点介绍药物合成中的CH键活化研究进展,探讨其在药物合成中的应用潜力以及面临的挑战。
2. CH键活化的重要性CH键是有机分子中最常见的键之一,其活化将使得有机合成更加高效、简洁。
CH键活化不仅可以通过在有机分子上直接引入功能基团,还可以实现碳氢键与其他键之间的选择性转化,有机分子的转化新途径使得合成药物的路径更加灵活。
因此,CH键活化研究具有重要的理论和应用价值。
3. CH键活化的反应类型目前,CH键活化研究已涉及多种反应类型。
其中包括碳—碳键活化、碳—氮键活化、碳—氧键活化等。
这些反应不仅可以实现原子经济性高的合成路线,还可以提高反应的选择性和速度,降低副反应的发生。
这些特点使得CH键活化成为一种非常有吸引力的反应。
4. 药物合成中的CH键活化研究案例4.1 药物合成中的碳—碳键活化碳—碳键活化是CH键活化研究的重要领域之一。
通过碳—碳键活化,可以在有机分子中引入新的碳—碳键,从而实现对药物结构的调整和优化。
例如,某些含有两个芳香环的药物分子,可以通过碳—碳键活化,在两个芳香环之间引入新的碳—碳键,增加药物分子的稳定性和活性。
其次,碳—碳键活化还可以在有机分子中引入稳定性较好的碳杂环结构,扩展药物分子的结构空间。
4.2 药物合成中的碳—氮键活化碳—氮键活化是另一个重要的研究方向。
通过碳—氮键活化,可以在有机分子中引入氮原子,从而实现对药物功能基团的引入和调整。
例如,一些药物中常见的酰胺结构,可以通过碳—氮键活化,在有机分子中引入酰胺基团。
这种方法不仅能提高药物的生物利用度和稳定性,还能增加药物分子与靶标的作用力。
4.3 药物合成中的碳—氧键活化碳—氧键活化是具有广泛应用前景的反应类型。
钯系催化剂加氢反应及应用开发

1 引 言
212 炔烃加氢
加氢还原是有机合成的一个重要单元操作。还 原催化剂主要有贵金属 ( Pt ,Rh , Pd) 催化剂 ,镍系催 化剂 ,铜系催化剂和钴系催化剂等 。贵金属催化剂 具有反应条件温和 , 活性高 , 选择性好等优点 , 得到 广泛的应用 。贵金属催化剂又分为固体催化剂和均 相催化剂 。固体催化剂不溶于反应介质 , 与产物易 分离 ,可循环使用 ,如 Pd/ C 催化剂 。均相催化剂溶 于反应介质 , 如威尔金森催化剂〔RhCl ( PPh 3 ) 3 〕 。 本文主要讨论 Pd/ C 催化剂在加氢反应中的开发应 用。 2 钯系催化剂加氢反应类型[ 1 ]
HO
θ
HO
CH = CHCOOH + H Pd/ C 2
θ
HO
CH2 CH2 COOH
OC4 H9 3 - 硝基 - 4 - 丁氧基苯甲酸 NO2 + H2
Pd/ Al2O3
OC4 H9 3 - 氨基 - 4 - 丁氧基苯甲酸 NHOH
3 ,4 —二羟基肉桂酸
3 ,4 —二羟基苯丙酸
硝基环己烷
・8 ・
专论与综述
化学工业与工程技术
2000 年第 21 卷第 5 期
钯系催化剂加氢反应及应用开发
吴鹤麟 ,朱新宝 ,张金龙 ,陆长峰
( 江苏省化工研究所 ,江苏 南京 210024)
[ 摘要 ] 将钯系催化剂催化加氢反应分成 11 个类型 , 分别作了简介 。Pd/ C 催化剂已应用于蒽醌 法双氧水 、 精对苯二甲酸及己内酰胺生产 。认为我国亟待开发的技术包括间苯二胺及同系产品 、 对氨基 苯甲醚及同系产品与对氨基酚 。指出 Pd/ C 催化剂催化加氢技术开发过程中 ,应注意解决氢气源及催化 剂开发与回收利用的问题 。
钯催化ch键活化 四种催化循环

钯催化ch键活化四种催化循环
钯催化CH键活化,是一种由钯催化实现的,可以帮助分子从低能量状态转变
到高能量状态的化学反应。
它的应用范围非常广泛,可以在合成有机化合物、生物活性分子等方面发挥重要作用。
由于钯催化CH键活化的发展,物理化学实验中的
应用越来越广泛。
钯催化CH键活化包括四种不同的催化循环:一是直接对CH键进行诱导活化
的催化循环;第二种是通过氨基R组装的CH键活化催化循环;第三种是反应碳氢
键活化催化循环;最后一种是由多元素体系实现CH键活化催化循环。
以上四种几
乎都是把氧化还原反应从普通状态提升到比较高能量状态,实现CH键活化。
以直接对CH键进行诱导活化的催化循环为例,它具体的反应机制是:钯催化
剂将直接作用于活性位点上的CH键,使CH键由其原有的平衡状态发生变化,产生出较高能量状态的中间体,从而促进反应的发生。
另外,氨基R组装的CH键活化
催化循环是在CH键上引入官能团的一种方法,通过引入R组装,使CH键发生变化,达到催化反应的目的。
碳氢键活化催化循环也可以简单地理解为,通过催化剂和反应物之间的作用,
使反应物在其他反应物的作用下,发生不同的反应,实现CH键活化。
最后一种是
由多元素体系实现CH键活化催化循环,它可以由有机化学反应体系实现CH键活化,具体的操作步骤也比较简单,也很常用。
总之,钯催化CH键活化是一种新型的反应机制,可以帮助从低能量状态转变
到高能量状态,从而促进反应发生,具有十分广泛的实际应用价值。
而当前,钯催化CH键活化催化循环已经被用于合成有机化合物、生物活性分子等领域,在物理
化学实验中也得到了广泛的应用。
有机化学中的C-H羧基化反应探讨

有机化学中的C-H羧基化反应探讨YU Hai-zhu;MA Xiang-yu;YE Li-na【摘要】羧基化反应是有机化学课程教学的一个基本内容(如格氏试剂与CO2的加成及Kolbe-Schmitt反应等),也是工业生产合成羧酸、酯等高附加值化学品的重要方法.然而这些反应需要使用格氏试剂或高压CO2,条件苛刻且底物官能团耐受性窄,在未来有机合成及工业生产中的应用前景有限.为解决这些问题,近年来以二氧化碳作为单碳源的碳-氢直接羧基化反应逐渐兴起,弥补了传统羧基化反应的诸多缺陷,反应的原子/步骤经济性显著提高,更符合“绿色化学”的理念.围绕有机化学课堂教学中的碳氢羧基化反应,分析近期科学研究在经典反应基础上的改进策略,用于丰富课堂教学内容,旨在达到开拓学生视野,引导学生跟进、了解前沿科技进展的目标.【期刊名称】《安徽化工》【年(卷),期】2019(045)003【总页数】5页(P3-7)【关键词】C-H键;羧基化;二氧化碳【作者】YU Hai-zhu;MA Xiang-yu;YE Li-na【作者单位】;;【正文语种】中文【中图分类】O643.32以CO2为碳源的有机物(如卤代烃、酚等)的羧基化是有机合成化学的传统反应,也是有机化学课程教学的一个重点。
初级有机化学课程中主要涉及到Kolbe-Schmitt反应和格氏试剂的羧基化两个典型反应。
以德国化学家Kolbe和Schmitt命名的Kolbe-Schmitt反应最早于1860年由Kolbe课题组提出[1],通过碱性条件下苯酚、金属钠与二氧化碳的羧基化反应生成水杨酸类化合物[2-3]。
1885 年,Schmitt课题组改进实验流程[4],通过将干燥的酚钠/钾在高温(100℃)、高压下与CO2封管,大大提高了羧基化效率,扩展了底物适用范围。
Kolbe-Schmitt反应由此得名,迄今已发展成工业合成水杨酸及其衍生物的重要方法。
相对于在酚上引入羧基的Kolbe-Schmitt反应,格氏(Gridnard)试剂与二氧化碳的反应不仅能够实现芳基羧酸的合成,而且可用于制备烷基羧酸[5]。
钯催化反应及其机理

钯催化反应及其机理研究摘要:目前过渡金属催化的有机反应研究一直是一个比较热的话题,其中由于钯催化的反应活性和稳定性等原因,使其在有机反应中得到了广泛的使用,被全球广泛关注。
本文主要列举了钯催化的交叉偶联反应的机理,及与偶联反应相关的钯催化的碳氢键活化反应、钯催化的脂肪醇的芳基化反应等的机理。
关键词:过渡金属催化偶联反应钯催化机理1.引言进入二十一世纪以后,钯催化的偶联反应已经建立了比较完整的理论体系,研究的侧重点也和以前有所不同化学键的断裂和形成是有机化学的核心问题之一。
在众多化学键的断裂和形成方式中,过渡金属催化的有机反应有着独特的优势:这类反应通常具有温和的反应条件,产率很高并有很好的选择性(包含立体、化学、区域选择性)。
很多常规方法根本无法实现的化学反应,采用了过渡金属催化后可以很容易地得到实现。
在众多过渡金属中,金属钯是目前研究得最深入的一个。
自上世纪七十年代以来,随着Kumada,Heck,Suzuki,Negishi [1]等偶联反应的陆续发现,钯催化的有机反应发展十分迅速,时至今日,钯催化的偶联反应作为形成碳-碳、碳-杂键最简洁有效的方法之一,已经得到了广泛应用。
2.钯催化各反应机理的研究2.1.钯催化的交叉偶联反应自上世纪七十年代以来,随着Kumada,Heck,Suzuki,Negishi 等偶联反应的陆续发现[1],钯催化的有机反应发展十分迅速,时至今日,钯催化的偶联反应作为形成碳-碳、碳-杂键最简洁有效的方法之一,已经得到了广泛应用[2]。
交叉偶联,就是两个不同的有机分子通过反应连在了一起(英文中交叉偶联为crosscoupling,同种分子偶联为homo coupling)。
2.1.1Heck反应Heck 反应是不饱和卤代烃和烯烃在强碱和钯催化下生成取代烯烃的反应,是一类形成与不饱和双键相连的新C—C 键的重要反应[3]。
反应物主要为卤代芳烃(碘、溴)与含有α-吸电子基团的烯烃,生成物为芳香代烯烃。
官能团保护1

在糖和核苷酸化学中,苄基广泛地用来保护羟基 制备: 1、醇/苄氯/强碱,加热
2、醇/苄氯/氧化银/DMF,室温反应
稳定性:
苄基醚对氧化剂(高碘酸盐、四乙酸铅等)、 还原剂(氢化铝锂等)、酸、碱都相当稳定。
脱保护:
1、Pd/H2(钯催化加氢)
2、Na/NH3(液)还原,Na/乙醇
举例:
O 碱 Cl3CH2CH2OC OR
脱去保护基:乙酸/Zn或乙酸/Zn-Cu, 室温
举例:
O
O O OH 1. Cl3C
O Cl 2. HCl/H2O/MeOH
HO 1.R HO OTceoc
O C Cl
RCOO
2.Zn/AcOH
RCOO
OH
成醚 1、苄基醚
通式:
碱 ROH + PhCH2Cl
RO-CH2Ph
五、胺基的保护
一、活波碳-氢键的保护
主要讨论的内容: 1)末端炔烃的碳-氢键的保护 2)环酮α-碳-氢键的保护
(一)端炔的C-H键
末端炔烃C-H易与活波金属、强碱、强氧化剂以
及有机金属化合物反应。因此,在某种情况下要
对炔氢进行保护
一、端炔的C-H键
例1:
Br Mg/THF BrMg C CSiMe3 C CH 1. RMgBr 2. Me2SiCl Br C CSiMe3
一、活化定位导向(主要手段)
例1 合成下列化合物
O
分析:
O
Ph
O +
Ph
Br
Ph
合成:
O B Ph Br O + Ph Ph
副产物
O Ph
本方法产率低,因为有二苄基酮产生。
钯催化总结

钯催化总结引言钯是一种重要的催化剂,广泛应用于有机合成、医药化学和材料科学等领域。
由于钯的高活性和选择性,钯催化反应已成为许多合成过程中不可或缺的工具。
本文将对钯催化反应的基本原理、常见催化剂和催化反应进行总结和概述。
基本原理1.氧化剂:钯催化反应通常需要使用氧化剂,常见的氧化剂有氧气和过氧化氢。
2.配体:钯醇配体和膦配体是常用的配体,可以对钯的活性和选择性进行调控。
3.活性位点:钯催化剂中的活性位点包括表面钯原子、钯纳米颗粒和钯合金等。
常见催化剂1.钯醇配体催化剂:常见的钯醇配体催化剂有Pd(PPh3)2Cl2、Pd(PPh3)4和Pd(OAc)2等。
这些催化剂在碳碳键形成、偶联反应和氢化反应等方面表现出较高的活性和选择性。
2.膦配体催化剂:常见的膦配体催化剂有PPh3、P(o-tolyl)3和BINAP等。
这些催化剂在不对称合成和氢化反应等领域具有重要的应用价值。
3.钯纳米颗粒催化剂:钯纳米颗粒催化剂具有较大的比表面积和高度分散性,因此在催化反应中具有较高的催化活性和选择性。
催化反应1.氢化反应:钯催化氢化反应是一种常见的反应,可以将烯烃或芳香化合物转化为相应的烷烃。
该反应在精细化学品合成和医药化学中具有广泛的应用。
2.偶联反应:钯催化偶联反应是一种重要的碳碳键形成方法,常见的偶联反应有Suzuki偶联、Heck偶联和Sonogashira偶联等。
这些反应可以高效地构建复杂有机分子骨架。
3.不对称合成:钯催化的不对称合成是一种重要的合成策略,可以合成手性分子。
常见的不对称合成反应有不对称氢化、不对称偶联和不对称加成等。
应用领域1.有机合成:钯催化反应在有机合成中广泛应用,可以高效地构建C-C和C-X键,为有机合成提供了重要的方法学。
2.医药化学:钯催化反应在药物合成中具有重要的应用,可以合成各种药物原料和药物中间体。
3.材料科学:钯催化反应在材料科学领域具有重要的应用,可以合成具有特殊结构和性能的功能材料。
钯催化的脱羧heck反应机理的理论研究

钯催化的脱羧heck反应机理的理论研究
近年来,钯催化的脱羧Heck反应(Pd-C Catalyzed Heck Reaction)在有机
合成中得到了广泛应用,因其适用性较强及反应条件较为温和而备受科学家们青睐。
为了使其反应性能进一步优化,对其反应机理的深入认识尤为重要、必不可少。
目前,就钯催化的脱羧Heck反应机理,理论和实验研究均达成了令人满意的
成果。
据认为,该反应以一种c-h活化机制发生,在催化剂钯(PdX _2及Pd(0))存在的条件下,X(此处指的是卤化物离子)的作用下,原料的c-h键通过紫外光
辐射及酸性19F谱仪的作用发生活化,形成高能中间体3-戊烯醛贵金属化合物
(M- 3-penten-2-one),表示为结构A.其中,羧基受到强烈的歧化作用,形成稳
定的歧化中间体4-戊二醛贵金属化合物(M-4-pentadien-3-ol),表示为结构B.
另外,催化剂作用介导着反应C,生成终产物(alkenyl-aryol ethers),表示为
结构C。
以上就是钯催化的脱羧Heck反应的一般机理,在实际的研究中,通过研究时
间的延长、加入分解剂发挥促进作用等方法进一步优化反应性能已取得了很好的效果。
除了以上提到的基本机理外,科研人员们还通过研究金属修饰剂,微观环境以及合成条件及抗歧化剂等更进一步改善反应性能,丰富了钯催化脱羧Heck反应技术。
实验和理论结果表明,钯催化的脱羧Heck反应机理是由一系列连续步骤组成,有较高的要求。
当前研究的重点是体现出高催化活性的最佳反应条件,尤其是找到有效的抗歧化剂,提高反应物的特异性,及其他优化反应性能的策略。
未来市场更广,也有更多的机会和应用。
钯催化反应机理汇总
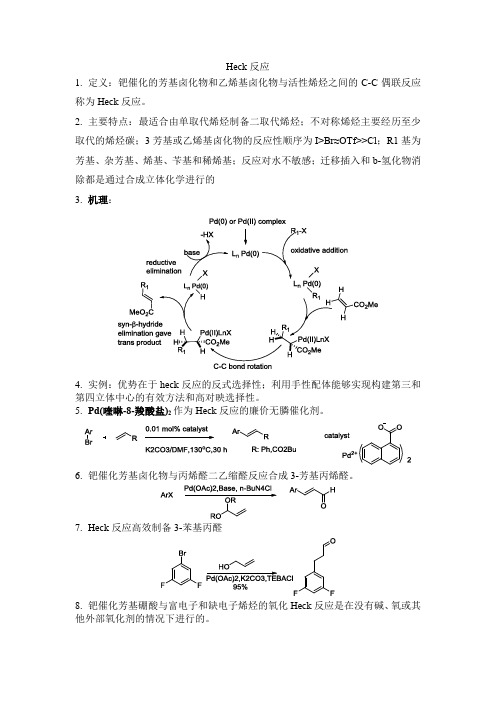
Heck反应1.定义:钯催化的芳基卤化物和乙烯基卤化物与活性烯烃之间的C-C偶联反应称为Heck反应。
2.主要特点:最适合由单取代烯烃制备二取代烯烃;不对称烯烃主要经历至少取代的烯烃碳;3芳基或乙烯基卤化物的反应性顺序为I>Br≈OTf>>Cl;R1基为芳基、杂芳基、烯基、苄基和稀烯基;反应对水不敏感;迁移插入和b-氢化物消除都是通过合成立体化学进行的3.机理:4.实例:优势在于heck反应的反式选择性;利用手性配体能够实现构建第三和第四立体中心的有效方法和高对映选择性。
5.Pd(喹啉-8-羧酸盐)2作为Heck反应的廉价无膦催化剂。
6.钯催化芳基卤化物与丙烯醛二乙缩醛反应合成3-芳基丙烯醛。
7.Heck反应高效制备3-苯基丙醛8.钯催化芳基硼酸与富电子和缺电子烯烃的氧化Heck反应是在没有碱、氧或其他外部氧化剂的情况下进行的。
Negishi reaction1.定义:钯或镍催化的有机锌试剂与有机卤化物、三氟化合物等的交叉偶联反应。
2.机理:3.优势:良好的官能团兼容性;条件温和;设备要求低;缺点:对水敏感4.有机锌试剂制备:5.实例:芳基溴化物与原位制备的2-吡啶锌的Negeshi偶联反应良好,分离收率为80%。
2-吡啶硼酸被证明是不稳定的,它的suzuki偶联剂与其他芳基硼酸一样有效。
文献:Org.Lett. 2002,4, 2385-23886.2005年开发了Pd-NHC催化体系,该体系能在室温下高收率地交叉偶联各种未活化的伯溴化物和烷基有机锌试剂。
文献:Org. Lett. 2007,9,4571-4574;Org. Lett. 2005,7,3805-38087.1977年首次报道了negishi交叉偶联反应的立体选择性反应。
目前,已开发出有效的催化剂和配体。
以下是含有机锌试剂的芳基卤化物的一个示例:文献:J. Am. Chem. Soc. 2006, 128, 3538-3539Sonogashira reaction1.定义:用钯催化剂、铜(I)助催化剂和胺基碱进行末端炔烃与芳基或卤化乙烯的偶联。
【国家自然科学基金】_c-h键活化_基金支持热词逐年推荐_【万方软件创新助手】_20140803

科研热词 推荐指数 c-h键活化 4 c-h活化 3 过渡金属 2 催化 2 高价金属配合物 1 非对称 1 间位 1 铜催化剂 1 钳形金属化合物 1 钯催化 1 醋酸三乙铵 1 酰基化 1 过渡金属催化 1 转化频率(tof) 1 衍生化 1 脱羧 1 肟醚导向 1 第ⅷ族过渡金属 1 离子液体 1 直接烃基化 1 甲苯 1 环羟基化 1 物理化学 1 氧化酰基化 1 机理 1 有机化学 1 手性 1 密度泛函理论方法 1 密度泛函理论(dft) 1 吡啶 1 双氧活化 1 双(咪唑啉)苯 1 卟啉类似物 1 二苄胺 1 二组氨酸一羧酸面式结构 1 二乙酸碘苯 1 ru催化 1 c-x键构筑 1 c-h键选择性活化 1 c-h官能团化 1 c-h,o-h键的活化 1 c-c键偶联反应 1 "一锅煮"的膦化/金属化反应 1
2014年 序号 1 2 3 4 5 6 7 8 9 10
2014年 科研热词 简化模型 离子对活性中心 甲烷氧化 晶面效应 微观动力学分析 密度泛函理论 rh 催化剂 c-h键活化 c-h 键活化 c-c 键偶联 推荐指数 1 1 1 1 1 1 1 1 1 1
2008年 序号 1 2 3 4 5 6 7 8 9
科研热词 邻位芳基化 甲烷c-h键活化 密度泛函理论 反应机理 从头算 二茂铁亚胺环钯化合物 g2m(+)方法 c-h活化 c-c偶联反应
推荐指数 1 1 1 1 1 1 1 1 1
2009年 序号 1 2 3 4 5 6 7 8 9 10 11 12
关于碳氢活化中含氮原子的导向基的综述

关于碳氢活化中含氮原子的导向基的综述摘要碳氢键活化反应因其具有原子经济性和环境友好的优点,引起了合成化学家的浓厚兴趣,已被广泛应用于药物分子、天然产物及功能材料的合成中。
其中,过渡金属催化的辅助基团导向的C-H键官能化由于区域选择性好,反应活性高而成为目前有机合成的热点研究领域,为高效和立体选择性地构筑药物分子和生物活性分子提供了新途径。
1 前言现代化学的宗旨是绿色可持续,但是在可预见的未来,化石资源等不可再生资源将逐渐枯竭,大自然对现代化学工业赖以存在的基本化学原料的供应也将会终止[1-3]。
所以碳氢键活化反应应运而生。
现在已成为现代有机合成化学最重要的合成工具,被广泛应用于药物、天然产物以及新型功能材料的合成中[4-8]。
一方面,石油、天然气以及煤的主要成分是含有惰性碳氢键的烷烃类化合物[9-11],而对这些宝贵的化石资源的利用,至今仍主要局限于将其燃烧而提供能源。
因此,对碳氢键进行有效的活化研究,可以大幅度提高资源的利用效率。
另一方面,目前为数不多的、已实现工业化的惰性烷烃碳氢键的转化存在着以下几个问题:(1)绝大多数需要在高温、高压以及强酸性的条件下进行,反应条件比较苛刻;(2)需要通过多步过程才能完成,反应步骤复杂;(3)转化效率不高,且通常存在选择性差的问题,得到人们所需产物的同时也会产生不必要的副产物[12-14]。
由于存在以上缺陷,目前化学化工领域对环境的污染已经成为严重影响人类生活质量及身体健康的社会问题。
因此,实现温和条件下惰性碳氢键的活化,可以减少能源消耗、降低污染物排放,使化学反应趋于“绿色化”[15-19]。
因而,从基础科学研究的角度讲,如何有效的对惰性碳氢键进行活化,如何发展新型高效,环境友好的构筑碳氢键方法一直是有机化学中研究的热点,研究具有极大的挑战性和重要的理论意义。
本文将从含氮原子导向基入手,从酰胺,吡啶,亚胺等[20-22]几个方面对碳氢键活化反应的发展进程进行综述。
钯和铜促进的芳香C-H键转化为碳卤键的最新研究进展

但是此类底物大多数 都存在 单双取 代无 法控制 的 问题 , 目 前主要是通过增加位 阻 的方 法来 控制单 双取代 。有 趣 的是 , 他 f , H 组还发现烯 烃也 可以发生 C—H活化 , 生成 C—c 产物 ( 1 方
程 式 7 。 )
Ae P ( c)f I dO^ 5 m0嘲
所 以判 断 C—H键 活 化 步 骤 是 决 速 步 骤 …。
H  ̄ 【O)em(m O' C PT(C】0O F N F d f f o (N 5 1 M 0% 。 1 P M )
。, g i f ,
这个反应也可 以用 C C, 为 氯源 , 个氯 源相 对 于 N S u1作 这 C 便宜 , 以也被很 多化学研 究工作 者关 注。最早报道 是 Sn r 所 af d o 小组使用催化量的钯对 2一苯基 吡啶类似物 邻位氯化 的反应 的
Aci a i n M e i td y Pal d um n Co tv to dae b l i a a d ppe r
M O o s ng
( ol eo i n n i n n cec s hn h i oma U i r t, h n hi 0 2 4 h a C l g f f adE v omet i e ,S ag a N r l nv s y S a g a 2 0 3 ,C i ) e Le r S n ei n
目前 , 家普遍认 为此类 反应是 氧化 剂对 P “ 大 d 进行 氧化 加 成, 生成 P 或者 P 一 P 二聚体 , d d d 然后再还原 消除生成 芳基 氯代物和 P “ d 。在对 3一甲基 一 2一苯基吡啶底物的钯催 化氯化 反应 的动力学研 究 中发现 , P ] 一级动 力学 , 对于 底物 和 [d是 而 N S是零级。同时动力学 同位素的研究 中发现 ( K 4 4 , C K / .= . )
过渡金属Pd催化的C-H键活化研究进展

山 东 化 工 收稿日期:2020-10-21作者简介:郭 艳(1979—),女,山东蓬莱人,经济学学士,烟台生产力促进中心(烟台市科技情报研究所),助理研究员。
过渡金属Pd催化的C-H键活化研究进展郭 艳,杨锦锦,贾挺挺(烟台生产力促进中心,山东烟台 264003)摘要:过渡金属催化的C-H键活化是构建C-C和C-X键(X为目标功能化基团,如O、N、S、B、卤素等)直接有效的方式,这为化学合成提供了更多选择性。
在众多过渡金属催化剂中,钯催化反应条件相对温和,具有良好的立体选择性和区域选择性。
因此,借助计算化学进一步了解钯催化的C-H活化反应的机理具有重要意义。
本文综述了在计算化学领域,钯催化的C-H活化反应的研究进展。
关键词:钯催化;C-H键活化;选择性;计算化学中图分类号:O643.36;O614.823 文献标识码:A 文章编号:1008-021X(2020)02-0082-07TransitionMetalPd-CatalyzedC-HBondActivationReactionGuoYan,YangJinjin,JiaTingting(YantaiProductivityPromotionCenter,Yantai 264003,China)Abstract:TransitionmetalcatalyzedC-Hbondactivationisaresearchhotspotinorganicsyntheticchemistry.ItisadirectandeffectivewaytoconstructC-CandC-Xbonds(Xisthetargetfunctionalgroup,suchasO,N,S,B,halogens).Itcanprovidemoreselectivityforchemicalsynthesis.MostC-Hbondactivationmainlydependsonnobletransitionmetals,andtheselectiveactivationofC-Hbondshasbeenthefocusandchallengeofresearch.Basedontheexamplesinrelatedliteratures,thisarticlereviewsthestudiesofPd-catalyzedselectiveC-Hbondactivation.Keywords:palladiumcatalysis;C-Hbondactivation;selectivity;C-Cbondconstruction;C-Xbondconstruction1 引言过渡金属催化剂具有抗毒性、热稳定性和使用寿命长等优点,过渡金属催化反应已成为高效、高选择性形成新键不可或缺的工具[1-10]。
过渡金属催化C-H活化插羰反应研究进展

过渡金属催化C-H活化插羰反应研究进展谢宁;金朝辉;高华晶;孙健【摘要】卤代芳烃的插羰反应是合成含羰基化合物的有效途径,使用未被官能化的C-H键实现插羰反应,具有更重要的价值.近年来,过渡金属催化C-H活化的偶联反应备受关注,但C-H键断裂需要很高的能量,对底物的结构和催化剂的类型要求苛刻.本文从反应中使用催化剂类型的角度,综述了过渡金属催化C-H键插羰反应的研究进展.【期刊名称】《吉林化工学院学报》【年(卷),期】2019(036)003【总页数】8页(P1-8)【关键词】过渡金属;C-H活化;插羰偶联反应【作者】谢宁;金朝辉;高华晶;孙健【作者单位】吉林化工学院石油化工学院,吉林吉林132022;吉林化工学院石油化工学院,吉林吉林132022;吉林化工学院石油化工学院,吉林吉林132022;吉林化工学院石油化工学院,吉林吉林132022【正文语种】中文【中图分类】O621.3一氧化碳(CO)是一种廉价的C1源,使用CO作为羰基源,来构筑含羰基的化合物是目前有机合成中引入羰基的一种重要手段.以卤代烃作为底物,与亲核性的物质插羰偶联的反应最为常见,比如:Heck反应、Suzuki反应、Sonogashira反应、Stille反应等,这类反应在较为温和的条件下就能发生.而C-H键断裂需要很高的能量,因此,碳氢键活化插羰反应不容易实现,具有一定的挑战性.这类反应目前主要是以酰胺或者氮原子作为定位基团,活化苯环邻位的碳氢键,实现插羰反应.为此,过渡金属催化的插羰反应吸引了广大研究者的兴趣,本文通过对近几年来的过渡金属催化下的插羰反应进行简要的综述.1 过渡金属在C-H活化插羰反应中的应用1.1 钌、铑等催化剂在C-H活化插羰反应中的应用早在1979年,Hong课题组报道了使用苯和乙烯作为底物,Rh4(CO)12作为催化剂,在CO的存在下,生成了少量苯丙酮[1].随后,有人使用Ir、Ru、Rh等催化剂活化了苯环上的碳氢键,实现了插羰反应[2].1992年,Moore发现钌催化剂可以催化一氧化碳和烯烃使吡啶环酰化[3].上述反应实现了碳氢键活化插羰的反应,但是体系中使用了苯或者吡啶,对人的身体有极大的危害,并且选择性和产率比较低,底物的兼容性差.1996年,Shinji Murai课题组使用咪唑衍生物作为底物,在Ru3(CO)12的作用下,与烯烃和一氧化碳作用,实现了插羰反应[4].随后,该课题组在相同的体系下,将咪唑环扩展到苯并咪唑、苯并恶唑、苯并噻唑等,苯并咪唑为底物的反应产率很高,而其他底物产率很低,作者对几种底物的pKa值做了测定,发现苯并咪唑的pKa值最大即酸性最弱,与过渡金属配位的能力最强,所以能更好的反应,而其他的底物则相反[5].2000年,该课题组使用[RhCl(CO)2]2催化剂,首次实现了sp3C-H键活化羰基化,所选的底物是N原子上连有吡啶环的四氢吡咯,四氢吡咯2位上碳氢键被活化,在一氧化碳的存在下,与乙烯发生插羰偶联反应,但该反应底物只能是这种结构,受到很大限制[6].随后,该课题组又将底物扩展到了N 原子上连有吡啶环的吲哚啉底物,在Rh3(CO)12作用下,与乙烯实现插羰偶联反应.同样的,此反应也受到底物结构限制,同时,必须在DMF溶剂中才能得到很好的收率,如图1所示[7].图1 杂环化合物与烯烃的插羰反应2004年,Naoto Chatani课题组分别使用Ru3(CO)12和Rh4(CO)12作为催化剂,实现了N-芳基吡唑与乙烯的插羰偶联反应.他们也发现底物的pKa值对反应的影响很大,吡唑环的pKa值较小,反应活性应该很低,与实验结果相反,作者推测是因为苯环的存在,增大了电子云密度,有利于还原消除的发生.所以在碳氢键活化插羰反应中,底物的pKa值和取代基的电子密度都会对实验结果造成影响[8].2009年,该课题组使用Ru3(CO)12作为催化剂,首次实现了底物中含有双氮原子配位的碳氢键活化羰基化反应,使用N-吡啶苯乙酰胺衍生物作为底物,成功合成了邻苯二甲酰亚胺的衍生物[9].2011年,该课题组使用Ru3(CO)12作为催化剂,酰胺和吡啶环作为定位基,成功的将甲基上的sp3C-H键羰基化,合成丁二酰亚胺的衍生物.作者尝试使用其他的定位基团,如苯基,以及改变取代基在吡啶环的位置对反应的影响很大,甚至不反应.他们还发现,使用苯环取代叔丁基上的一个甲基作为底物,只少量生成五元环,大量生成了六元环,说明sp2C-H键更容易断裂,同时用氘带实验证明了碳氢键的断裂步骤是决速步骤,随后,他们在之前的工作上,想要实现手性的诱导,合成手性分子,但是没有成功,如图2所示[10,11].图2 吡唑与吡啶衍生物的C-H活化插羰反应在他们工作的基础上,Tomislav Rovis等人在2011年,使用了较为复杂的RhCp*(MeCN)3(ClO4)2作为催化剂,苯甲酰胺作为底物,成功的合成了丁二酰亚胺的衍生物[12].近年来,在国内钌、铑等过渡金属对C-H键及C-C键的活化研究也受到关注并取得了一定的研究进展[13,14].1.2 钯催化剂在C-H活化插羰反应中的应用上述的反应都是使用Ru、Rh等催化剂,并且定位基团是杂环上的杂原子.Kazuhiko Orito等人在2004年,首次使用Pd(OAc)2作为催化剂,实现了苄胺衍生物的邻位碳氢键活化插羰成环反应,合成了一系列五元或者六元环,如图3所示[15].施章杰等人用PdCl2作为催化剂,N,N-二甲基苄胺和脂肪醇作为底物,成功的实现插羰偶联反应,与Kazuhiko Orito等人工作的区别是,氮原子上由于没有氢原子,所以不能发生成环反应(如图4所示)[16].2011年,MatthewJ.Gaunt等人使用N-取代的苯乙胺衍生物作为底物,使N-H和苯环上的C-H键发生断裂而发生插羰成环反应(如图5所示)[17].图3 苄胺衍生物的邻位C-H活化插羰成环反应图4 N,N-二甲基苄胺的插羰反应图5 以氨基为定位基团的钯催化C-H活化1.3 不同的定位基在C-H活化插羰反应中的应用2008年,于金权课题组使用Pd(OAc)2作为催化剂,首次实现了苯甲酸和苯乙酸衍生物邻位碳氢键活化羧化.这是第一次使用羰基做定位基实现碳氢键活化羰基化的反应,作者首次得到了Pd配合物的中间体,为机理的证明提供了有力证据.随后,作者又将底物扩展到了酰胺做定位基团的衍生物,活化sp3C-H键羰基化,成功的合成了丁二酸衍生物.2011年,该课题组使用苯乙醇作为底物,Pd(OAc)2作为催化剂,在手性配体N-叔丁氧羰基-L-丙氨酸的作用下,实现了用羟基做定位基团合成内酯的反应(如图6所示)[18-20].2009年,Booker-Milburn课题组使用芳基取代的脲衍生物作为底物,Pd(OTs)2(MeCN)2作为催化剂,成功的活化了苯环邻位的碳氢键,在分子内插羰成环合成了内酯类衍生物,在甲醇的存在下,则可以一锅法合成苯甲酸甲酯的衍生物.图6 以不同定位基团的钯催化C-H活化该反应对底物的依赖性很强,必须是这种脲的结构,酰胺基作定位基是不反应的.该反应的应用价值很高,可以合成喹唑酮类衍生物,内酯类衍生物可以在中性条件下水解成邻氨基苯甲酸甲酯,可以用于制备各种氨基取代的苯甲酸[21].有意思的是,于金权课题组使用Pd(OAc)2作为催化剂,在TsOH·H2O的作用下,用酰胺类衍生物成功合成了羧化的产物.与Booker-Milbur等人的工作不同的是,底物可以使用酰胺做定位基团,而不再仅仅局限与复杂的脲的衍生物,体系中少量的水会使酸酐和苯并噁嗪酮中间体水解成羧酸(如图7所示)[22].图7 C-H活化邻位羧化2 过渡金属催化C-H活化插羰反应其他的应用2009年张绪穆等人使用[Rh(COD)Cl]2作为催化剂,2-苯基吡啶与脂肪醇作用实现了插羰偶联反应,合成了酯类衍生物.但是该反应仅限于直链的脂肪醇,异丙基、叔丁基醇或者酚类的衍生物作为底物,反应的产率会<5%,反应中过硫酸氢钾复合盐是必须的(如图8所示)[23].2013年,史炳峰课题组使用钯催化体系,同样实现了2-苯基吡啶与戊醇的插羰偶联反应,但是仅限于戊醇[24].该课题组使用2-苯氧基吡啶作为底物,与醇生成了羧化产物(如图9所示)[25].2013年,Matthias Beller课题组使用Ru(COD)nCl2作为催化剂,使2-苯基吡啶与碘代芳烃实现了插羰偶联,该反应使用水做溶剂,具有一定的环保意义(如图10所示)[26].图8 铑催化芳香族C-H活化插羰反应图9 钯催化芳香族C-H活化羧化反应图10 钌催化C-H活化插羰反应2011年,江焕峰课题组使用烯丙基苯衍生物作为底物,在Pd(OAc)2的作用下与醇类发生插羰偶联反应,该反应使用混合氧化剂,其中摩尔分数BQDDQ(41),选择性可达85%,而收率可以达到77%(如图11所示)[27].Jaume Granell课题组使用没有活化的苯乙胺衍生物作为底物,成功的实现了插羰环合,并且作者巧妙的证明了没有活化的这种仲胺,容易选择性的生成六元环,而并非是五元环(如图12所示)[28].图11 钯催化氧化插羰反应图12 苯乙胺的插羰反应2014年,江焕峰课题组选用喹啉酮的衍生物作为底物,实现了稠环芳烃的合成,作者使用零价钯Pd2(dba)3作为催化剂,在醋酸铜和对甲苯磺酸的作用下,原位生成Pd(OTs)2,进入催化循环(如图13所示)[29].图13 多环芳烃的合成雷爱文课题组在2011年使用吲哚作为底物,三苯基膦作为配体,在空气存在下,成功的活化了3位的碳氢键实现了与醇的衍生物的插羰偶联反应,有意思的是,作者使用噻吩环作为底物只在邻位发生反应,而若是将吲哚的N上无取代基,插羰反应则发生在N-H键[30].而李福伟等人使用[Rh(COD)Cl]2作为催化剂,过硫酸钾作为氧化剂,得到了与雷爱文课题组相同的产物(如图14所示)[31].图14 吲哚类化合物的C-H活化2012年,该课题组使用Pd(OAc)2作为催化剂,也得到的吲哚3位的羧化产物,不同的是,在反应体系中引入了I2,从而提出的机理也有所不同.首先,二价钯在一氧化碳的作用下还原成零价钯,吲哚在I2的作用下在3位碘代,然后经过氧化加成、还原消除,得到目标产物(如图15所示)[32].2013年,雷爱文课题组使用N,N-二甲基苯胺作为底物,使用Pd/Cu的需氧催化体系,与对甲基苯乙烯衍生物反应,得到了插羰产物,首次将Cu(Ⅱ)/O2活化C-N键的体系引入到插羰偶联反应中[33].2014年,Troels Skrydstrup等人使用2-羟基吲哚和溴代芳烃作为底物,Xantphos作为配体,在羟基吲哚的3位实现了插羰偶联反应.作者推测的机理是底物的羰基和MgCl2配位,在三乙胺的作用下去质子化,得到了中间体B,随后中间体A与其发生金属交换,可能生成了一种类似π-烯丙基钯的中间体,再经过还原消除生成了中间体C,在经过与MgCl2、三乙胺的作用,最后质子化,得到最终产物(如图16所示)[34].图15 吲哚C-H活化插羰反应机理图16 2-羟基吲哚C-H活化插羰反应机理3 结论与展望综上所述,过渡金属催化C-H键活化的反应已经备受到科研人员的关注.碳氢键活化构筑含有羰基结构的化合物是一种重要的有机合成方法.目前的研究主要集中在以下几个方面:(1)底物的选择.包括酰胺、胺、杂环、甲苯等等,未来的研究需要巧妙的设计底物.(2)催化剂的选择.最初的研究使用了Ru、Rh等复杂的催化剂,但是最近几年的研究基本上都是围绕这简单易得Pd催化剂,所以未来的研究更倾向于简单易得的金属或非金属催化剂.(3)溶剂的选择.从上述的研究中可以看出,有些反应对溶剂很敏感,不同的溶剂对反应结果影响很大,而且混合溶剂也可达到意想不到的效果.(4)机理的研究.目前还没有π-烯丙基钯这样的机理提出,在未来的研究中可能会出现.总而言之,过渡金属催化C-H活化插羰反应已经成为有机合成领域中一大研究热点,并且在未来的发展中也会成为不可或缺的一环.【相关文献】[1] Hong P,YamazakiH.Reactions of ethylene and benzenes catalyzed by rhodium carbonyls under carbon monoxide.The formation of styrenes and 3-pentanone[J].Chemistry Letters,1979(8):1335-1336.[2] Sakakura T,Sodeyama T,Sasaki K,et al.Carbonylation of hydrocarbons via carbon-hydrogen activation catalyzed by RhCl(CO)(PMe3)2 under irradiation[J].Journal of the American Chemical Society,1990(112):7221-7229.[3] Moore E,Pretzer W,O’Connell T,et al.Catalytic and regioselective acylation of aromatic heterocycles using carbon monoxide and olefins[J].Journal of the AmericanChemical Society,1992(114):5888-5890.[4] Chatani N,Fukuyama T,Kakiuchi F,et al.Ru3(CO)12-Catalyzed Coupling of Heteroaromatic C-H/CO/Olefins.Regioselective Acylation of the Imidazole Ring[J].Journal of the American Chemical Society,1996(118):493-494.[5] (a) Fukuyama T,Chatani N,Tatsumi J,et al.Ru3(CO)12-Catalyzed Site-Selective Carbonylation Reactions at a C-H Bond in Aza-Heterocycles[J].Journal of the American Chemical Society,1998(120):11522-11523;(b) Wu X,Anbarasan P,Neumann H,et al.Palladium-Catalyzed Carbonylative C-H Activation of Heteroarenes[J].Angewandte Chemie International Edition,2010(49):7316-7319.[6] Chatani N,Asaumi T,Ikeda T,et al.Carbonylation at sp3 C-H Bonds Adjacent to a Nitrogen Atom in Alkylamines Catalyzed by Rhodium Complexes[J].Journal of the American Chemical Society,2000(122):12882-12883.[7] Chatani N,Yorimitsu S,Asaumi T,et al.Ru3(CO)12-Catalyzed C-H/CO/Olefin Coupling of N-Pyridylindolines.Direct Carbonylation at a C-H Bond δ to the Pyridine Nitrogen[J].The Journal of Organic Chemistry,2002(67):7557-7560.[8] Asaumi T,Matsuo T,Fukuyama T,et al.Ruthenium- and Rhodium-Catalyzed Direct Carbonylation of the Ortho C-H Bond in the Benzene Ring of N-Arylpyrazoles[J].The Journal of Organic Chemistry,2004(69):4433-4440.[9] (a) Inoue S,Shiota H,Fukumoto Y,et al.Ruthenium-Catalyzed Carbonylation at Ortho C-H Bonds in Aromatic Amides Leading to Phthalimides:C-H Bond Activation Utilizing a Bidentate System[J].Journal of the American Chemical Society,2009,131,6898-6899;(b) Wrigglesworth J,Cox B,Lloyd-Jones G,et al.New Heteroannulation Reactions of N-Alkoxybenzamides by Pd(Ⅱ) Catalyzed C-H Activation[J].Organic letters,2011(13):5326-5329.[10] Hasegawa N,Charra V,Inoue S,et al.Highly Regioselective Carbonylation of Unactivated C(sp3)-H Bonds by Ruthenium Carbonyl[J].Journal of the American Chemical Society,2011(133):8070-8073.[11] Hasegawa N,Shibata K,Charra V,et al.Ruthenium-catalyzed cyclocarbonylation of aliphatic amides through the regioselective activation of unactivated C(sp3)-Hbonds[J].Tetrahedron,2013(69):4466-4472.[12] Du Y,Hyster T,Rovis T.Rhodium(III)-catalyzed oxidative carbonylation of benzamides with carbon monoxide[J].Chemical Communications,2011(47):12074-12076.[13] 于晓波,陈连发,王巍.钌配合物催化C—H活化反应合成10-乙氧酰基苯并喹啉的研究[J].吉林化工学院学报,2015,32(11):104-106.[14] 于晓波,庄德吉,陈佳特.铑催化无水茚三酮和炔烃脱羰环加成C-C键活化反应的研究[J].吉林化工学院学报,2018,35(11):64-68.[15] Orito K,Horibata A,Nakamura T,et al.Preparation of Benzolactams by Pd(OAc)2-Catalyzed Direct Aromatic Carbonylation[J].Journal of the American Chemical Society,2004(126):14342-14343.[16] Li H,Cai G,Shi Z.LiCl-Promoted Pd(Ⅱ)-catalyzed ortho carbonylation of N,N-dimethylbenzylamines[J].Dalton Transactions,2010(39):10442-10446.[17] Haffemayer B,Gulias M,Gaunt M.Amine directed Pd(Ⅱ)-catalyzed C-H bond functionalization under ambient conditions[J].Chemical Science,2011(2):312-315. [18] Giri R,Yu J.Synthesis of 1,2- and 1,3-Dicarboxylic Acids via Pd(Ⅱ)-Catalyzed Carboxylation of Aryl and Vinyl C-H Bonds[J].Journal of the American Chemical Society,2008(130):14082-14083.[19] Yoo E,Wasa M,Yu J.Pd(Ⅱ)-Catalyzed Carbonylation of C(sp3)-H Bonds:A New Entry to 1,4-Dicarbonyl Compounds[J].Journal of the American Chemical Society,2010(132):17378-17380.[20] Lu Y,Leow D,Wang X,et al.Hydroxyl-directed C-H carbonylation enabled bymono-N-protected amino acid ligands:An expedient route to 1-isochromanones[J].Chemical Science,2011(2):967-971.[21] Houlden C,Hutchby M,Bailey C,et al.Room-Temperature Palladium-Catalyzed C-H Activation:ortho-Carbonylation of Aniline Derivatives[J].Angewandte Chemie International Edition,2009(48):1830-1833.[22] Giri R,Lam J,Yu J.Synthetic Applications of Pd(Ⅱ)-Catalyzed C-H Carboxylation and Mechanistic Insights:Expedient Routes to Anthranilic Acids,Oxazolinones,and Quinazolinones[J].Journal of the American Chemical Society,2010(132):686-693.[23] Guan Z,Ren Z,Spinella S,et al.Rhodium-Catalyzed Direct Oxidative Carbonylation of Aromatic C-H Bond with CO and Alcohols[J].Journal of the American Chemical Society,2009(131):729-733.[24] Liu B,Shi B.Efficient Synthesi s of Carboxylic Esters via Palladium(Ⅱ)-Catalyzed Direct Alkoxycarbonylation of Arenes with CO and Alcohols[J].Synlett,2013(24):2274-2278. [25] Liu B,Jiang H,Shi B.Pd(Ⅱ)-catalyzed oxidative alkoxycarbonylation of 2-phenoxypyridine derivatives with CO and alcohols[J].Organic & Biomolecular Chemistry,2014(12):2538-2542.[26] Tlili A,Schranck J,Pospech J,et al.Ruthenium-Catalyzed Carbonylative C-C Coupling in Water by Directed C-H Bond Activation[J].Angewandte Chemie International Edition,2013(52):6293-6297.[27] Chen H,Cai C,Liu X,et al.Highly regioselective palladium-catalysed oxidative allylic C-H carbonylation of alkenes[J].Chemical Communications,2011(47):12224-12226.[28] López B,Rodriguez A,Santos D,et al.Preparation of benzolactams by Pd(Ⅱ)-catalyzed carbonylation of N-unprotected arylethylamines[J].Chemical Communications,2011(47):1054-1056.[29] Ji F,Li X,Wu W,et al.Palladium-Catalyzed Oxidative Carbonylation for the Synthesis of Polycyclic Aromatic Hydrocarbons(PAHs)[J].The Journal of Organic Chemistry,2014(79):11246-11253.[30] Zhang H,Liu D,Chen C,et al.Palladium-Catalyzed Regioselective Aerobic Oxidative C-H/N-H Carbonylation of Heteroarenes under Base-Free Conditions[J].Chemistry-A European Journal,2011(17):9581-9585.[31] Lang R,Wu J,Shi L,et al.Regioselective Rh-catalyzed direct carbonylation of indoles to synthesize indole-3-carboxylates[J].Chemical Communications,2011(47):12553-12555.[32] Lang R,Shi L,Li D,Xia,et al.A General Method for Palladium-Catalyzed Direct Carbonylation of Indole with Alcohol and Phenol[J].Organic letters,2012(14):4130-4133.[33] Shi R,Lu L,Zhang H,et al.Palladium/Copper-Catalyzed Oxidative C-H Alkenylation/N-Dealkylative Carbonylation of Tertiary Anilines[J].Angewandte Chemie International Edition,2013(52):10582-10585.[34] Lian Z,Friis S,Skrydstrup T.Palladium-Catalyzed Carbonylative α-Arylation of 2-Oxindoles with(Hetero)aryl Bromides:Efficient and Complementary Approach to 3-Acyl-2-oxindoles[J].Angewandte Chemie International Edition,2014(53):9582-9586.。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
肟醚类化合物作为导向基团在钯催化C-H 活化中的应用作者:程肖丁来源:《当代化工》2019年第01期摘 ;;;;;要:肟醚结构中氮原子可与过渡金属配位成金属螯合物,诱导整个反应体系的定向和化学选择性催化。
目前,肟醚衍生物作为导向基团广泛应用于过渡金属催化的C-H官能化,构建C-C键、C-N键、C-X键、C-O键等。
肟醚除可诱导sp2/sp3 C-H活化,实现C-H键的官能化外,还可被脱去生成酮醇类,与过渡金属合成稳定配合物,亦可作为氧化剂参与反应。
介绍了肟醚类化合物作为导向基团在钯催化C-H活化领域中的应用和进展。
关 ;键 ;词:肟醚;C-H活化;钯催化;导向基团中图分类号:TQ 032 ;;;;;;文献标识码: A ;;;;;;文章编号: 1671-0460(2019)01-0092-06Abstract: Oxime ethers can coordinate with transition metals to form the metal chelates,which can induce the directional and chemoselective catalysis of the entire reaction system. As directing groups, oxime ethers are widely used in C-H functionalization catalyzed by transition metals,constructing C-C bonds, C-N bonds, C-X bonds, C-O bonds,C-N bonds and so on. In this paper, the application and research progress of oxime ethers as directing groups in the palladium-catalyzed C-H activation were described. It's pointed out that oxime ethers can be used to directsp2/sp3 C-H activation.Key words: Oxime ethers; C-H activation; Pd-catalysis; Directing groups (DG)一直以来,肟醚类化合物在有机化学以及植物化学领域中占据着重要地位。
肟和肟醚的基本结构单元(C=NO-R)是很多药物的关键骨架,具有抗菌、抗肿瘤和消炎等生物活性,被广泛应用于医药领域[1-3]。
此外,肟醚类化合物还是多功能的化学中间体和前体,可通过化学反应如亲电加成、环化加成、过渡金属催化交叉偶联等生成一系列含氮化合物[4-6]。
由于肟醚结构中氮原子孤对电子的存在,使得该结构呈现出一定的路易斯碱特性,因此可与过渡金属配位络合,是一些过渡金属催化的有机转化中重要的诱导基团,尤其是在钯催化的C-H键的活化方面。
C-H键活化是构建C-C键的重要策略之一,最早可以追溯到19世纪末人们发现的金属与碳氢化合物之间的的反应,然而真正实现过渡金属参与催化的C-H键活化是在20世纪60年代,以Fujiwara[7]的烯烃芳基化为典型代表。
一般地,底物的C-H键因其所处位置不同、取代基不同而导致带电性不同,因此,对C-H键进行选择性的官能团化是可以控制并实现的。
对于某些底物,可以通过路易斯碱诱导基团对金属催化剂配位,从而控制分子内的碳氢活化。
常见的导向基团有烷基甲酰基、烷基甲酰胺基、烷基甲酰氧基、羧基、2-吡啶基、亚胺、肟醚、恶唑啉以及N-甲氧基胺基甲酰基等。
目前,关于肟醚作为导向基团在C-H活化方面的应用已成为C-H官能化中的重要研究领域和方向。
本文根据近几年来肟醚作为导向基团在碳氢活化方面的应用,系统地概述肟醚的诱导作用并作出自己的见解和展望,主要从肟醚参与诱导的C-H官能化类型以及产物的延伸方面做出详细阐述。
1 ;C-H键的官能化1.1 ;C-H键的乙酰氧基化目前,关于sp3 C-H键的常规氧化催化方法已经在合成化学中广泛应用。
然而,由于sp3 C-H 键的键能较大,且氧化产物易于被氧化剂过度氧化以及在某些复杂有机分子的存在下难以实现区域选择性官能化,所以sp3 C-H 键的催化氧化仍然具有挑战性。
甲烷和一些更复杂烷烃的C-H键的催化氧化条件往往比较苛刻,且底物耐受性较差。
2004年,Sanford[8]报道了一种选择性钯催化氧化芳烃和苄型C-H键的新方法。
他发现在含有肟基团和吡啶骨架的底物中,未取代的sp3 C-H键可在氧化剂PhI(OAc)2存在下进行高区域化学选择性的Pd(II)催化。
该催化同时具备反应可行性和空间诱导选择性,Pd(II)与底物生成的螯合物诱导和促进sp3 C-H键的激活,同时,螯合物依据烷烃底物的空间和电子性质定向活化C-H键,实现C-O键的构建。
Sanford研究的烷烃催化氧化主要集中在频哪醇酮O-甲基肟的官能化,在催化剂Pd (OAc)2 (5 mol%)和氧化剂PdI(OAc)2 (1.1 eq)作用下得到了β-含氧产物(图1)。
随后,Sanford在相同的反应条件下,探索底物适用性范围及转化的区域选择性。
实验得出,一系列O-甲基肟底物分别以0%~%的产率得到相应的β-含氧产物(表1)。
此外,Sanford发现取代环酮肟也是良好的氧化底物和诱导基团,并且转化的速率对构象效应极其敏感。
Sanford[9]在2010年报道了以O-乙酰肟为导向基团的Pd(II)催化的sp2和sp3 C-H官能化反应,且C-H官能化产物可转化为邻位或者β-官能化的酮、醇、胺和杂环。
酮类化合物是有机合成中通用且应用广泛的合成中间体。
然而,酮是Pd(II)的不良配体,通常在Pd催化的C-H官能化中是无效的导向基团。
在之前的研究中,Sanford[10]利用肟醚(3)为有效的引导基团用于PhI(OAc)2/Pd(OAc)2催化氧化的sp2和sp 3 C-H乙酰氧基化反應,然而,从官能化产物(4)中除去肟醚保护基是比较困难的(图2)。
因此,Sanford试图在C-H官能化时用乙酰基修饰肟基,使其转化为具有更多配位点的O-乙酰肟醚衍生物,然后在碱性环境下脱去该修饰基团得到相应的酮产物。
实验证明,原位产生的O-乙酰肟(6)作为Pd(II)催化C-H官能化中的导向基团在催化条件下稳定存在,且可在一定条件下脱去乙酰保护基,破坏肟醚骨架得到酮(7)。
而端位的sp3 C-H键经乙酰氧基化构建C-OAc(6),后经脱乙酰基作用生成了醇(7),间接实现了C-H键的羟基化(图3)。
Sanford尝试了14个例子,涉及脂肪族肟、芳香族肟以及环烷基肟等,在钯催化氧化C-H键活化诱导中以33-86%的产率得到O-乙酰肟衍生物。
随后,得到的O-乙酰肟衍生物通过各种方法(K2CO3/NaHSO3,K2CO3/NaHSO3/H2,K2CO3/p- TsOH/ZnCl2等)除去乙酰修饰基团得到相应的醇、胺类、酮和含氮杂环。
2012年,Ren 和Dong[11]通过肟醚诱导和钯催化氧化作用活化sp3 C-H键,生成乙酰氧基取代的肟醚衍生物(10),随后肟醚10经两种不同氧化途径得到化学结构不同的1,2-二醇衍生物(11和12)。
他们指出脂肪族sp3 C-H键进行选择性官能化,从一元醇衍生物经过碳氢活化再氧化得到两种化学结构不同的1,2-二醇衍生物(图 4)。
其中,肟用作该转化的外导向基团(DG)和醇替代物。
由此可知,在C-H活化中使用外导向基团(exo-DG)可能会发现新的转化和易消除的诱导基团。
值得一提的是,活化产物经脱乙酰基后得到相应的醇(12),与Sanford除去乙酰修饰基团的研究有着异曲同工之妙,但是Ren 和Dong采用的条件更加温和,保护了肟基团骨架,只是选择性地脱去乙酰基团。
同样地,该反应可认为是C-H键的间接羟基化。
Ren 和Dong对sp3 C-H键活化的反应条件进行了优化和探索,以肟衍生的2-丁醇(A:2,6-二甲基苯基,B:H,8)为底物,发现Pd(OAc)2 (10 mol%)/PhI(OAc)2/AcOH/Ac2O体系是构建C-OAc键产率最高的反应条件。
在此基础上,他们对该官能化反应进行了底物适用性范围探索(12个例子),发现均可以与肟醚的sp3 C-H键发生选择性氧化(44%~%)。
除了-CH3的C-H键可以活化之外,Ren 和Dong发现环状肟醚中的亚甲基(-CH2-)和桥头位上的次甲基(-CH)同样可以被官能化。
另外,薄荷醇衍生的肟醚在进行钯催化氧化时发生了氧化骨架重排。
关于C-H活化产物中残留诱导基团的消除问题,Johnson[12]在合成甾体生物碱Paspaline 的研究中也进行了探讨。
与Sanford的O-乙酰肟醚经诱导基团转移生成酮醇的研究类似,Johnson利用HCl和DMP使O-Bn和O-Ac键断裂,除去导向基团,生成相应的醛酮(图5)。
Johnson将Sanford报道的C-H键乙酰氧基化条件直接用于肟(13),以79%的产率得到了单一非对映异构体的单乙酸酯(14),该转化完成了最终季铵中心的组装,随后经脱保护和氧化作用生成酮醛化合物(15)。
1.2 ;C-H键的酰胺化2006年,Che[13]报道了钯催化级联活化的sp2 C-H键和sp3 C-H键的分子间酰胺化反应,研究并开发了一种基于级联螯合-定向环化钯催化C-H键的酰胺化方案。
该方案可实现未激活的sp2和sp3 C-H键的活化催化并构建C-N键,具有显著的区域和化学选择性。
Che发现以O-甲基肟为诱导基团的钯催化C-H键活化可以得到极高转化率和良好收率的邻位酰胺化产物(图6)。
由对位取代的苯甲醛衍生的O-甲基肟有效地转化为相应的邻位C-H 酰胺化产物,且具有良好的区域选择性。
1.3 ;C-H键的芳基化2008年,Yu和Shi[14]等人实现了以钯催化、肟醚基团导向的O-甲基-(E)-2甲基苯甲醛肟与芳基硼酸之间的交叉偶联反应,构建C-Ar键。
以Cu(OTf)2及苯醌(BQ)为氧化剂,通过2,6-二甲氧基吡啶(DMOP)抑制芳基硼酸与C=N键的直接加成,从而实现邻位C-H键的活化,构建C-Ar键(图7)。
Shi还进行了取代硼酸的底物耐受性探索,发现各类电性的芳基硼酸均可以得到较高产率的目标化合物,且具有良好的官能团耐受性。
值得一提的是,对于空间位阻较大的芳基酮肟,在相同的条件下,其活化转化率较低,然而在当体系中去除2,6-二甲氧基吡啶时,则可以得到良好的转化率。
1.4 ;C-H键的酰化2010年,Chan 和Yu[15]开发了一种新型的钯催化C-H键活化方案,以叔丁基过氧化氢(TBHP)作为氧化剂使芳基酮肟和醛发生交叉偶联,进行直接C-H键酰化。