干扰载体构建SOP
RNA干扰载体的构建

C hapter 40 C onstruction of Hairpin RNA-Expressing Vectorsfor RNA-Mediated Gene Silencing in FungiShaobin Zhong,Yueqiang Leng,and Melvin D.BoltonAbstractR NA-mediated gene silencing is one of the major tools for functional genomics in fungi and can be achieved by transformation with constructs that express hairpin (hp) RNA with sequences homologous to the target gene(s). To make an hpRNA expression construct, a portion of the target gene can be amplifi ed by PCR and cloned into a vector as an inverted repeat. The generic gene-silencing vectors such as the pSilent1 and pSGate1 have been developed and are available for RNA-mediated gene-silencing studies. In this protocol, we describe construction of hpRNA-expressing constructs using both pSilent1 and pSGate1. With pSilent1, the PCR products of the target gene are inserted into the vector by conventional cloning (i.e., restriction enzyme digestion and ligation). For pSGate1, the PCR products of the target gene are inserted into the vector through the Gateway-directed recombination system. In this chapter, we describe the construction of RNAi vectors for RNA-mediated gene silencing using both pSilent1 and pSGate1.K ey words:R NAi ,G ene silencing ,G ateway cloning system ,H airpin RNA ,p Silent1 ,p SGate11.IntroductionR NA-mediated gene silencing or RNA interference (1)has beenfound to be a common gene-silencing phenomenon in eukaryoticorganisms. In this process, double-stranded RNA (dsRNA),which can be induced by viral RNA, hairpin RNA (hpRNA), orvirus-induced gene-silencing encoded RNA (2), is degraded intosmall interfering RNAs (siRNAs). These siRNAs are incorporatedinto the RNA-induced silencing complex to target and degrademRNA of genes with complementary sequence (2–5). RNAi hasbeen demonstrated to silence genes in a number of fungi (forreview see ref. (6)). Examples include C ryptococcus neoformans(5),M agnaporthe oryzae(7–9),V enturia inaequalis(10),H istoplasma Melvin D. Bolton and Bart P.H.J. Thomma (eds.), Plant Fungal Pathogens: Methods and Protocols,Methods in Molecular Biology,vol. 835, DOI 10.1007/978-1-61779-501-5_40, © Springer Science+Business Media, LLC 2012623624S. Zhong et al.capsulatum ( 11 ) , S chizophyllum commune ( 12 ) , C oprinopsiscinerea ( 13,14 ) , M ortierella alpina ( 15 ) , D ictyostelium discoi-deum (16 ) , A spergillus and F usarium species ( 4 ) , B ipolaris oryzae ( 17 ) , C ladosporium fulvum ( 18– 20 ) , and O phiostoma novo-ulmi( 21 ) . Most of the studies mentioned above used RNAi vectors containing inverted repeats of the target gene or its partialsequence separated by a spacer for transformation. Constructionof these RNAi vectors often involves several cloning steps andthus is laborious and time-consuming. Nakayashiki et al. (9 ) devel-oped the pHANNIBAL-like silencing vector, pSilent-1 (see Fig.1 ), for RNA silencing studies in fi lamentous fungi. To make anRNAi construct expressing the hpRNA of the target gene witha ) A schematic map of the vector pSilent1 . Unique restriction sites are indicated.A mpR ampicillin-resistant gene; H phR hygromycin-resistant gene; I ntron intron 2 of thecutinase (CUT) gene from M agnaporthe oryzae; PtrpC Aspergilus nidulans trpC promoter;T trpC A. nidulans trpC terminator. ( b ) Restriction sites are u nderlined .A sterisks indicateunique sites in the silencing vector. I talic nucleotides indicate sequence from the CUTgene and b old letters represent 5 and 3 splice sites ( 9 ).62540 Construction of Hairpin RNA-Expressing Vectors …pSilent-1, a DNA fragment from a target gene is amplifi ed byPCR with primers containing appropriate restriction sites andcloned sequentially into each side of the intron spacer in the vec-tor (see Fig.2 ). The applicability of pSilent-1 was demonstrated in several phytopathogenic ascomycete fungi, including M . oryzae ,C olletotrichum lagenarium , and B . oryzae (9, 17 ) . However, pSi-lent-1 is a restriction enzyme-based cloning vector and thereforemay not be suitable for the construction of large numbers ofRNAi vectors for high-throughput gene-silencing studies.Recently, a pHELLSGATE-like RNAi vector (pTroya) based onInvitrogen’s Gateway technology has been developed and shownto be useful in C . gloeosporioides (22 ) . A similar type of RNAi vec-tor (pSGate1) (see Fig.3 ) based on the Gateway system has also been developed and used for gene silencing in the spot blotchfungal pathogen of barley and wheat C ochliobolus sativus (23 ) . To make an RNAi construct with pSGate1, a portion of the targetgene is amplifi ed by PCR with primers containing the a tt B 1 anda tt B 2 sites, and cloned into an entry plasmid (pDONR221) tocreate an entry clone by BP reaction (see Fig.4 ). The insert in the entry clone is then recombined into both sides of the intronspacer in pSGate1 in opposite orientation with the LR reaction,resulting in an RNAi construct expressing the hpRNA of the tar-get gene (see Fig.4 ). In this protocol, we describe the construc-tion of RNAi vectors for RNA-mediated gene silencing usingboth pSilent1 and pSGate1.F ig. 2. A fl owchart showing the construction of a hairpin (hp) RNA-expressing RNAi vector using pSilent1. A DNA fragment from a target gene was amplifi ed by PCR with primers containing appropriate restriction sites. The PCR product was cloned into each side of the intron in pSilent1 sequentially, resulting in an RNAi construct expressing the hpRNA of the target gene.626S. Zhong et al.1. B ench top microcentrifuge.2. E nvironmental incubator shaker.3. p H meter.2.Materials2.1.Equipmentand Consumables F ig. 4. F lowchart showing the construction of a hairpin (hp) RNA-expressing RNAi vector using pSGate1. A DNA fragment from a target gene is amplifi ed by PCR with primers containing the a ttB1and att B 2 sites. The PCR product is cloned into the plasmid pDONR221 to create an entry clone by BP reaction. The insert in the entry clone is then replaced by the c cdB genes of pSGate1 by LR reaction, resulting in an RNAi construct expressing the hpRNA of the target gene.Hind III F ig. 3. A schematic map of the Gateway cloning system-based RNAi vector, pSGate1 .The ccdB cassettes were derived from pHellsgate-12 ( 24 ) and cloned into the silencing vector pSilent1 ( 9 ) . Unique restriction sites are indicated. A mpR ampicillin-resistant gene; H phR hygromycin-resistant gene; I ntron intron 2 of the cutinase (CUT) gene from M agnaporthe oryzae; PtrpC Aspergilus nidulans trpC promoter; T trpC A. nidulans trpC terminator.62740 Construction of Hairpin RNA-Expressing Vectors … 4. T hermal cycler. 5. E lectrophoresis equipment. 6. W ater bath. 7. P etri plates. 8. C ell culture tubes. 9. M icrocentrifuge tubes. 10. P CR tubes. 1. L B medium: 10 g tryptone, 5 g yeast extract, 10 g NaCl, 20 g of agar (for plates only) to 900 mL deionized H 2 O . Adjust pH to 7.0 with NaOH solution. Add deionized H 2O to a fi nalvolume of 1 L. Autoclave. Cool to 55–60°C and add 1 mL of ampicillin (100 mg/mL; fi lter-sterilized with 0.2- m m mem-brane fi lter) or kanamycin (50 mg/mL; fi lter-sterilized with 0.2- m m membrane fi lter) as necessary.2. S OC medium: 2% (w/v) tryptone, 0.5% (w/v) yeast extract,10 mM NaCl, 2.5 mM KCl, 10 mM MgCl 2 , 10 mM MgSO 4, 20 mM glucose. Autoclave. 3. E lectrophoresis-grade agarose.4. X ho I and H ind I II restriction enzymes, including 10× reactionbuffers as provided by supplier.5. T 4 ligase, including 10× ligation buffer as provided by supplier.6. P CR-grade Taq polymerase, including 10× PCR amplifi cationbuffer as provided by supplier.7. P CR-grade dNTPs.8. C hemically competent E scherichia coli cells.9. P roteinase K: supplied with Gateway ® BP or LR kits.10. T E (10 mM Tris–HCl pH 8.0, 1 mM EDTA).11. 30%PEG/MgCl 2 : supplied with Gateway ®BP or LR kits.12. D NA purifi cation Kit.13. P lasmid miniprep Kit.14. G ene-specifi c primers with appropriate restriction sites at the5 ¢ end (for construction of RNAi vectors with pSilent1) (see Note 2).15. p Silent1 (for construction of RNAi vectors with pSilent1; seeNote 3).16. G ene-specifi c primers with a tt B 1 and a tt B 2 sequences at the5 ¢ end (for construction of RNAi vectors with pSGate1; see Note 4).17. p SGate1 (for construction of RNAi vectors with pSGate1; seeNote 5).2.2.Mediaand Reagents(see N ote 1 )628S. Zhong et al.18. p DONR221 (Invitrogen, Carlsbad, CA).19. G ateway ® BP Clonase ® II enzyme mix (Invitrogen).20.G ateway ® LR Clonase ® II enzyme mix (Invitrogen). 1. D esign the fi rst set of primers containing appropriate restric-tion sites added to the 5 ¢ end of each primer that amplify the sense sequence of the target gene (see Note 2). For examplepurposes, we have chosen the forward primer to contain the X ho I restriction site and the reverse primer to contain H ind I II restriction site.2. A mplify the targeted gene using the primer pair designed above in a standard PCR reaction following Taq polymerase suppli-er’s recommendations. The thermal cycling conditions are as follows: initial denaturation (95°C, 2 min), followed by 35 cycles of denaturation (94°C, 30 s), annealing (58°C, 30 s; see Note 6), and extension (72°C, 1 min), and then one fi nal cycle of extension (72°C, 10 min).3. D igest the PCR product with X ho I and H ind I II following restriction enzyme supplier’s digestion recommendations and buffers. Digestion is performed in a 50 m L reaction which con-tains 40 m L of PCR product, 5 m L of 10× restriction enzyme buffer, 0.5 m L 100× bovine serum albumin (BSA), 5 units of each enzyme, and water up to 50 m L . The reaction is incubated at 37°C overnight (see Note 7).4. P urify the PCR product using a commercial PCR purifi cation kit following the manufacturers’ recommendations.5. L inearize pSilent-1 by H ind I II and X ho I double-digestion fol-lowing restriction enzyme supplier’s digestion recommenda-tions and buffers.6. P urify the linearized pSilent-1 vector using a commercial puri-fi cation kit following supplier’s recommendations.7. L igate the X ho I / H ind I II-digested PCR product in the X ho I / H ind I II-digested pSilent1. Ligation is performed in a 10 m L reaction which contains 50 ng of purifi ed PCR product, 100 ng of pSilent-1, 1 m L of 10× ligation buffer, 1 m L of T4 Ligase, and water up to 10 m L . Incubate the reaction at room temperature for 1–2 h (see Note 8).8. A dd 2–5 m L of the ligation reaction to 50 m L of competent E . coli cells and incubate on ice for 30 min. Heat-shock cells by incubating at 42°C for 60 s without shaking. Remove the vial from 42°C water bath and place on ice for 2 min. 3.Methods3.1.Construction of RNAi Vectors with pSilent162940 Construction of Hairpin RNA-Expressing Vectors … 9. A dd 250 m L of SOC medium and incubate at 37°C for 1 h with shaking at 150 rpm. 10. S pread an aliquot of the transformation reaction onto LB agar plates with 100 m g /mL ampicillin (see Note 9). Incubate plates at 37°C for 24 h. 11. P ick single colonies and inoculate culture tubes containing 2 mL of LB containing 100 m g /mL ampicillin and incubate at 37°C with shaking at 250 rpm overnight (around 14–16 h). 12. I solate the plasmid DNA using a commercial plasmid purifi ca-tion kit following the manufacturer’s recommendations. 13. V erify the integration of the PCR product into pSilent1 by double digestion of the plasmid with H ind I II and X ho I follow-ing restriction enzyme supplier’s digestion recommendations and buffers. Alternatively, use PCR to confi rm integration. 14. D esign the second set of primers containing appropriate restric-tion sites to amplify the sense sequence of the target gene (see Note 2). For example purposes, we have chosen the forward primer to contain the K pn I restriction site and the reverse primer to contain B gl I I restriction site. 15. R epeat steps 2–13 above but utilize K pn I and B gl I I for the design, integration, and confi rmation of the anti-sense copy of the gene to be silenced into pSilent1. 16. V erify the hpRNA expression construct by double digestion ( X ho I / H ind I II or B gl I I/ K pn I ) and visualize pattern in stan-dard gel electrophoresis (see Note 10). 17. T he verifi ed silencing construct is linearized by S pe I and can be used for RNAi via PEG-mediated transformation (see Note 11). 1. D esign a primer pair with the appropriate attB sequences added at the 5 ¢ end of each primer (see Note 4). 2. A mplify the targeted gene using the primer pair designed abovein a standard PCR reaction following Taq polymerase suppli-er’s recommendations. The thermal cycling conditions are as follows: initial denaturation (95°C, 2 min), followed by 35 cycles of denaturation (94°C, 30 s), annealing (58°C, 30 s; see Note 6), and extension (72°C, 1 min), and then one fi nal cycle of extension (72°C, 10 min).3. A dd 75 m L of TE and 50 m L of 30% PEG/MgCl 2solution to25 m L of the PCR reaction to purify the PCR product. Mix well and centrifuge for 15 min at 14,000 rpm in a microcentrifuge. 4. R emove the supernatant carefully and re-suspend the pellet in10 m L TE.5. Q uantify the PCR product. The concentration of the purifi edPCR product should be more than 10 ng/ m L (see Note 12).3.2.Constructionof RNAi Vectorswith pSGate1630S. Zhong et al.6. M ix 1–7 m L (15–150 ng) of the purifi ed PCR products with1 m L of pDONR221, and add TE to the fi nal volume of 8 m L.Keep the reaction on ice and add 2 m L BP Clonase II enzymemix (see Note 13). Mix well and incubate at 25°C for 2 h.7. A dd 1 m L Proteinase K solution to the sample and incubate at37°C for 10 min to terminate the reaction.8. A dd 3–5 m L of BP reaction to 50 m L of competent E. coli cellsand incubate on ice for 30 min. Heat-shock cells by incubatingat 42°C for 60 s without shaking. Remove the vial from 42°Cwater bath and place on ice for 2 min.9. A dd 250 m L of SOC medium and incubate at 37°C for 1 hwith shaking at 150 rpm.10. S pread an aliquot of the transformation reaction onto LB agarplates containing 50 m g/mL kanamycin. Incubate plates at37°C for 24 h (see Note 9).11. P ick single colonies and inoculate culture tubes containing2 mL of LB with 50 m g/mL kanamycin and incubate at 37°Cwith shaking at 250 rpm overnight (around 14–16 h).12. I solate the plasmid DNA using a commercial plasmid purifi ca-tion kit following the manufacturer’s recommendations.13. C arry out PCR using the isolated plasmid DNA as templateand the primers designed in step 1 to verify whether the plas-mid contains the DNA insert from the target gene. The veri-fi ed plasmid is then used as entry clone for the LR reaction.14. P erform LR reaction by mixing 1 m L entry clone (150 ng/ m L),1 m L destination clone (pSGate1, 100 ng/ m L), and 6 m L TE.Keep the reaction on ice and add 2 m L LR clonase enzymemix (see Note 14). Mix the reaction well and incubate at25°C for 2 h.15. A dd 1 m L Proteinase K solution to the sample and incubate at37°C for 10 min to terminate the reaction.16. A dd 3–5 m L of the BP reaction to 50 m L of competent E. colicells and incubate on ice for 30 min. Heat-shock cells by incu-bating at 42°C for 60 s without shaking. Remove the vial from42°C water bath and place on ice for 2 min.17. A dd 250 m L of SOC medium and incubate at 30°C (see Note15) for 1 h with shaking at 150 rpm. Spread 100 m L of trans-formation reaction onto LB agar plates containing 50 m g/mLampicillin. Incubate plates at 30°C (see Note 15) for 24 h.18. P ick 6–10 single colonies and inoculate them individually toculture tubes containing LB medium with 50 m g/mL ampicil-lin. Incubate the cultures at 30°C (see Note 15) with shakingat 250 rpm overnight (around 14–16 h).63140 Construction of Hairpin RNA-Expressing Vectors…19. I solate the plasmid DNA using a commercial plasmid purifi ca-tion kit following the manufacturer’s recommendations.20. V erify the hpRNA-expressing construct by single restrictionenzyme ( X ho I) and double restriction enzyme ( B gl I I and K pn I) digestions (see Note 16).21. T he verifi ed silencing construct is linearized by S pe I and can beused for RNAi via PEG-mediated transformation (see Note 11).1. A ll media used for culturing bacteria should be sterilized byautoclaving at 121°C for 20 min.2. T wo sets of primers must be designed to amplify the targetgene. The fi rst primer pair amplifi es a fragment that is cloned into the H ind I II -Sna B I -Xho I polylinker of pSilent1 and the second primer pair amplifi es the same fragment, which is cloned into the B gl I I -Sph I-Stu I-Kpn I-Apa I polylinker of pSilent1 (see Fig. 1).The region of the target gene to be amplifi ed should not contain the same restriction enzyme sites as those used in primers. Gene-specifi c primers should be 18–25 bp with melt-ing temperature ( TM ) ranging from 55 to 60°C. The size offragment to be amplified from the target gene should rangefrom 300 to 600 bp to achieve effective gene silencing.3. p Silent-1 is available from the Fungal Genetics Stock Center(FGSC#634).4. T he forward primer is 5 ¢-GGGGACAAGTTTGTACAAAAAAGCAGGCT- g ene-s pecifi c primer-3 ¢and the reverse primer is5 ¢-GGGGACCACTTTGTACAAGAAAGCTGGGT- g ene-s pecifi c primer-3 ¢. Gene-specifi c sequences should be 18–25 bpwith annealing temperature ranging from 55 to 60°C. The sizeof fragment to be amplifi ed from the target gene should rangefrom 300 to 600 bp to achieve effective gene silencing.5. p SGate1 is available from the Fungal Genetics Stock Center.6. A nnealing temperature depends on TMof the primers used.7. D igestion of PCR productions may take as little as 2 h.However, if a large amount of DNA is digested, an overnightdigestion is recommended.8. F or more effi cient ligation, the ligation reaction can be incu-bated at 16°C overnight. The following formula shows how tocalculate the amount of insert and vector used in the ligationreaction: amount of insert/100 ng vector = 100 × size of insert(Kb) × 3/size of vector (Kb).4.Notes632S. Zhong et al.9. W hen plating E. coli cells on LB agar plates, plate with two dif-ferent volumes (50 and 100 m L) to ensure that at least oneplate has well-separated colonies.10. D ouble-digestion by H ind I II and X ho I or B gl I I and K pn Ishould generate a fragment with the same size as the PCRproduct amplifi ed from the target gene. Sequencing could beused to further confi rm the insert in the vector construct.11. T he hpRNA-expressing construct cassette can also be releasedwith appropriate restriction enzymes and cloned into an appropri-ate T-DNA vector for A grobacterium-mediated transformation.12. A fter purifi cation, 10 m L of TE is usually added to the purifi edPCR products and 3 m L of the DNA solution is used for BPreaction. If the concentration of the purifi ed PCR product istoo low, a larger volume (50 m L or more) of the PCR reactioncan be purifi ed to achieve a higher concentration.13. T haw the BP clonase enzyme mix on ice before use and storeat −20 or −80°C immediately after use.14. T haw the LR clonase enzyme mix on ice before use and storeat −20 or −80°C immediately after use.15. I ncubation and culture of bacteria containing the hrRNA-expressing plasmid construct at 30°C is highly recommendedto optimize bacterial growth and stabilize the construct.16. D igestion by single restriction enzyme ( X ho I) and doublerestriction enzymes ( B gl I I and K pn I) should generate the samesize of fragments. Sequencing could be used to further confi rmthe vector. Approximately 50% of the colonies are likely tocontain the right construct.AcknowledgmentsT he authors thank Dr. H. Nakayashiki (Kobe University, Japan)for generously providing pSilent-1.References1. C oppin E, Debuchy R, Arnaise S, Picard M (1997) Mating types and sexual development in fi lamentous ascomycetes. Microbiol Mol Biol Rev 61:411–4282. W aterhouse PM, Helliwell CA (2003) Exploring plant genomes by RNA-induced gene silencing. Nat Rev Genet 4:29–383. N akayashiki H,Nguyen QB (2008) RNA inter-ference: roles in fungal biology. Curr Opin Microbiol 11:494–5024. M cDonald T, Brown D, Keller NP, Hammond TM (2005) RNA silencing of mycotoxin pro-duction in A spergillus and F usarium species. Mol Plant-Microbe Interact 18:539–5455. L iu H, Cottrell TR, Pierini LM et al (2002) RNA interference in the pathogenic fungus C ryptococcus neoformans. Genetics 160:463–4706. Kück U, Hoff B (2010) New tools for the genetic manipulation of fi lamentous fungi. Appl Microbiol Biotechnol 86:51–62633 40 Construction of Hairpin RNA-Expressing Vectors…7. J eon J, Park SY, Chi MH et al (2007) Genome-wide functional analysis of pathogenicity genesin the rice blast fungus. Nat Genet 39: 561–5658. K adotani N, Nakayashiki H, Tosa Y, Mayama S(2003) RNA silencing in the phytopathogenicfungus M agnaporthe oryzae. Mol Plant-MicrobeInteract 16:769–7769. N akayashiki H, Hanada S, Quoc NB et al(2005) RNA silencing as a tool for exploringgene function in ascomycete fungi. Fungal Genet Biol 42:275–28310. F itzgerald A, van Kan JAL, Plummer KM(2004) Simultaneous silencing of multiple genes in the apple scab fungus, V enturiainaequalis, by expression of RNA with chimericinverted repeats. Fungal Genet Biol 41: 963–97111. R appleye CA, Engle JT, Goldman WE (2004)RNA interference in H istoplasma capsulatumdemonstrates a role for a-(1, 3)-glucan in viru-lence. Mol Microbiol 53:153–16512. D e Jong JF, Deelstra HJ, Wosten HAB, LugonesLG (2006) RNA-mediated gene silencing inmonokaryons and dikaryons of S chizophyllumcommune. Appl Environ Microbiol 72: 1267–126913. N amekawa SH, Iwabata K, Sugawara H et al(2005) Knockdown of L IM15/DMC1in themushroom C oprinus cinereus by double-strandedRNA-mediated gene silencing. Microbiology 151:3669–367814. W alti MA, Villalba C, Buser RM et al (2006)Targeted gene silencing in the model mush-room C oprinopsis cinerea( C oprinus cinereus)by expression of homologous hairpin RNAs.Eukaryot Cell 5:732–74415. T akeno S, Sakuradani E, Tomi A et al (2005)Improvement of the fatty acid composition ofan oil-producing filamentous fungus, M ortierellaalpina1S-4, through RNA interference withD12-desaturase gene expression. Appl EnvironMicrobiol 71:5124–512816. M artens H, Novotny J, Oberstrass J et al (2002)RNAi in D ictyostelium: the role of RNA-directed RNA polymerases and double-strandedRNase. Mol Biol Cell 13:445–45317. M oriwaki A, Ueno M, Arase S, Kihara J (2007)RNA mediated gene silencing in the phyto-pathogenic fungus B ipolaris oryzae. FEMS Microbiol Lett 269:85–8918. v an Esse HP, Bolton MD, Stergiopoulos I et al(2007) The Chitin-Binding C ladosporium ful-vum effector protein Avr4 is a virulence factor.Mol Plant-Microbe Interact 20:1092–1101 19. B olton MD, van Esse HP, Vossen JH et al(2008) The novel C ladosporium fulvum lysinmotif effector Ecp6 is a virulence factor withorthologues in other fungal species. Mol Microbiol 69:119–13620. v an Esse HP, van’t Klooster JW, Bolton MDet al (2008) The C ladosporium fulvum viru-lence protein Avr2 inhibits host proteases required for basal defense. Plant Cell 20:1948–196321. C arneiro JS, de la Bastide PY, Chabot M et al(2010) Suppression of polygalacturonase geneexpression in the phytopathogenic fungus O phiostoma novo-ulmi by RNA interference.Fungal Genet Biol 47:399–40522. S hafran H, Miyara I, Eshed R et al (2008)Development of new tools for studying genefunction in fungi based on the Gateway system.Fungal Genet Biol 45:1147–115423. L eng Y, Wu C, Liu Z et al (2011) RNA-mediated gene silencing in the cereal fungalpathogen C ochliobolus sativus. Mol Plant Pathol12:289–29824. W esley SV, Helliwell CA, Smith NA et al (2001)Construct design for efficient, effective and high throughput gene silencing in plants. Plant J27:581–590。
FPR基因shRNA干扰载体的合成与构建

FPR基因shRNA干扰载体的合成与构建目的设计并构建靶向甲酰肽受体FPR基因的shRNA 慢病毒表达载体并鉴定。
方法根据GenBank数据库提供的FPR基因序列,设计合成针对FPR的shRNA序列,构建PDS019_pL/shRNA/GFP/F-FPR慢病毒载体,并通过测序鉴定。
结果成功构建的重组质粒测序结果与Genebank 中的FPR cDNA 序列相符。
结论构建FPR基因shRNA干扰载体,为该基因的相关实验研究提供载体。
Abstract:Objective To construct and identify FPR shRNA lentiviral vector. Methods Genome sequences of FPR gene was retrieved from Genebank. The shRNA sequences for FPR were synthesized and cloned into PDS019_pL/shRNA/GFP/F to generate shRNA lentiviral vector. The recombinant vectors was identified by sequencing.Results The sequence identified by sequencing were the same as the targeting one. Conclusion The constructed FPR shRNA lentiviral vectors were constructed,which may be used for the further research the role of FPR in the malignant behavior of the cancer.Key words:FPR;Vector construction甲酰肽受体(Formyl peptide receptor,FPR)在肿瘤的发生、演进和转移过程中可能起重要作用。
载体构建流程

载体构建SOP流程:GenBank查询目的基因序列→根据ORF序列利用引物设计软件设计引物→表达目的基因的组织或细胞总RNA提取→RT-PCR获取目的基因→酶切目的基因和载体→分别纯化酶切的目的基因和载体并建立连接反应→转化→初步筛选阳性克隆→阳性克隆测序→测序正确的质粒保种并重提质粒I.获取目的基因/序列片段一.获取序列信息通过GENBANK数据和生物信息的方法设计目的基因或目的片段引物(shRNA、miRNA)。
PCR引物的设计原则:①引物应用核酸系列保守区内设计并具有特异性。
②产物不能形成二级结构。
③引物长度一般在15~30碱基之间。
④ G+C含量在40%~60%之间。
⑤碱基要随机分布。
-⑥引物自身不能有连续4个碱基的互补。
⑦引物之间不能有连续4个碱基的互补。
⑧引物5′端可以修饰。
⑨引物3′端不可修饰。
⑩避免在引物的3’端使用碱基A。
在实际设计引物中由于ORF两末端序列本身的限制,不能完全按照上述理想的设计原则,但也切记引物不能过长或过短。
过长的引物不容易打开其二级结构,与模板结合缓慢,也容易形成引物二聚体,通常不超过35bp(不包括酶切位点和保护碱基)。
过短的引物特异性差,扩出其它不相关片段,最终很难得到目的片段,通常不短于18bp(不包括酶切位点和保护碱基)。
要将目的基因定向克隆至相应载体,需要在上下游引物两端设计不同的酶切位点,由于酶切位点位于线性末端时酶对其识别切割能力大大降低,需依据NEB目录添加相应保护碱基,酶切时可相应增加时间。
二.制备模板1.分离高质量RNA:成功的cDNA合成来自高质量的RNA。
高质量的RNA至少应保证全长并且不含逆转录酶的抑制剂,如EDTA或SDS。
RNA的质量决定了能够转录到cDNA上的序列信息量的最大值。
现在实验室通常使用Trizol试剂法提取总RNA,可以从多种组织和细胞中提取高质量的非降解RNA。
Trizol试剂法可以从最少100个细胞或1mg组织中提取RNA。
gateway构建干涉载体步骤
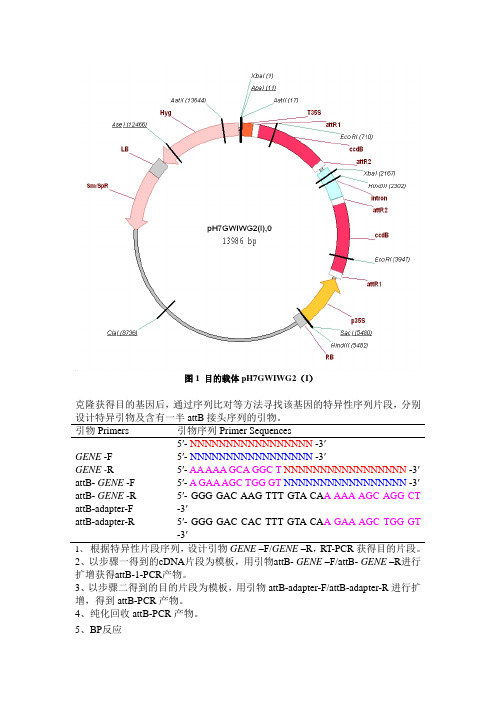
图1 目的载体pH7GWIWG2(I )克隆获得目的基因后,通过序列比对等方法寻找该基因的特异性序列片段,分别设计特异引物及含有一半attB接头序列的引物。
引物Primers 引物序列Primer SequencesGENE -F GENE -R attB- GENE -F attB- GENE -R attB-adapter-F attB-adapter-R 5′- NNNNNNNNNNNNNNNNN -3′5′- NNNNNNNNNNNNNNNNN -3′5′- AA AAA GCA GGC T NNNNNNNNNNNNNNNNN -3′ 5′- A GAA AGC TGG GT NNNNNNNNNNNNNNNNN -3′ 5′- GGG GAC AAG TTT GTA CA A AAA AGC AGG CT -3′5′- GGG GAC CAC TTT GTA CA A GAA AGC TGG GT-3′1、 根据特异性片段序列,设计引物GENE –F/GENE –R ,RT-PCR 获得目的片段。
2、以步骤一得到的cDNA 片段为模板,用引物attB- GENE –F/attB- GENE –R 进行扩增获得attB-1-PCR 产物。
3、以步骤二得到的目的片段为模板,用引物attB-adapter-F/attB-adapter-R 进行扩增,得到attB-PCR 产物。
4、纯化回收attB-PCR 产物。
5、BP 反应(1)室温下,在1.5 ml eppendorf管中加入下列反应体系,并混匀,25℃温育过夜。
试剂名称体积(反应体系:10 µL)attB-PCR 回收产物 2 µLpDONRTM vector(150 ng/ µL) 1 µLBP Clonase TM enzyme mix 1 µL5 × BP ClonaseTM Reaction Buffer 2 µLTE Buffer (pH8.0) 4 µL(2)加1 µL Proteinase K solution,37℃温育10 min,以终止BP反应。
干扰慢病毒设计、合成及包装的具体方法及步骤

干扰慢病毒设计、合成及包装的具体方法及步骤
1.shRNA序列设计与合成
(1)客户提供干扰序列;
(2)针对目的基因中洪代为设计并合成三对特异性的siRNA序列,根据siRNA序列设计对应茎环结构的shRNA序列,然后合成三对shRNA序列(结果中保证其中一条干扰效率不低于70%)。
2.shRNA慢病毒载体构建
将三个shRNA分别克隆入慢病毒载体,构建shRNA干扰慢病毒载体,并进行测序鉴定构建成功。
二、目的基因干扰慢病毒载体的包装和浓缩
1.慢病毒载体的大量包装
大量扩增效率三组shRNA慢病毒载体和慢病毒的辅助质粒,将三个质粒按一定比例大量转染293T细胞,进行慢病毒颗粒的大量包装。
2. 慢病毒的浓缩
包装48小时后收集病毒,并对病毒进行体外大比例浓缩,得到有效滴度为10^8PFU/ml的慢病毒颗粒。
Spl基因RNA干扰载体的构建及鉴定

Spl基因RNA干扰载体的构建及鉴定
目的:构建干扰载体pSilencer3.1-spl,并初步研究其对Spl基因的干扰作用.方法:根据SplcDNA编码序列,设计并合成针对Spl基因的特异*RNA干扰片段,并将其克隆入pSilencer3.1-Hlneo干扰载体中,构建Spl基因小干扰RNA(siRNA)真核表达载体pSilencer3.1-Spl;分别将**对照载体pSilencer3.1与重组载体pSilencer3.1-Spl经脂质体LipofectAMINE2000介导转染HeLa细胞,采用RT-PCR、Westernblot方法分别检测Spl基因的转录与表达水平.结果:构建了Spl基因siRNA真核表达载体pSilencer3.1-Spl,经酶切、测序鉴定*实克隆正确,并在mRNA水平和蛋白水平*实了载体的干扰效果.结论:特异*siRNA能明显抑制Spl基因在HeLa细胞中的表达,为进一步研究Spl 的生物学功能和作用机制奠定了实验基础.
陈苏宁,CHENSu-Ning(第四*医大学,基础部生物化学与分子生物学教研室,国家肿瘤生物学重点实验室,西京医院*剂科,陕西,西安,710032)。
vigs载体构建的基本流程

vigs载体构建的基本流程下载温馨提示:该文档是我店铺精心编制而成,希望大家下载以后,能够帮助大家解决实际的问题。
文档下载后可定制随意修改,请根据实际需要进行相应的调整和使用,谢谢!并且,本店铺为大家提供各种各样类型的实用资料,如教育随笔、日记赏析、句子摘抄、古诗大全、经典美文、话题作文、工作总结、词语解析、文案摘录、其他资料等等,如想了解不同资料格式和写法,敬请关注!Download tips: This document is carefully compiled by theeditor. I hope that after you download them,they can help yousolve practical problems. The document can be customized andmodified after downloading,please adjust and use it according toactual needs, thank you!In addition, our shop provides you with various types ofpractical materials,such as educational essays, diaryappreciation,sentence excerpts,ancient poems,classic articles,topic composition,work summary,word parsing,copy excerpts,other materials and so on,want to know different data formats andwriting methods,please pay attention!VIGS载体构建的基本流程。
1. 选择目标基因,确定要干扰的靶基因。
干扰载体的构建原理

干扰载体的构建原理干扰载体是一种用来传输干扰信号的工具,它通过与目标信号进行叠加或干扰的方式,使目标信号无法正确地传输或接收。
干扰载体的构建原理主要包括选择合适的干扰信号源、设计有效的干扰信号处理电路以及采用适当的传输方式。
首先,干扰载体的构建需要选择合适的干扰信号源。
常见的干扰信号源包括信号发生器、噪声源和扫频源等。
信号发生器可以产生单一频率或多频率的干扰信号,噪声源可以产生宽频带的随机干扰信号,而扫频源可以产生一定范围内的频率扫描信号。
根据实际需要选择合适的干扰信号源,确保其频率范围和信号特性与目标信号相适应。
其次,干扰载体的构建需要设计有效的干扰信号处理电路。
干扰信号处理电路常见的功能包括信号放大、频率调整、幅度调整和频谱调制等。
信号放大可以增加干扰信号的强度,频率调整可以使干扰信号的频率与目标信号接近或重叠,幅度调整可以调节干扰信号的强弱,频谱调制可以改变干扰信号的频谱特性。
这些功能的设计需要根据目标信号的特点和干扰效果的需要进行调整和优化,以达到最佳的干扰效果。
最后,干扰载体的构建需要采用适当的传输方式。
常见的传输方式有有线和无线两种。
有线传输方式包括电缆和光纤传输,其特点是传输稳定可靠,但受距离和传输介质等限制。
无线传输方式包括电磁波传输和红外线传输,其特点是传输范围广,适用于远距离传输,但易受干扰和信号衰减等影响。
根据实际需求选择合适的传输方式,确保干扰信号能够有效地传输到目标信号位置。
除了以上的基本原理,干扰载体的构建还需要考虑一系列的技术要求和应用需求。
例如,对于军事应用中的干扰装备,需要具备高功率、宽频带、快速调频调幅、较高的抗干扰能力等特点,以应对敌方现代化通信设备和电子战装备的挑战。
而对于民用应用中的干扰装置,如防止无人机干扰航空器、阻止手机在银行等场所使用等,需要具备小型化、低功耗、灵活可控、安全可靠等特点,以满足不同场景下的需求。
综上所述,干扰载体的构建原理主要包括选择合适的干扰信号源、设计有效的干扰信号处理电路以及采用适当的传输方式。
RNAi干扰测序结果分析SOP

测序结果分析SOP一、目的:确认重组质粒中是否含有目的基因二、步骤实验室将目的基因和质粒酶切连接后,将重组质粒送到测序公司测序,测序公司将测序结果发回来之后,就需要验证重组质粒的序列中是否包含目的基因。
1.将.Seq文件运行回车符,去掉序列中包含的回车符。
这一步很重要,直接影响测序结果。
2.根据克隆号去查找对应的合同号,然后用合同号找出目的序列的碱基序列。
而目的基因与质粒连接又分为直接退火连接和转库连接。
①对于直接退货连接的重组质粒,复制整个目的序列,在重组质粒的序列中查找目的序列。
②对于转库的目的基因,需根据框架结构,选择序列,在重组质粒的序列中查找对应的的序列。
3.在查询过程中会遇到以下问题(针对未测通的重组质粒):(1).对于突变,包括缺少碱基,增加碱基和错读,此时需要根据峰图判断在此位置处的峰图情况,如果峰图正常,则可判断为突变,需要再送2个重新测序,如果出现套峰,重峰和峰图不好,这种情况下往往会出现错读,少读,多读的现象,这种情况往往出现在目的基因序列的末端,此时需要根据峰图的情况,结合测序结果和目标序列来判断。
判断结果可能会是无法判断,这时需要再送一个,并且再接一个;也可能是无法判断,需要再送2个重新测序。
也可能判断为测通ok,这种情况也会出现。
(2).非目的序列。
这种情况下在重组质粒序列中找不到目的序列的任何片段,查看峰图可能很差,也可能很好。
若是峰图正常则该目的序列可能是其他克隆的目的序列。
若是峰图很乱,则需要再送。
若是第一个克隆,需要引物重合。
对于非目的序列,需要确定该目的序列是否是其他克隆的目的序列,针对这种情况需要去实验室了解情况。
(3).中断ok。
这种情况下由于测序时信号衰减,而得到目的序列的部分序列,而峰图也是这样,那么需要再送2个;或者是只有前面一部分连续的目的序列,后半部分找不到,查看峰图,后半部分峰图杂乱,也需要再送两个。
(4).乱峰。
整个测序峰图杂乱,且找不到目的基因,则需要再送(5).对于一个目的基因已经测序很多次,时间较长,仍未测通ok但是存在中断ok,需要进行引物重合。
基因干扰载体构建方法

基因干扰载体构建方法我在基因干扰载体构建这事儿上可费了不少劲呢,今天就跟你好好唠唠。
我一开始真的是瞎摸索。
就像在黑暗中找东西,啥也看不见。
我知道构建这个载体首先得有合适的模板,我当时就想当然地找了一个看着相关度挺高的,结果呢,搞了半天发现不行,这才明白合适的模板可不是那么容易找的,得非常精准才行。
就拿设计shRNA(小发夹RNA)来说吧,这就像给目标基因量身定制一个干扰工具。
我刚开始自己设计的时候,那些各种规则把我搞得晕头转向的。
什么碱基配对原则,还有避免某些特定序列之类的。
我试过根据一些文献上的范例来改,可总是不成功。
后来我才发现,那些软件工具真的很重要。
我就用了几个专门设计shRNA的软件,像RNAi Designer之类的,把基因序列输进去,然后软件会给你一些候选的shRNA序列。
然后就是载体的选择啦。
这就好比你要给一个东西找个合适的存放容器。
有很多种载体可以选,像质粒载体啊之类的。
我一开始没太在意载体的特性,随便拿了一种就用。
结果发现它的一些功能和我后续的实验不配套,没办法只能重新换。
所以选载体的时候得把自己后续要干嘛想清楚喽,像要不要加标签啊,对转录启动子有什么要求啊这类的问题得提前考虑好。
在把shRNA连接到载体上这个环节也不容易。
我那时候手都哆嗦,生怕搞砸了。
操作就跟搭积木一样,得把各个部分按照正确的顺序和方式组合起来。
我之前不好好做连接反应的对照实验,结果出问题了都不知道是哪一步的错。
后来就知道连接反应一定要做各种对照,像阴性对照、阳性对照之类的,这样才能判断到底有没有连接成功。
还有呢,转化到细胞里后确认干扰效果也是头疼。
细胞里各种复杂的情况就像一个大杂烩。
有时候你觉得构建好了肯定行,但是细胞环境一作用,结果达不到预期。
我有次等了好久才去检测干扰效果,结果发现已经错过最佳时间了。
所以得按时检测,而且要多检测几次来确保。
我还会同时用不同的方法检测,像RT - PCR啊还有蛋白免疫印迹之类的,这样结果就更可靠。
shRNA干扰慢病毒载体构建技术原理

shRNA干扰慢病毒载体构建技术原理
shRNA干扰慢病毒载体由慢病毒基因组序列和细菌质粒序列构成。
细菌质粒序列含有一个Ampicillin抗性基因和和一个高拷贝复制子pUCori。
慢病毒基因组序列是从元件5'LTR开始,到元件3'LTR结束。
慢病毒基因组序列可容纳两个外源基因表达盒子:一个是shRNA 表达盒子,另外一个是marker基因表达盒子。
Marker基因表达盒子已经克隆在载体上,无需重新构建。
我们提供了荧光marker、抗药性marker或者荧光/抗药性双markerft您选择。
shRNA表达盒子则包括三部分序列:U6启动子、shRNA和终止子。
U6启动子已经克隆在载体上,每次构建载体,只需通过annealing-ligation技术将感兴趣的shRNA和终止子克隆到载体上即可。
RNA干扰载体的构建的实验流程

RNA干扰载体的构建的实验流程RNA干扰载体主要用来研究基因表达调控,RNA干扰技术已已被广泛用于基因结构功能研究和传染性疾病及基因治疗领域,进行RNA干扰实验首先是构建RNA干扰载体,本文以pRI 系列载体为例论述了干扰载体的构建的实验流程。
产品技术背景pRI系列载体是基于III类rna聚合酶启动子:人类H1启动子的专用于哺乳动物细胞RNA干扰的载体。
H1启动子在哺乳动物细胞内合成类似siRNA分子的小分子RNA。
由于H1启动子有精确的转录起始位点和终止信号,H1启动子转录产物精确生成人工设计的shRNA,shRNA 经过RISC剪切后形成有2个U突出末端的成熟siRNA。
由于H1启动子对转录产物长度的严格限制,基本上杜绝了非特异性干扰片段的产生,将载体转染细胞后对其它基因的影响降到最低。
pRI系列载体已经成功用于多种哺乳动物细胞进行基因的RNA干扰。
本系列中含有新霉素抗性基因的载体用于稳定表达siRNA,可以在更长时间内对基因表达抑制后的细胞功能和生理现象进行观察和分析。
插入寡核苷酸设计pRI系列载体的使用需要将人工设计的寡核苷酸片段插入pRI系列载体中特定的酶切位点之间,寡核苷酸片段中包含了针对目标基因的mRNA设计的长度为19nt的干扰片段。
合成时需要化学合成正向和反向两条寡核苷酸。
正、反向寡核苷酸退火后与载体连接,插入载体XhoI,BglII位点之间,位于载体上H1启动子下游正确的位置上。
连接后的载体转入哺乳动物细胞在H1启动子作用下转录产生shRNA。
1. 选择干扰序列在RNA干扰实验中,RNA干扰序列的选择会显著影响RNA干扰效果。
我们建议您按照以下几点指导原则选择RNA干扰序列:推荐长度为19 nt,采用21 nt序列也可以取得良好效果。
RNA干扰序列中不包含大于3 nt的连续相同碱基。
RNA干扰序列的GC含量为低到中等水平(推荐GC含量在35%到50%之间)。
不要将RNA干扰序列设计在已知的RNA-蛋白质结合位置附近。
rna干扰载体设计原则

rna干扰载体设计原则RNA干扰(RNA interference,简称RNAi)是一种广泛应用于生物学研究和基因治疗领域的技术。
RNA干扰通过介导小分子RNA (siRNA或miRNA)与靶向基因的mRNA结合,从而降低或抑制该基因的表达。
为了实现高效、可靠的RNA干扰作用,科学家们设计了一系列RNA干扰载体。
本文将介绍RNA干扰载体设计的原则和策略。
RNA干扰载体的设计需要考虑siRNA或miRNA的选择和合成。
siRNA 通常由21到23个核苷酸组成,而miRNA则由60到100个核苷酸组成。
在选择siRNA或miRNA时,需要注意序列的选择和靶向基因的选择。
合适的siRNA或miRNA序列可以通过生物信息学工具进行预测和选择,确保其能够与靶向基因的mRNA特异性结合。
RNA干扰载体的设计需要考虑载体的构建和转染效率。
一般而言,RNA干扰载体可以采用质粒或病毒载体进行构建。
质粒载体的优点是方便构建和操作,但其转染效率较低。
病毒载体则可以实现高效的转染,但需要注意病毒的选择和安全性。
在构建RNA干扰载体时,还需要考虑启动子的选择和siRNA或miRNA的插入位置。
RNA干扰载体的设计还需要考虑表达水平的调控。
为了实现RNA干扰的最佳效果,需要调控siRNA或miRNA的表达水平。
常用的调控策略包括使用不同强度的启动子、添加启动子的启动子和使用外源基因的转录调控序列。
RNA干扰载体的设计还需要考虑递送方式和细胞特异性。
RNA干扰载体可以通过转染、转导或转化等方式递送到目标细胞。
在选择递送方式时,需要考虑载体的稳定性、细胞毒性和细胞特异性。
为了实现特异性的RNA干扰作用,可以在载体中加入细胞特异性启动子、细胞特异性表达调控元件或细胞特异性的运载体。
RNA干扰载体的设计还需要考虑评价和验证。
设计好的RNA干扰载体需要进行验证和评价,确保其能够实现预期的RNA干扰效果。
常用的评价方法包括实时荧光定量PCR、Western blot、免疫组化和功能实验等。
人Spsb1基因慢病毒干扰载体的构建及病毒包装

粒。
[ 关键词 ]肝肿瘤; 慢病毒干扰载体 ; 脂质体; 转染 ; s 6 1 基因 [ 中 图分 类 号 ]R 3 6 1 . 3 [ 文献 标识 码 ]A [ 文章编 号 ]1 0 0 0 — 2 7 0 7 ( 2 0 1 7 ) 0 6 0 6 2 1 - 0 5
LI Ho n g me i ,XU Gu o q i a n g
( S c h o o l o f B a s i c Me d i n e , G u i z h o u Me d i c a l U n i v e r s i t y , G u i y a n g 5 5 0 0 2 5 , G u i z h o u , C h i n a )
第4 2卷 第 6期 2 0 1 7年 6月
贵 州 医 科 大 学 学 报
J OURNAL OF GUI Z H oU M EDI CAL UNI VER S I TY
Vo 1 . 4 2 NO . 6 2 01 7. 6
・
专题 研 究 1・
人 S p s b l 基 因慢 病 毒 干 扰 载 体 的 构 建 及 病 毒 包 装
Ge ne a n d i t s Le nt i v i r u s Pa r t i c l e Pa c k a g i ng
S UN Da q u a n,S HI S o n g,XU Yi n g y i n g,L EI Ti n g we n,MO Xi a o c h u a n,W ANG Z h u t i n g,
干扰载体构建SOP

shRNA干扰载体构建SOP目录第一部分RNA干扰原理及shRNA设计 2第二部分酶切与回收 6第三部分退火与连接7第四部分转化与涂平板9第五部分挑菌与菌液PCR 11第六部分质粒提取13第一部分RNA干扰原理及shRNA设计标准操作规程(SOP)1.目的:了解RNA干扰原理,设计shRNA干扰序列2.仪器与设备:需要能够连接Internet的个人计算机,引物设计相关软件(如Primer Premier 5).3.操作步骤3.1RNA干扰原理1)RNAi概述:RNA干扰(RNA interference,RNAi)是指在进化过程中高度保守的、由双链RNA(double—stranded RNA,dsRNA)诱发的、同源mRNA高效特异性降解的现象。
简单的说是指一种分子生物学上由双链RNA诱发的基因沉默现象.当细胞中导入与内源性mRNA编码区同源的双链RNA时,该mRNA发生降解而导致基因表达沉默,是一种特异性的转录后基因沉默(post-transcriptional gene silencing).由于RNAi具有高度的序列专一性和有效的干扰,可以特异地将特定的基因沉默,从而获得基因功能丧失或基因表达量的降低,因此可以作为功能基因组学的一种强有力的研究工具。
RNAi技术可广泛应用到包括功能学,药物靶点筛选,细胞信号传导通路分析,疾病治疗等等。
2)RNAi原理:RNA干扰包括起始阶段和效应阶段(inititation and effector steps)。
在起始阶段,小分子RNA被切割为21—23核苷酸长的小分子干扰RNA片段(small interfering RNAs, siRNAs).证据表明;一个称为Dicer的酶,是RNase III家族中特异识别双链RNA的一员,它能以一种ATP依赖的方式逐步切割由外源导入或者由转基因,病毒感染等各种方式引入的双链RNA,形成19-21bp的双链RNAs(siRNAs),每个片段的3’端都有2个碱基突出。
干扰质粒的构建 2

干扰质粒的构建所需试剂:1.Primer合成2.限制性内切酶:Xho I & Hpa I3.20 bp DNA ladder marker (具有300 – 1000 bp的条带)4.Middle DNA Marker (具有7000 bp左右的条带)5.T4 DNA ligase6.DNA胶回收试剂盒7.DNA载体(pll3.7质粒)5 -10 μg/reaction8.质粒抽提试剂盒操作步骤:1.引物退火(形成互补的DNA双链):取合成的引物S链和A链(用去离子水配制成100 μM)各2.5 μl,加入15 μl退火缓冲液,按照如下步骤进行退火:94 o C, 4 min; 85 o C, 4 min; 82 o C, 4 min; 80 o C, 4 min; 78 o C, 4 min;75 o C, 4 min; 70 o C, 4 min; 37 o C, 15 min; 10 o C, 4 min; 4 o C, 10 min。
退火后,测定DNA浓度及OD260/280的读数,评价DNA质量。
或者吸取1 μl 样品上样于4%琼脂糖胶。
2.载体双酶切所需限制性内切酶:Xho I & Hpa I按照限制性内切酶使用说明书进行酶切,需要设置的对照及样品组如下:双酶切组、Xho I 单酶切组、Hpa I 单酶切组和未酶切组(注:双酶切组一般做2管,以避免胶回收时样品损失过多)。
按照说明书推荐的酶切时间,水浴中酶切,然后将酶切产物上样于0.8%琼脂糖凝胶。
预计酶切结果:未酶切组一般有三条带(但依据DNA螺旋程度的不同,通常会观察到1-3条带),单酶切和双酶切组均有一条带(双酶切组本应有两条带,另外一条为切下的小片段,可能因为片段过小而跑出胶外)。
图中左边三个为双酶切产物,右边分别为XhoI单酶切、HpaI单酶切和为经过酶切的pll3.7质粒确认酶切完全正确后,割胶回收,得到酶切后的pll3.7载体3.连接将退火产物和双酶切后的pll3.7载体依据T4连接酶的说明书进行连接,通常用量为摩尔比(酶切后的pll3.7载体:退火后的DNA)=1:10由于退火产物和双酶切后的pll3.7载体分子量之比为60:7000,因此二者的质量比约为1:11。
siRNA干扰载体构建与引物设计的一些小建议

siRNA干扰载体构建与引物设计的一些小建议
siRNA干扰载体构建过程中,针对siRNA设计的引物是怎么设计的?
一般是合成一对引物,然后退火,再与线性化的载体做连接。
由于引物比较长,所以合成时一般都会有比较多的碱基突变,因此,做shRNA表达载体构建的主要难度,在于筛选出正确的克隆,有时候可能要选很多个克隆测序才能筛到想要的。
具体的序列组成,一般是sense-loop-antisense。
不同的公司会有不同的设计原则的。
经验:我们一般不去考虑构建载体来得到siRNA!
载体表达siRNA方法主要存在以下缺点:
1、外源导入shRNA的表达会干扰细胞本身miRNA的正常表达,而miRNA在基因的精确表达调控方面有着至关重要的作用,目前已经证实很多疾病的产生通miRNA的表达异常存在确切关系,已有很多文献报道和证实;
2、载体构建的周期长,操作较为复杂,试验重复性不好,做过的人应该大多都知道;
3、载体用于体内试验的毒性非常大,容易激发机体强烈的免疫应答反应,副作用大,已有多篇文献证实;
4、通量低,影响因素更多,体内试验的效果很差,存在安全性的问题;
5、shRNA的表达在时间和表达量上都较难控制等。
这也是为什么现在10多种进入临床试验的RNAi药物几乎都是用化学合成的siRNA来做的原因!。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
shRNA干扰载体构建SOP目录第一部分RNA干扰原理及shRNA设计2第二部分酶切与回收6第三部分退火与连接8第四部分转化与涂平板9第五部分挑菌与菌液PCR11第六部分质粒提取13第一部分RNA干扰原理及shRNA设计标准操作规程(SOP)1.目的:了解RNA干扰原理,设计shRNA干扰序列2.仪器与设备:需要能够连接Internet的个人计算机,引物设计相关软件(如Primer Premier 5)。
3.操作步骤3.1RNA干扰原理1)RNAi概述:RNA干扰(RNA interference, RNAi)是指在进化过程中高度保守的、由双链RNA(double-stranded RNA,dsRNA)诱发的、同源mRNA高效特异性降解的现象。
简单的说是指一种分子生物学上由双链RNA诱发的基因沉默现象。
当细胞中导入与内源性mRNA编码区同源的双链RNA时,该mRNA发生降解而导致基因表达沉默,是一种特异性的转录后基因沉默(post-transcriptional gene silencing)。
由于RNAi具有高度的序列专一性和有效的干扰,可以特异地将特定的基因沉默,从而获得基因功能丧失或基因表达量的降低,因此可以作为功能基因组学的一种强有力的研究工具。
RNAi技术可广泛应用到包括功能学,药物靶点筛选,细胞信号传导通路分析,疾病治疗等等。
2)RNAi原理:RNA干扰包括起始阶段和效应阶段(inititation and effector steps)。
在起始阶段,小分子RNA被切割为21-23核苷酸长的小分子干扰RNA片段(small interfering RNAs, siRNAs)。
证据表明;一个称为Dicer的酶,是RNase III家族中特异识别双链RNA的一员,它能以一种ATP依赖的方式逐步切割由外源导入或者由转基因,病毒感染等各种方式引入的双链RNA,形成19-21bp的双链RNAs(siRNAs),每个片段的3’端都有2个碱基突出。
在RNAi效应阶段,siRNA双链结合一个核酶复合物从而形成所谓RNA诱导沉默复合物(RNA-induced silencing complex, RISC)。
激活RISC需要一个ATP依赖的将小分子RNA解双链的过程。
激活的RISC通过碱基配对定位到同源mRNA转录体上,并在距离siRNA 3’端12个碱基的位置切割mRNA。
尽管切割的确切机制尚不明了,但每个RISC都包含一个siRNA和一个不同于Dicer的RNA酶。
3)RNAi示意图(见图1)3.2shRNA设计1)shRNA: 即short hairpin RNA(短发卡RNA) ,包含两个短反向重复序列(其中一个与目的基因互补),中间由一个loop序列分隔,组成发夹结构。
shRNA在体内可以被加工成siRNA 从而降解目的基因.。
示意图见图2。
2)shRNA作用原理:见图3。
3)shRNA设计原则:●克隆到shRNA表达载体中的shRNA包括两个短反向重复序列,中间由一茎环(loop)序列分隔的,组成发夹结构,由polⅢ启动子控制。
随后在连上5-6个T作为RNA 聚合酶Ⅲ的转录终止子;●两个互补的寡核苷酸两端须带有限制性酶切位点;●StrataGENE发现29个寡核苷酸较之原先推荐的23个寡核苷酸可以更有效的抑制目的基因;●在启动子下游的酶切位点下方紧连一个C,使插入片段和启动子有一定空间间隔以确保转录的发生;●shRNA目的序列的第一个碱基必须是G以确保RNA聚合酶转录。
如果选择的目的序列不以G开头,必须在紧连正义链的上游加一个G;●ShRNA插入片段中的茎环应当靠近寡核苷酸的中央。
不同大小和核苷酸序列的茎环都被成功的运用过。
其中包含一个独特的限制性酶切位点的茎环利于检测带有shRNA插入片段的克隆。
在比较了众多不同长度和序列的茎环,5'TCAAGAG3'序列最为有效(AMBION use);●5-6个T必须放置在shRNA插入片段尾部以确保RNA聚合酶III终止转录(stop);●在正义链和反义链序列上不能出现连续3个或以上的T,这可能导致shRNA转录的提前终止;●从转录本(mRNA)的AUG起始密码开始,寻找“AA或者NA”二连序列,并记下其3'端的19个碱基序列,作为潜在的siRNA靶位点。
正义链和反义链都采用这19个碱基(不包括AA或者NA重复)来设计。
图1 RNAi原理图2 shRNA示意图图3 shRNA作用原理4)shRNA设计举例●以人源基因VEGFC为例,选取干扰靶点序列为:GCCGATGCATGTCTAAACT●在该序列两端加上酶切位点,中间插入Loop环,设计shRNA片段:BamH I 靶点序列环结构靶点反义序列Hind IIIGATCC GCCGATGCATGTCTAAACT TTCAAGAGA AGTTTAGACATGCATCGGCTTTTTT AG CGGCTACGTACAGATTTGA AAGTTCTCT TCAAATCTGTACGTAGCCGAAAAAA TTCGA●正反向Oligo序列分别为:H-VEGFC-sh-F:5’-GATCC GCCGATGCATGTCTAAACTTTCAAGAGAAGTTTAGACATGCATCGGCTTTTTT A-3’H-VEGFC-sh-R:5’-AGCTT AAAAAAGCCGATGCATGTCTAAACTTCTCTTGAAAGTTTAGACATGCATCGGC G-3’将该Oligo片段正反向一起混合退火后,即可直接与载体进行连接,构建shRNA干扰载体。
第二部分干扰载体pRNAT-U6.1概述1.目的:了解干扰载体pRNAT-U6.1的特点2.pRNAT-U6.1载体概述●带有U6启动子,保证shRNA的高表达;●带有GFP标签,可用于检测转染效率;●带有多克隆位点;●带有新霉素(Neomycin)抗性,可用于筛选稳定细胞株3.pRNAT-U6.1载体图谱图1 pRNAT-U6.1载体图谱第三部分酶切与回收标准操作规程(SOP)实验名称shRNA干扰载体构建SOP S3:酶切与回收起草人/修订人何章荣修订日期2015.7审核人审核日期批准人批准日期颁发部门1.目的:通过酶切可以获得带酶切位点的线性化质粒载体,使用试剂盒对其进行回收。
2.试剂与材料名称公司货号限制性内切酶Thermo1.5mL tube Axygen0.2mL PCR tube AxygenCycle Pure Kit OMEGA D6492-013.仪器与设备名称公司货号MAXYGENE Thermal Cycler Axygen MaxyGeneGradient 移液器大龙掌上离心机其林贝尔LX-200恒温水浴锅精宏DK-804.操作步骤4.1酶切以Thermo内切酶为例,准备好冰盒,将所需试剂从冰箱中取出后置于冰上,待试剂融化后,在0.2mL离心管中加入:10×buffer Tango 5μL内切酶HindIII 1μL内切酶BamHI 1μLshRNA干扰质粒pNAT-U6.1 1μgddH2O up to 50μL盖紧管盖,离心15s。
将0.2mL离心管置于离心管架上,放入37℃水浴锅中,酶切1h(时间长短根据酶的反应效率而异,可参考产品说明书)。
也可以直接使用PCR仪进行酶切反应。
Notes:●根据酶的特点,可能要求用到BSA,可根据说明书进行操作。
双酶切时,如果仅其中一种酶需要用到BSA,则也需要在酶切体系中加入BSA,因为BSA不会影响内切酶的活性。
●若两个位点的内切酶使用相同缓冲液(即在该种缓冲液里酶活性最高),可加入两种酶同时进行反应●若使用不同缓冲液,则可先进行单酶切,回收后再进行第二次酶切,再次回收。
●由于载体上的两个酶切位点一般相距较近,有些酶需要更多的保护碱基,则同时进行双酶切可能效率不高。
可先加入需要更多保护碱基(或是酶切效率较低)的内切酶,反应结束后回收(如果两种酶使用同一类缓冲液,则可省去回收步骤),再加入第二种酶进行反应。
酶切后的载体通过凝胶电泳分离然后回收纯化,去除切割下来的小片段,从而降低假阳性。
4.2酶切产物回收以OMEGA Cycle Pure Kit试剂盒为例。
1)将酶切产物转移至1.5mL 离心管,加入4-5倍酶切产物体积的Buffer CP;如果PCR片段长度小于200bp,则加入6倍体积的Buffer CP;2)漩涡震荡混匀,短暂离心,收集管壁上的液体;3)将混合物加入至试剂盒提供的HiBind DNA吸附柱,10000g,室温离心1min;4)弃残液,加入700μL DNA WASH Buffer,10000g,室温离心1min;5)重复步骤4;6)弃残液后,13000,室温离心2min;7)将HiBind DNA吸附柱置于干净的1.5mL 离心管,加入15-30μL Elution Buffer或ddH2O,室温放置1-2min,13000,室温离心2min,收集DNA产物。
第四部分退火与连接标准操作规程(SOP)1.目的:将单链的shRNA上下游片段进行退火,形成双链DNA片段,随后连接至酶切后的载体。
4.操作步骤4.1退火2)将合成好的Oligo片段用蒸馏水溶解稀释至100μM,在0.2mL 离心管中按下表加入各成95℃5min80℃4min75℃4min70℃4min65℃2min60℃2min55℃2min50℃2min45℃2min40℃2min37℃2min20℃20min程序结束后,可4℃短时间保存,或-20℃长期保存。
4.3连接由于shRNA片段已带有酶切后的粘性末端,故可直接与酶切后的载体进行连接。
退火后的混合物稀释10倍,使用1 ~ 10 μL移液枪在0.2mL离心管中加入:10×buffer 1μLT4 DNA ligase 0.1μL退火混合物(已稀释10倍) 1μL酶切后载体pRNAT-U6.1 3μLH2O up to 10μL一共10μL,轻微吹吸混匀,盖紧管盖,离心15s,PCR仪上22℃反应30min。
第五部分转化与涂平板1.目的:将连接好的质粒转化大肠杆菌,然后涂平板进行筛选。
4.1配制LB培养基♦液体LB培养基:在大烧杯中加入900 ml去离子水,称取以下试剂加入烧杯中:胰化蛋白胨10g酵母提取物5gNaCl 10g用搅拌器搅拌混匀,直至全部溶解。
用去离子水定容至1L,121℃高温蒸汽灭菌20min。
♦固体LB培养基:配置好液态LB培养基后,加入琼脂粉至终浓度1.5%,121℃高温蒸汽灭菌20min。
4.2转化插入片段和载体连接后,可直接进行转化。
1)打开水浴锅,调至42℃。