美国cGMP-中英文对照
CGMP中英文对照

C G M P中英文对照Table of Contents 目录• SUBPART B 111.10 – 111.14: Personnel 人员• SUBPART C 111.15 – 111.23: Physical Plant and Grounds 工厂与场所• SUBPART D 111.25 – 111.35: Equipment and Utensils 设备与器具• SUBPART E 111.55-111.95: Production and Process Control System 生产与过程控制系统• SUBPART F 111.103-111.140: Production and Process Control System:• Requirements for Quality Control 生产与过程控制系统对质量控制的要求• SUBPART G 111.153 – 111.180: Production and Process Control System:• Requirements for components, packaging, and labels 生产与过程控制系统对成分,包装与标签的要求• SUBPART H 111.205-111.210: Production and Process Control System:• Requirements for the Master Manufacturing Record. 生产与过程控制系统对主要制造记录的要求• SUBPART I 111.255 – 111.260: Production and Process Control System:• Require ments for the Batch Production Record. 生产与过程控制系统对批生产记录的要求• SUBPART J 111.303 – 111.325: Production and Process Control System:• Requirements for Laboratory Operations 生产与过程控制系统对实验室操作的要求• SUBPART K 111.353 – 111.365: Production and Process Control System• Requirements for Manufacturing Operations 生产与过程控制系统对制造过程的要求• SUBPART L 111.403 – 111.425: Production and process control system• Requirements for Packaging and Labels Operation 生产与过程控制系统对包装与标签操作的要求• SUBPART M 111.453 – 111.475: Holding and Distributing 扣留与分发• SUBPART N 111.503 – 111.535: Return of Dietary Supplements 膳食补充剂的退货• SUBPART O 111.553 – 111.570: Product Complaints 产品投诉• SUBPART P 111.605 – 111.610: Records and Recordkeeping 记录与记录保留• 11.10 – 11.50: Electronic Records 电子记录• N SF/ANSI 173 Section 8.2:• Compliance with the Public Health Security and Bioterrorism 符合公共健康安全与生物反恐• NSF/ANSI 173 Section 8.3: Adverse Event Reporting 不利事件的报告• NSF/ANSI 173 Section 8.4: Recall Procedures 回收程序• Appendix 1 NSF 229 – Functional Food Guideline 功能性食品导则• Appendix 2 NSF 306 – GMP for Sport 运动食品的GMP要求• Appendix 3 NSF 306 – GMP for Sport & NSF 229 – Functional Food Guideline• 运动食品与功能性食品的GMP要求• PERSONNEL• B人员Question 1 CFR 111.10Procedures have been established that define work requirements for personnel to prevent microbial contamination from illnesshygienic practices.建立员工工作要求的程序以预防疾病的微生物污染• a. A written procedure shall exist and be current stating that personnel with medical conditions such as open lesions or infected• 规定员工身体状况如开放性伤害或感染的书面程序必须存在并且为当前的• wounds will be removed from the manufacturing process so as to prevent product adulteration during manufacturing or storage.• 在制造或存储时有伤口的员工远离制造流程,以避免产品被掺杂• The procedure shall state that such health conditions will be reported to supervision.• 必须建立上述身体情况向主管汇报的程序,以便于监管• b. Inspection verifies that such workers are not in areas where adulteration could occur.• 有检查的验证表明上述的员工不在掺杂可能发生的区域• c. Personnel shall be trained on the written procedure and knowledgeable of the disease cont rol policies • 员工必须进行疾病控制方针的书面程序与知识的培训Question 2 CFR 111.10Hygienic practices have been established to include appropriate garments, personal hygiene, hand washing and sanitization, etc. prior to starting work and at any time whereby personnel can become soiled/contaminated.在工作前或员工被污染时需要建立包括适当的服装,员工卫生,洗手与消毒等的卫生操作• a. A written dress code shall exist and be current stating appropriate attire for workers, supervisors, managers and visitors to all parts of the production, storage, packaging and testing facilities.•表明员工,主管,经理,参观者进入生产,储存,包装与测试区域穿着适当服装的书面服装规则必须存在且为当前的• b. Outer garments shall be donned prior to entering the facility and shall not be worn outside the production facility or home. Therefore proper changing areas are required. Outer garments shall have long sleeves and have secured fasteners. Above waist pockets (or carrying items in pockets) should be avoided. •进入工厂前必须穿工作衣,并且不能在生产工厂外面或家里穿工作衣。
美国药品生产质量管理规范(cGMP)

GOOD MANUFACTURE PRACTICE 美国药品生产质量管理规范(CGMP)二○○三年十二月目录210.1 cGMP法规的地位 (2)210.2 cGMP法规的适用性 (2)210.3 定义 (2)211-A- 总则 (4)211-B- 组织与人员 (4)211-C- 厂房和设施 (5)211-D- 设备 (7)211-E- 成份、药品容器和密封件的控制 (8)211-F- 生产和加工控制 (10)211-G- 包装和标签控制 (11)211-H- 贮存和销售 (13)211-I- 实验室控制 (14)211-J- 记录和报告 (16)211-K- 退回的药品和回收处理 (20)210部分—人用及兽用药品的生产、加工、包装或贮存的CGMP210.1 cGMP法规的地位(a) 在本部分及21CFR 211—226部分中陈述的法规是在药品生产、加工、包装或贮存中使用的现行生产质量管理规范及使用的设施或控制的最低标准,以保证该药品符合联邦食品、药品及化妆品法对安全性的要求,具有均一性和效价(或含量)并符合或代表其生产过程的质量及纯度等特征。
(b) 凡是在药品生产、加工、包装或贮存过程中存在任何不符合本部分及21CFR 211—226部分中陈述的法规的药品,依据联邦食品、药品及化妆品法501 (a)(2)-(B),该药应被视为劣药,同时导致该事故发生的负责人应受相应的法规的制裁。
210.2 cGMP法规的适用性(a) 本部分及21CFR 211—226适用于普通药品,21CFR 600—680适用于人用生物制品,除非另有明确规定,否则上述两者之间应该是相互补充而不是相互取代。
如有上述两部分的法规不适用的药品,则可用特定的具体法规来替代。
210.3 定义(a) 在联邦食品、药品及化妆品法201部分中包含的定义和解释、说明适用于21CFR 211—226部分中的术语。
(b) 下面定义的术语适用于本部分及21CFR 211—226。
中英文-美国FDA GMP检查

• Quality System 质量体系 • Facilities and Equipment System
设施和设备系统 • Materials System 物料体系 • Production System 生产体系 • Packaging and Labeling System 包装和标签体系 • Laboratory Control System 实验室控制体系
/AboutFDA/CentersOffices/CDER/ucm095598.htm
What type of inspection can be done? 可以进行什么类型的检查?
• Full Inspection Option 全面检查方案 – Quality System plus (at least) three other systems 质量体系加(至少)三个其他系统
• Facilities设施 Major considerations when evaluating aseptic processing operations… 对无菌工艺操作评估需要重要考虑的因素…
• Building Management System (BMS) 建筑管理系统(BMS)
• Environmental Controls (Temp, RH, DP) 环境控制(温度、湿度、压差)
China - June 2011
Inspections… 检查 …
• Are FACT finding in nature 是事实性的调查结果
• Require EVIDENCE 需要证据
• Are REGUALTORY in nature 实质上是一种监管 – What is said could end up in court 所说的内容可能最终会在法庭上裁决
美国现行GMP(中文版)

美国现行药品生产质量管理规范(cGMP)目录A-总则 (3)B-组织与人员 (3)C-厂房与设施 (4)D-设备 (7)E-成份、药品容器和密封件的控制 (8)F-生产和加工控制 (11)G-包装和标签控制 (13)H-贮存和销售 (16)I-实验室控制 (17)J-记录和报告 (20)K-退回的药品和回收处理 (25)A.总则211·1 范围(a) 本部分的条例包含人用或兽用药品制备的现行最低限度的药品生产质量管理规范(GMP)。
(b) 在本章里的这些针对药品的现行GMP条例和本章600至800的所有部分针对人用生物制品的现行GMP条例,除非明确另有说明者外,应认为是对本部分条例的补充,而不是代替。
本章其他部分或本章600至680各部分和本部分均可适用的条例,前部分的条例可代替本部分条例。
(c) 在考虑经提议的,发表在1978年9月29日联邦注册表(FR)上一项免除时,若产品及其所有成份是以人用物品形式作一般销售和消费,且这些产品根据其预期用途,亦可列入药品的范围内,则不应对这些非处方药(OTC)实施本部分条例,直至进一步的通知为止。
本章110部分和113至119部分的条例用于鉴别这些亦是食品的OTC药品是否按照GMP的要求生产、加工、包装和贮存。
211·3 定义本章210·3的定义适用于本部分。
B. 组织与人员211.22 质量控制部门的职责(a) 本部门有批准和拒收所有成份、药品包装容器、密封件、中间体、包装材料、标签及药品的职责与权力。
复查生产记录的权力,保证不产生差错,或若发生差错,保证他们充分调查这些差错。
本部门负责根据合同,批准或拒收由其它公司,生产、加工、包装或贮存的产品。
(b) 适当的实验室检验设备、批准(或拒收)的各种成份、药品容器、密封件、包装材料及药品,质量控制部门是可以获得的。
(c) 本部门有批准或驳回影响药品的均一性、效价或含量、质量及纯度的所有程序或规格标准的职责。
USP中文版

GOOD MANUFACTURE PRACTICE 美国药品生产质量管理规范(CGMP)二○○三年十二月目 录210.1 cGMP法规的地位 (2)210.2 cGMP法规的适用性 (2)210.3 定义 (2)211-A- 总则 (4)211-B- 组织与人员 (4)211-C- 厂房和设施 (5)211-D- 设备 (7)211-E- 成份、药品容器和密封件的控制 (8)211-F- 生产和加工控制 (10)211-G- 包装和标签控制 (11)211-H- 贮存和销售 (13)211-I- 实验室控制 (14)211-J- 记录和报告 (16)211-K- 退回的药品和回收处理 (20)210部分—人用及兽用药品的生产、加工、包装或贮存的CGMP210.1 cGMP法规的地位(a) 在本部分及21CFR 211—226部分中陈述的法规是在药品生产、加工、包装或贮存中使用的现行生产质量管理规范及使用的设施或控制的最低标准,以保证该药品符合联邦食品、药品及化妆品法对安全性的要求,具有均一性和效价(或含量)并符合或代表其生产过程的质量及纯度等特征。
(b) 凡是在药品生产、加工、包装或贮存过程中存在任何不符合本部分及21CFR 211—226部分中陈述的法规的药品,依据联邦食品、药品及化妆品法501 (a)(2)-(B),该药应被视为劣药,同时导致该事故发生的负责人应受相应的法规的制裁。
210.2 cGMP法规的适用性(a) 本部分及21CFR 211—226适用于普通药品,21CFR 600—680适用于人用生物制品,除非另有明确规定,否则上述两者之间应该是相互补充而不是相互取代。
如有上述两部分的法规不适用的药品,则可用特定的具体法规来替代。
210.3 定义(a) 在联邦食品、药品及化妆品法201部分中包含的定义和解释、说明适用于21CFR 211—226部分中的术语。
(b) 下面定义的术语适用于本部分及21CFR 211—226。
美国cGMP-中英文对照

PART 210 CURRENT GOOD MANUFACTURING PRACTICE IN MANUFACTURING, PROCESSING, PACKING, OR HOLDING OF DRUGS;GENERAL210部分有关于生产、加工、包装和药品的储存的现行GMP —般准则Sec. 210.1 Status of current good manufacturing practice regulations.(a) The regulations set forth in this part and in parts 211 through 226 of this chaptercontain the minimum current good manufacturing practice for methods to be used in, and the facilities or controls to be used for, the manufacture, processing, packing, or holding of a drug to assure that such drug meets the requirements of the act as to safety, and has the identity and strength and meets the quality and purity characteristics that it purports or is represented to possess.(b) The failure to comply with any regulationset forth in this part and in parts 211 through 226 of this chapter in the manufacture, processing, packing, or holding of a drug shall render such drug to be adulterated undersection 501(a) (2)(B) of the act and such drug, as well as the person who is responsible forthe failure to comply, shall be subject to regulatory action.(c) Owners and operators of establishments engaged in the recovery, donor screening, testing (including donor testing), processing, storage, labeling, packaging, or distributionof human cells, tissues, and cellular andtissue-based products (HCT/Ps), as defined in 1271.3(d) of this chapter, that are drugs (subject to review under an application submitted under section 505 of the act or under a biological product license application under section 351 of the Public Health Service Act), are subject to the donor-eligibility and applicable current good tissue practice procedures set forth in part 1271 subparts C and D of this chapter, in addition to the regulations in this part and in parts 211 through 226 of this chapter. Failure to comply with any applicable210.1 cGMP 的法规地位。
美国制剂cGMP简介

说明
具体内容可以从网上下载。中文版仅供参 考,以英文为准。 211规定了要达到哪些要求,但是没有规定 怎样去做。
违反cGMP的后果
• 产品被定为劣药 • 产品查封 • 被媒体曝光 • 影响其他产品 • 成为被告 • 工厂关停 • 产品召回 •企业竞争力下降 • 罚款
三、举例说明(483/警告信)
J. 记录和报告
在批生产记录中使用涂改液。 检验记录上没有填写收到的样品的名称, 样品量,样品和标准品的称取量,稀释过 程。 在培养基灌装过程中,作了录象,以备发 现发生问题后使用。 但是,录象后来被丢 掉了。 录象时,没有把配液和无菌过滤过程录下来。 没有关于录象的书面规程。
J. 记录和报告(续)
I. 实验室控制
所建立的质量标准不完善,例如缺乏对 “有关物质”的控制;缺乏针对某些剂型 的崩解或释放度的标准和方法。 分析方法没有经过恰当验证。例如微生物 检验方法。 没有采用USP的方法,且没有恰当验证。 未能证明:经过100%的灯检,能够去除所 有不合格的安瓿(有异物、炭化现象)。 灯检的标准不够严格。
第210部分的简介(续)
210.2 cGMP法规的适用性 本部分和第21CFR 211—226部分适用于药品, 21CFR 600—680适用于人用生物制品,除非另有 明确规定,否则上述两者之间应该是相互补充而 不是相互取代。如有药品不可能适用于这些部分 的所有相关法规,则可用具体的适用法规来替代 通用法规。 如果一个人仅从事一些业务(这些业务受本部分 及211—226部分和600—680部分的管辖),那么 他只需要符合适用于这些业务的相关法规。
搅拌机和压片机的外表面涂了漆。 除热原隧道加热段的HEPA过滤器没有定期 检测完整性。
FDA工业指南--CGMP的质量体系(中文译稿)

Guidance for IndustryQuality Systems Approach to Pharmaceutical CGMP Regulations业界指南——制药企业CGMP规范的质量体系U.S. Department of Health and Human ServicesFood and Drug AdministrationCenter for Drug Evaluation and Research (CDER)Center for Biologics Evaluation and Research (CBER)Center for Veterinary Medicine (CVM)Office of Regulatory Affairs (ORA)September 2006Pharmaceutical CGMPs目录Ⅰ简介Ⅱ背景和目的A 背景B 指南的目标C 指南适用范围D 指南的组织结构Ⅲ CGMP和现代质量系统的概念A 质量B 质量设计和产品开发C 质量风险管理D CAPA(纠偏和预防措施)E 变更控制F 质量部门G 六系统检查模式IV 质量系统模型A 管理责任1 授予领导权2 构建组织3 建立符合要求的质量系统4 建立方针政策、目标和计划5 系统审核B资源1 授予领导权2人员提高3厂房和设备4对外包工作的控制C生产制造1产品和生产工艺的设计、开发和文件化2检查输入3运作的执行和监控4 非一致性D 评估活动1 数据的趋势分析2 内部审核3 质量风险管理4 纠偏措施5 预防措施6 推动改善V 结论术语表(本指南代表了FDA对质量体系当前的思考。
它既没有为任何人也不是为赋予任何人利益而制定。
并且其操作也不对FDA或公众形成约束。
只要能够满足对现行法规和规范的要求你可以采用其它方法。
如果你有兴趣讨论其它方法,可以联系负责执行本指南的FDA人员。
如果你无法确认恰当的FDA人员,可以拨打本指南公布的电话。
《美国FDA化妆品良好操作规范(FDA-CGMP)》中文版

美国食品和药品管理局Cosmetic Good Manufacturing Practice Guidelines化妆品良好生产规范指南联邦食品、药品和化妆品法案(The Federal Food, Drug and Cosmetic Act, 以下简称FD&C 法案)禁止在州际直接贸易的化妆品是掺杂的或贴假标签的情况。
(Sec. 301)以下4种情况下,化妆品被认为是可能掺杂的:1.在用户使用过程中,由于化妆品本身含有或在包装容器中有潜在的、对人体有害的成分而使用户受到伤害的;2.本身含有不洁成分的;3.本身含有禁用成分,例如:未认可的色素添加剂;4.在不卫生条件下生产的、或保留的,可导致产品伤害用户有害或被不洁成分所污染。
以下几种情况下,化妆品被认为可能会认为贴假标签(Sec. 602):1.虚假的标签或存在误导信息的标签2.显著违反了联邦食品、药品和化妆品法案的要求在标签上声明的信息要求3.在容器上有误导的信息为了确定化妆品生产厂家是否保留或发货了掺杂的、或是贴假标签的化妆品,和防止这些违反了FD&C 法案生产的化妆品流入市场,法律给了FDA进入这些化妆品工厂检查的权利,包括检查相关工厂的设备,成品,原料,容器和标签。
(见Sec. 704(a) of the FD&C Act.)如果工厂严格的根据良好的操作规范(GMP)的要求生产,将最低限度的减少掺杂的,或贴假标签的情况。
随后的化妆品指导,引用于FDA检查操作手册(FDA's Inspection Operations Manual),可以作为指南,用来有效的进行自我检查除。
良好的检查得分则意味着工厂执行了良好的操作规范(GMP)的要求。
指南1.建筑物和设施:检查是否a.用于生产或存放化妆品的建筑物应大小合适,设计和结构应保证设备进出不受阻碍,材料存放整洁,操作卫生以及正确的清洁和维护;b.地面,墙壁和天花板结构表面应光滑,易于清洁,并保持干净和良好状况;c.安装的固定装置,管道的滴水或者冷凝水不会污染化妆品原料,器具,以及与化妆品原料,散装产品或成品接触的设备的表面;d.照明和排风系统应满足预期员工操作和舒适的要求;e.供水,清洗和卫生设施,地面排水和废水系统应充分满足清洁操作的要求,和设备、器具的清洁要求,以及满足员工的需要并易于让员工保持个人清洁2.设备:检查是否a.加工、盛放、中转和灌装过程使用的设备和器具应设计合理,使用的材料和工艺能防止腐蚀、污垢的堆积、以及被润滑油、灰尘或者消毒剂污染;b.器具,运送管道以及和化妆品接触的设备表面应维护良好,并定期清洁和消毒;c.清洁和消毒后的便携式设备和器具应妥善放置,与化妆品接触的设备表面应罩住,以防止飞溅,灰尘或其他污染物3.员工: 检查是否a.监督化妆品的生产或者控制的员工应具有一定的教育背景,培训和/或经验来执行指定的监督工作;b.为防止化妆品掺杂,与化妆品原料,散装成品或化妆品接触表面直接接触的员工,应穿戴适合的工作服,手套,头套等,并保持良好的个人清洁;c.吃东西,喝水,或者抽烟都应严格限制在制定的区域。
美国FDA的cGMP

本指南不包括无菌原料药的消毒和灭菌工艺,但是
,应当符合地方当局所规定的药品(医疗用品)生
产的GMP指南。
2.生产部门的责任
书面描述 根据书面规程,准备、审查、批准、传达生产 指令 根据书面规程,进行生产 审查BPR:完整并签名 报告、评估生产偏差;调查和记录关键偏差 清洁、消毒生产设施
动态药品生产管理规范 英文:Current Good Manufacture Practices 简称:cGMP cGMP要求在产品生产和物流的全过程都必须验 证。美国FDA执行cGMP在国际上有较高的权威性,所 以产品若得到美国cGMP认证,在国际市场上常常身价 倍增。在药品市场日趋国际化的今天,企业获得美国 的cGMP认证已是大势所趋。
4. 生产设备-维护和清洗
定期清洗计划与规程 责任分配 目的:预防性维护 清洗书面规程 放行标准 足够详细 清洗结果具重现性和良好效果
4. 生产设备-维护和清洗
清洗书面规程内容 责任分配 计划表,消毒计划 清洗方法,清洗剂: 完整描述:如何稀释清洗剂 拆卸与装配规程 消除上批号标识步骤 清洗后、使用前保护措施 使用前检查步骤 上工序结束与清洗设备的时间规定
美 国 FDA 之 cGMP
2014年9月
美国食品和药物管理局 英文全称:Food and Drug Administration
简称:FDA
职责:确保美国本国生产或进口的食品、化妆品、药
物、生物制剂、医疗设备和放射产品的安全。
在国际上,FDA被公认为是世界上最大的食品与 药物管理机构之一;其它许多国家都通过寻求和接收 FDA 的帮助来促进并监控其该国产品的安全。
ICH Q7A原料药生产的GMP指南
cGMP中文版

GOOD MANUFACTURE PRACTICE 美国药品生产质量管理规范(CGMP)二○○三年十二月目 录210.1 cGMP法规的地位 (2)210.2 cGMP法规的适用性 (2)210.3 定义 (2)211-A- 总则 (4)211-B- 组织与人员 (4)211-C- 厂房和设施 (5)211-D- 设备 (7)211-E- 成份、药品容器和密封件的控制 (8)211-F- 生产和加工控制 (10)211-G- 包装和标签控制 (11)211-H- 贮存和销售 (13)211-I- 实验室控制 (14)211-J- 记录和报告 (16)211-K- 退回的药品和回收处理 (20)210部分—人用及兽用药品的生产、加工、包装或贮存的CGMP210.1 cGMP法规的地位(a) 在本部分及21CFR 211—226部分中陈述的法规是在药品生产、加工、包装或贮存中使用的现行生产质量管理规范及使用的设施或控制的最低标准,以保证该药品符合联邦食品、药品及化妆品法对安全性的要求,具有均一性和效价(或含量)并符合或代表其生产过程的质量及纯度等特征。
(b) 凡是在药品生产、加工、包装或贮存过程中存在任何不符合本部分及21CFR 211—226部分中陈述的法规的药品,依据联邦食品、药品及化妆品法501 (a)(2)-(B),该药应被视为劣药,同时导致该事故发生的负责人应受相应的法规的制裁。
210.2 cGMP法规的适用性(a) 本部分及21CFR 211—226适用于普通药品,21CFR 600—680适用于人用生物制品,除非另有明确规定,否则上述两者之间应该是相互补充而不是相互取代。
如有上述两部分的法规不适用的药品,则可用特定的具体法规来替代。
210.3 定义(a) 在联邦食品、药品及化妆品法201部分中包含的定义和解释、说明适用于21CFR 211—226部分中的术语。
(b) 下面定义的术语适用于本部分及21CFR 211—226。
美国 CGMP--中英文对照.12

Subpart A-General Provisions§211.1 Scopea)The regulations in this part contain theminimum current good manufacturing practice for preparation of drug products for administration to humans or animals.b)The current good manufacturing practiceregulations in this chapter, as they pertain to drug products, and in parts 600 through 680 of this chapter, as they pertain to biological products for human use, shall be considered to supplement, not supersede, the regulations in this part unless the regulations explicitly provide otherwise. In the event it is impossible to comply with applicable regulations both in this part and in other parts of this chapter or in parts 600 through 680 of this chapter, the regulation specifically applicable to the drug product in question shall supersede the regulation in this part.c)Pending consideration of a proposedexemption, published in the Federal Register of September 29, 1978, the requirements in this part shall not be enforced for OTC drug products if the products and all their ingredients are ordinarily marketed and consumed as human foods, and which products may also fall within the legal definition of drugs by virtue of their intended use. Therefore, until further notice, regulations under part 110 of this chapter, and where applicable, parts 113 to 129 of this chapter, shall be applied in determining whether these OTC drug products that are also foods are manufactured, processed, packed, or held under current good manufacturing practice.§211.3 Definitions.The definitions set forth in §210.3 of this chapter apply in this part.A.总则211.1 范围(a)本部分的条例包含人用或兽用药品制备的现行最低限度的药品生产管理规范(GMP)。
CGMP-中英文对照

C G M P中英文对照Table of Contents 目录• SUBPART B 111.10 – 111.14: Personnel 人员• SUBPART C 111.15 – 111。
23: Physical Plant and Grounds 工厂与场所• SUBPART D 111。
25 – 111.35: Equipment and Utensils 设备与器具• SUBPART E 111。
55—111.95: Production and Process Control System 生产与过程控制系统• SUBPART F 111.103-111。
140: Production and Process Control System:• Requirements for Quality Control 生产与过程控制系统对质量控制的要求• SUBPART G 111。
153 – 111.180: Production and Process Control System:• Requirements for components, packaging, and labels 生产与过程控制系统对成分,包装与标签的要求• SUBPART H 111.205—111。
210: Production and Process Control System:• Requirements for the Master Manufacturing Record。
生产与过程控制系统对主要制造记录的要求• SUBPART I 111.255 – 111。
260: Production and Process Control System:• Require ments for the Batch Production Record. 生产与过程控制系统对批生产记录的要求• SUBPART J 111.303 – 111。
美国FDACGMP英汉对照版

美国FDACGMP英汉对照版Subpar t A-Genera l Provis ions§ 211.1 Scopea)The regula tions in this part contai n theminimu m curren t good manufa cturi ng practi ce for prepar ation of drug produc ts foradmini strat ion to humans or animal s.b)The curren t good manufa cturi ng practi ceregula tions in this chapte r, as they pertai n to drug produc ts, and in parts600 throug h 680 of this chapte r, as they pertai n to biolog icalproduc ts for humanuse, shallbe consid eredto supple ment,not supers ede, the regula tions inthis part unless the regula tions explic itlyprovid e otherw ise. In the eventit is imposs ibleto comply with applic ableregula tions both inthis part and in otherpartsof this chapte r or inparts600 throug h 680 of this chapte r, the regula tionspecif icall y applic ableto the drugproduc t in questi on shallsupers ede theregula tionin this part.c)Pendin g consid erati on of a propos edexempt ion, publis hed in the Federa l Regist er of Septem ber 29, 1978, the requir ement s inthis part shallnot be enforc ed for OTC drug produc ts if the produc ts and all theiringred ients are ordina rilymarket ed andconsum ed as humanfoods,and whichproduc ts may also fall within the legaldefini tionof drugsby virtue of theirintend ed use. Theref ore, untilfurthe r notice, regula tions underpart 110 of this chapte r, and whereapplic able,parts113 to 129 of this chapte r, shallbe applie d in determ ining whethe r theseOTC drug produc ts that are also foodsare manufa cture d, proces sed, packed, or heldundercurren t good manufa cturi ng practi ce.§ 211.3 Defini tions.The defini tions set forthin §210.3 of this chapte r applyin this part.A.总则211.1 范围(a)本部分的条例包含人用或兽用药品制备的现行最低限度的药品生产管理规范(GMP)。
FDA工业指南--CGMP质量体系(中文译稿)

Guidance for IndustryQuality Systems Approach to Pharmaceutical CGMP Regulations业界指南——制药企业CGMP规范的质量体系U.S. Department of Health and Human ServicesFood and Drug AdministrationCenter for Drug Evaluation and Research (CDER)Center for Biologics Evaluation and Research (CBER)Center for Veterinary Medicine (CVM)Office of Regulatory Affairs (ORA)September 2006Pharmaceutical CGMPs目录Ⅰ简介Ⅱ背景和目的A 背景B 指南的目标C 指南适用范围D 指南的组织结构Ⅲ CGMP和现代质量系统的概念A 质量B 质量设计和产品开发C 质量风险管理D CAPA(纠偏和预防措施)E 变更控制F 质量部门G 六系统检查模式IV 质量系统模型A 管理责任1 授予领导权2 构建组织3 建立符合要求的质量系统4 建立方针政策、目标和计划5 系统审核B资源1 授予领导权2人员提高3厂房和设备4对外包工作的控制C生产制造1产品和生产工艺的设计、开发和文件化2检查输入3运作的执行和监控4 非一致性D 评估活动1 数据的趋势分析2 内部审核3 质量风险管理4 纠偏措施5 预防措施6 推动改善V 结论术语表(本指南代表了FDA对质量体系当前的思考。
它既没有为任何人也不是为赋予任何人利益而制定。
并且其操作也不对FDA或公众形成约束。
只要能够满足对现行法规和规范的要求你可以采用其它方法。
如果你有兴趣讨论其它方法,可以联系负责执行本指南的FDA人员。
如果你无法确认恰当的FDA人员,可以拨打本指南公布的电话。
美国cGMP-中英文对照
PART 210 CURRENT GOOD MANUFACTURING PRACTICE IN MANUFACTURING,PROCESSING,PACKING, OR HOLDING OF DRUGS;GENERAL210部分有关于生产、加工、包装和药品的储存的现行GMP;一般准则Sec. 210.1 Status of current goodmanufacturing practice regulations。
210.1 cGMP的法规地位。
(a)The regulations set forth in this part and in parts 211 through 226 of this chapter contain the minimum current good manufacturing practice for methods to be used in, and the facilities or controls to be used for, the manufacture,processing, packing, or holding of a drug to assure that such drug meets the requirements of the act as to safety, and has the identity and strength and meets the quality and purity characteristics that it purports or is represented to possess. (a)在本部分及本章第211-226部分中所陈述的法规,为现行GMP最低要求,适用于药品制造、加工、包装或贮存中所采用的方法及所使用的设施或控制手段,以保证该药品符合《联邦食品、药品及化妆品法案》(以下简称法案)对安全性的要求,具有均一性和效价(或含量)并符合或代表其生产过程的质量及纯度等特征。
美国GMP中文稿(CGMP)

----------------------- Page 1-----------------------GOOD MANUFACTURE PRACTICE美国药品生产质量管理规范CGMP二三年十二月----------------------- Page 2-----------------------目录210.1 cGMP法规的地位2210.2 cGMP法规的适用性2210.3 定义 2211-A- 总则 4211-B- 组织与人员4211-C- 厂房和设施5211-D- 设备7211-E- 成份药品容器和密封件的控制8211-F- 生产和加工控制10211-G- 包装和标签控制11211-H- 贮存和销售13211-I- 实验室控制14211-J- 记录和报告16211-K- 退回的药品和回收处理201----------------------- Page 3-----------------------210 部分人用及兽用药品的生产加工包装或贮存的CGMP210.1 cGMP 法规的地位(a) 在本部分及21CFR 211 226 部分中陈述的法规是在药品生产加工包装或贮存中使用的现行生产质量管理规范及使用的设施或控制的最低标准以保证该药品符合联邦食品药品及化妆品法对安全性的要求具有均一性和效价(或含量)并符合或代表其生产过程的质量及纯度等特征(b) 凡是在药品生产加工包装或贮存过程中存在任何不符合本部分及21CFR 211226 部分中陈述的法规的药品依据联邦食品药品及化妆品法501 (a)(2)-(B) 该药应被视为劣药同时导致该事故发生的负责人应受相应的法规的制裁210.2 cGMP 法规的适用性(a) 本部分及21CFR 211 226 适用于普通药品21CFR 600 680 适用于人用生物制品除非另有明确规定否则上述两者之间应该是相互补充而不是相互取代如有上述两部分的法规不适用的药品则可用特定的具体法规来替代210.3 定义(a) 在联邦食品药品及化妆品法201 部分中包含的定义和解释说明适用于21CFR 211226 部分中的术语(b) 下面定义的术语适用于本部分及21CFR 211 226(1) 法(Act)指联邦食品药品及化妆品法修订版(21 U.S.C 301 et seq.)(2) 批(Batch)指在规定限度内按照某一生产指令在同一生产周期内生产出来的具有同一性质和质量的一定数量的药品或其它物料(3) 组分(Component)指用于药品生产的所有成份包括那些未在药品中出现的成份(4) 药品(Drug Product)指成品制剂(如片剂胶囊剂口服液等) 通常含有一种活性成份并伴有非活性成份(但不是必需的) 本术语也包括不含有活性成份但作为安慰剂使用的成品制剂(5) 纤维(Fiber)指长度大于其宽度的3 倍的任何微粒状污染物(6) 无纤维脱落的过滤器(Non-fiber-releasing filter)指任何经过适当的预处理(如清洗或冲洗)后不会将纤维脱落到已过滤的组分或药品中的所有过滤器所有含石棉过滤器均被认为是有纤维脱落的过滤器(7) 活性成份(Active Ingredient)2----------------------- Page 4-----------------------是指所有用于保证药物活性或其他在疾病的诊断治愈缓解治疗或预防中起直接作用或影响人或其他动物身体结构或功能的组分本术语包括那些能承受药品生产中的化学变化和为了保证其指定的活性或作用以一种经调整的形式存在于药品中的组分(8) 非活性成份(Inactive ingredient)指不同于活性成份的其他组分(9) 中间产品(In-process material)是指所有经制备复合混合或由化学反应得到的用于药品生产或制备的物料(10) 批lot指一批或是一批中特定的均一部分在指定的范围内具有相同的性质和质量或者若为由连续的生产过程制造出的药品批指在单位时间或单位数量生产出的特定的均一的部分并且确保该部分在指定的范围内具有均一性质与质量(11) 批号(Lot number, control number batch number)指由字母数字符号或他们的组合组成由此可确定某批药品或物料的生产加工包装贮存或销售的情况(12) 药品的生产加工包装或贮存(Manufacture, processing, packing, or holding of adrug product)包括药品的包装和标签操作检验质量控制(13) 药用物料(medicated feed)指在21CFR 558.3 中定义的B 型和C 型药用物料该物料含有联邦食品药品及化妆品法201(g)部分中定义的一种或一种以上的药物药用物料的生产应符合21CFR 226 部分中的要求(14) 药用预混合料(medicated premix)指21CFR 558.3 中定义的A 型药用物质该预混合料含有联邦食品药品及化妆品法201(g)部分中定义的一种或一种以上的药物药用预混合料生产应符合21CFR 226 部分中的要求(15) 质量控制部门(Quality control unit)指由企业任命负责质量控制相关责任的任何人员或组织机构(16) 含量或效价Strength指( ) 原料药的浓度(如以重量/重量重量/体积单位剂量/体积为基础) 和/(或)( ) 活性(效价)也即由适当的实验室检测或由足够的临床数据得出的指定的药品治疗活性(如可表达为对照于某标准的单位的术语)(17) 理论产量(Theoretical yield)指在生产加工或包装某种药品的任一适当阶段中并且基于所使用的组分的数量在实际生产中无任何损失或错误的情况下应能生产的数量(18) 实际产量(Actual yield)指某种药品在生产加工包装的任一适当的阶段实际生产出的数量(19) 比率(Percentage of theoretical yield)3----------------------- Page 5-----------------------实际产量(生产加工或包装某种药品的适当阶段)与理论产量(在相同阶段)的比率以百分数表示(20) 验收标准(Acceptance criteria)建立在相应的取样方法基础上的药品的质量检验标准和合格不合格标准(如合格质量水平和不合格的质量水平) 是决定批准或拒收一批(或其他生产单元的小组)药品的必需因素(21) 代表性样品(Representative sample)指一个样品按合理的标准抽取如随机取样法并包含若干单位元以能保证样品准确描绘被取样品的物料A 总则211·1 范围a 本部分的条例包含人用或兽用药品制备的现行最低限度的药品生产质量管理规范GMPb 在本章里的这些针对药品的现行GMP 条例和本章600 至800 的所有部分针对人用生物制品的现行GMP 条例除非明确另有说明者外应认为是对本部分条例的补充而是不代替本章其他部分或本章600 至680 各部分和本部分均可适用的条例前部分的条例可代替本部分条例c 在考虑经提议的发表在1978 年9 月29 日联邦注册表FR 上一项免除时若产品及其所有成份是以人用物品形式作一般销售和消费且这些产品根据其预期用途亦可列入药品的范围内则不应对这些非处方药OTC 实施本部分条例直至进一步的通知为止本章110 部分和113 至119 部分的条例用于鉴别这些变是食品的OTC 药品是否按照GMP 的要求生产加工包装和贮存211·3 定义本章210·3 中的定义适用于本部分B 组织与人员211·22 质量控制部门的职责a 本部门有批准和拒收所有成份药品包装容器密封件中间体包装材料标签及药品的职责与权力复查生产记录和权力保证不产生差错或若发生差错保证他们充分调查这差错本部门负责根据合同批准或拒收由其它公司生产加工包装或贮存的药品b 适当的实验室检验设备批准或拒收各种成份药品容器密封件包装材料及药品质量控制部门是可以获得的c 本部门有批准或驳回影响药品的均一性效价或含量质量及纯度的所有程序或规格标准的职责d 适用于本部门的职责与程序应成文字材料并应遵循211·25 人员资格4----------------------- Page 6-----------------------a 每位从事药品生产加工包装或仓贮工作人员应接受培训教育及有实践经验完成委派的各项职务培训是按照现行GMP 包括本章中的现行GMP 条例和这些条例要求的成文程序中涉及雇员的内容邀请合格人员指导并连续多次培训保证雇员熟悉现行GMP 对他们的要求b 负责监督药品的生产加工包装或仓贮工作的每一个工作人员应受教育培训及有经验完成委派的各项职务以此作为提供药品具有安全性均一性效价或含量质量及纯度的保证c 有足够量招待和监督每种药品的生产加工包装或仓贮的合格人员211·28 人员职责a 从事药品生产加工包装或仓贮的人员应穿着适合于其履行职责的清洁衣服按需要头部脸部手部臂部另外罩防止药物受污染b 人员保持良好的个人卫生和健康c 未经监督人员允许其他人员不能进入限制进入的建筑物和设施d 任何人在任何时间明显地表现出现有影响药品安全性和质量的疾病或开放性`损伤应避免接触各种成份药品容器包装设备密封件中间体直至监督人员结对药品有不利影响的健康情况211·34 顾问为了对问题提出意见聘请顾问顾问应对药品生产加工包装或仓贮提出建议他们受过足够的教育培训且有丰富的实践经验保留他们的姓名地址任何的顾问资格及服务形式等履历资料C 厂房和设施211·42 设计与建造特征a 任何用于某类药品生产加工包装或贮存的厂房或建筑群大小适宜结构与位置使其易于清洁保养适合操作b 建筑物有足够空间来有条理地安装设备和放置材料避免不同类的成份药品容器密封件标签中间体或药品等相互混放防止污染通过厂房的上述物料其流向在设计时要防止污染c 操作应在明确规定的大小适中的地区内进行这些地区内进行这些地区按规定各自分隔开以防止污染下列操作须在单独的地区内进行1 发放给生产或包装前质量控制部门取样期间成份药品容器密封件及标签的签收鉴别贮存及拒收2 在处理前拒收的成份药品容器密封件及标签的贮存3 已发放的成份药品容器密封件及标签的贮存4 中间体的贮存5 生产与加工操作6 包装和贴标签操作5----------------------- Page 7-----------------------7 药品发放前的隔离贮存8 发放后药品的贮存9 控制室与实验室操作10 无菌操作及有关操作地板墙壁和天花板平滑坚硬表面易清洁温度与湿度控制空气经高效过滤器在正压下过滤层流或非层流均可环境监测系统创造无菌环境房间和设备清洁消毒系统控制无菌环境的设备维修系统d 青霉素生产加工及包装设备与生产其他人用药品的设备分开211·44 照明所有地区均须提供充足的照明211·46 通风空气过滤空气加热与冷却a 提供足够的通风b 提供足够能控制空气正压微生物尘土温度和湿度的设备适应药品生产加工和贮存需要c 空气过滤系统包括预过滤器和微粒物质空气过滤器空气经过滤才送至生产区如果空气是再循环到生产区应测量尘埃含量控制从生产区带来的尘埃在生产区生产中发生空气污染应以排气系统或其他系统充分抽出空气控制污染d 青霉素生产加工和包装的空气输送系统应与其他人用药品的空气输送系统完全分开211·48 管件a 在持续正压下应对药品无污染的管道系统内供应饮用水饮用水应符合环境保护机构制订的基本饮用水条例标准40CFR141 部分不符合该标准的水不许进入水系统b 排水设备应有足够的大小可直接连接排水管及安装防止虹吸倒流的空气破坏设备或其他机械设备43FR45077 1978 年9 月29 日修正于48FR11426 1983 年3月18 日211·50 污水和废料来自水厂和附近建筑物的污水垃圾及其他废料用安全卫生的方法处理211·52 洗涤和盥洗设备提供洗涤和盥洗设备包括热冷水肥皂清洁剂空气干燥器或专用毛巾及进入厕所的清洁设备211·56a 所有用作药品生产加工包装及贮存的三房应保持清洁卫生的环境且不受啮齿动物鸟类及其他害虫侵害扰实验动物除外垃圾和有机废料定时以卫生的方法控制处理6----------------------- Page 8-----------------------b 填写分配卫生清洁任务的详细的清洁项目方法设备用于清洁厂房和设施的材料的一览表c 填写适用的杀鼠剂杀昆虫剂杀真菌剂熏蒸剂去垢剂和消毒剂一览表防止这些物品对设备成份药品容器密封件包装材料标签或药品污染除依据联邦杀虫剂杀真菌剂及杀鼠剂法规7U.S.C135 已登记和使用的品种外其他的不用211·58 保养任何用于药品生产加工包装或贮存的厂保持良好状态D 设备211·63 设备的设计尺寸及位置药品生产加工包装或贮存设备设计合理大小适当布置合理便于操作清洁和保养211·65 设备制造a 设备表面与各种成人中间体或药品接触不产生化学反应和作用保证药品的安全性均一性效价或含量质量或纯度改变b 操作所需之物质如滂沱剂冷却剂等不能进入设备里与成人药品容器封口物品中间体或药品接触保证药品的安全性均一性效价或含量质量或纯度不变211·67 设备清洁与保养a 相隔一定时间对设备与工具进行清洁保养和消毒防止出故障与污染影响药品的安全性均一性效价或含量质量或纯度b 制订药品生产加工包装或贮存设备包括用具的清洁和保养文字程序并执行这些程序包括但不一定限于以下内容1 分配清洁保养任务2 保养和清洁细目一览表3 详细说明用于清洁和保养的设备物品和方法拆卸和装配设备的方法必须保证适合清洁和保养的要求4 除去或擦去前批遗留物的鉴定5 已清除了污染的清洁设备的保护6 使用前检查清洁的设备7 保留保养清洁消毒的记录按211·180 及211·182 的说明检查211·68 自动化设备机械化设备和电子设备a 用于药品生产加工包装和贮存的自动化机械化或电子包括计算机或其它类型的设备按惯例对其设计之成文条款作标定检查或核对保证其工作性能良好保留检查标定核对等文字记录b 对保障重要生产变化的计算机或有关系统进行操作培训操作记录或其他记录只能7----------------------- Page 9-----------------------由被认可的人员制订向计算机或有关系统输入或从中输出的各种方案其他记录或资料应核查其准确性输入计算机或关系统内的档案资料除与实验室共同分析计算的结果可消除外其他的应保留文字记录与相应的证明资料一起保存事先设计好的硬件复制品或多各选择系统台复印件磁带或微型胶卷等保证其支持资料正确可靠及完整出现资料改动非人为消除或遗失时应维修211·72 过滤器用于生产加工的液体过滤器或人用注射药品的包装材料不许释放出纤维的进入产品除非不得以不在生产加工中使用释放纤维的过滤器或注射药品的包装材料若必须使用一种能释放纤维素的过滤器最后应使用一非释放纷纷物平均最大孔径为0.22μm(如实际生产条件限制可用0.45μm)的附加过滤器过滤降低注射剂内微粒量使用含石板的过滤器最后用或不用特殊非释放纤维过滤器均可以但要根据FDA 有关部门提供的该非释放纤维过滤器会或可能损害注射剂的安全性和有效性的证据而定E 成分药品容器和密封件控制211·80 总要求a 有文字详细说明成份药品容器密封件的签收鉴定贮存装运取样检验和批准或拒收程序并遵循b 成份药品容器和密封件应专人管理和在防止污染的环境下贮存c 药品容器的包装袋或包装箱或密封件应离地面放置保持适当间隔全球清洁和检查d 用明显的已接收的每装货量中的批号代码对成分药品容器或密封件加以鉴别此代码用于记录每批货的放置地方对每批货的情况如隔离批准或拒收等作检查211·82 未检验的成份药品容器和密封件的接收与贮存a 接收时和验收前对每个或编组的成份容器药品容器和密封件进行目检给内容物容器损坏或拆封和污染等情况作适当的标志b 成份药品容器各密封件应隔离贮存直至经检验为止合格可发放在符合211·80要求的地区中贮存211·84 成份药品容器和封口物品的试验批准或拒收a 每批成份药品容器和封口物品在未经质量部门取样检查合格前不准使用检验合格后发放使用b 收集每批的每一装货量的代表性样品供检验用容数目和每一容器里物质的取样量是有适当的标准的例如成份的变异性统计学标准可信限要求的精密度供应商过去的质量历史21·170 要求分析和留样所需的数量等c 收集样品程序1 用适当的方法清洁选出成份容器2 打开容器取样重新封口防止其内容物受污染和其他成分药品容器或密封件的污染8----------------------- Page 10-----------------------3 必要时使用灭菌设备和无菌取样技术4 如果需要从容器顶部中部和底部的成分中取样样品须混合5 鉴定样品容器目的是确定如下资料被取样的材料名称批号被取样的容器取样日期及样品收集人的名字等6 已取样的容器应作标志表示样品已取出d 样品检验程序1 一个药品的每个成分最少做一个特性试验如有专一特性实验就应采用2 依照所有成文的规格标准检验每个成份的纯度含量和质量生产厂家代替上述试验规定生产厂家最少要做个成份特别试验可承认这些成分的供应者扫提供的分析报告规定隔一定时间生产厂家定期验证供应午的试验结果证明供应者的分析结果是正确的3 依照成文规程检验容器和密封件生产厂家代替上友谊赛试验规定生产厂家对这些容器或封口物品最少做一次目检可承认供应者的检验证书规定生产厂家定期验证供应者的试验结果证明其试验结果是正确的4 必要时用显微镜检测成分5 每批易受污物昆虫或其他外来杂物污染的某一成份药品容器或密封件鉴于其预期用途在使用前应做微生物试验e 任何批号的成份药品容器或密封件若符合已成文的均一性效价或含量质量纯度等的规格标准和本部分 d 的有关试验可批准使用任何批号的上述材料不符合这些规格应拒收211·86 获准作用的成份药品容器和密封件先入库者先用若产生的偏差是暂时的和适当这种偏差是容许的211·87 获准的成份药品容器和密封件的复检经质量控制部门批准或拒收的成份药品容器密封件若长期贮存或曝露在空气热或其他可能对其产生不良影响的环境后应依照211·84 对均一性效价或含量质量纯度等复检211·89 拒收的成份药品容器和封口物品拒收的成份药品容器和封口物品应经鉴定和在隔离系统下加以控制防止在生产和加工使用211·94 药品密封容器和密封件a 药品包装容器和密封件应不起反应不吸着不吸附不致改变药品的安全性均一性含量或效价质量和纯度而超出制定的或其它颁布的规定要求b 容器封口系统应对贮藏和使用过程中可预见的能引起药品变质或污染的外部因素提供足够的防护c 药品容器和密封件应清洁灭菌和除热原保证其适用于预期目的d 药品容器和密封件的标准或规格检验方法指清洁和消毒方法除热原过程应成文并遵循9----------------------- Page 11-----------------------F .生产和加工控制211·100 成文的规程偏差a 编写为保证药品的均一性含量或效价质量及纯度而设计的生产和加工控制程序这些程序包括本部内全部要求这些成文程序包括变化须经有关部门起草复查和批准然后再经质量控制部门复查与批准b 在实施各种生产和加工控制功能中遵循已制定的生产和加工控制程序并在招待时以文件加以证明程序中出现的任何偏差应作记录并提出证据211·101 成分的进料成文的生产和控制程序包括下面的内容其设计应保证所生产的药品具有核武器原有的均一性含量和效价质量和纯度a 按处方配制的药品保证其活性成份含量不低于100%标示量或规定量b 生产药品用的成份应称量测量或适当粉碎若一种成份从原来容器转移到另一容器内用下列资料以鉴别(1) 成份名称或项目代码(2) 接收或控制号(3) 在新容器中的重量或份量(4) 使用此成分2 配制的一批药品包含其产品名称含量和批号c 成份的称重测量或粉碎操作应受到严密的监督所盛成份已用于生产的每一容器须经第二人检查保证1 此成份是由质量控制人员发放的2 重量或份量正确批生产记录一致3 容器经严格鉴别d 每一成份投料时一人操作另一人核对211·103 产量计算在药品生产加工或贮存的每一适当阶段结束时测算实际产量与理论产量的百分比211·105 设备鉴别a 在整个生产周期内同批药品生产使用的全部混合和贮存容器生产线和主要设备应严格识别标示出药品的成份需要时不须标出所处的加工阶段b 一种药品每批生产使用的主要设备以一鉴别性识别号或代号加以识别此鉴别号或代号记录在该批号产品的记录本若生产中只使用一种特殊型号的设备可用该设备名字代替鉴别性识别或代号211·110 中间体和药品的取样与检验a 制订和遵循说明每批的加工过程控制及对加工过程中材料的适当样品实行检验或检查的成文程序保证药品的一致性和完整性上述控制程序包括但不限于如下内容1 片剂或胶囊的重量变化2 崩解时间10----------------------- Page 12-----------------------3 充分混和保证均匀4 溶解时间和溶解速率5 溶液的澄明度溶解完全性及PH 值b 考虑上述特性而制定的有效中间加工规格与药品最终规格一致此中间加工规格应在以前可靠的加工方法稳定性评估和经应用统计学程序断定认为合适的基础上制定的样品测试保证药品和中间体符合规格标准c 在生产加工期间如在重要阶段的开始由质量控制部门审定决定联取舍d 不合格的中间体在隔离系统下鉴别及控制防止其在征税或加工操作中使用211·111 生产时间限制在适当时候制定完成每一生产阶段的时间限制保证药品质量制定的时间限制产生偏差如这些偏差不损害药品质量是可以接受的这些偏差应有文字文件证明是正当的211·113 微生物污染的控制a 制订和遵循预防不需消毒药品有害微生物的适当程序b 制订和遵循预防已消毒药品微生物污染的适当程序这些程序包括所有消毒过程的验证211·115 返工a 制订和遵循指导人合格批号返工及保证返工批号达标的程序b 没有质量控制部门复检与批准不许进行返工G.包装和标签控制211·122 材料的检查和使用标准a 制订详细标签和包装材料的接收鉴别贮存半年取样检验的程序并遵循这些成文程序在接收用于药品包装和贴标签前有代表性地对其取样与检验b 符合成文规格标准的标签和包装材料可批准发放使用不符合规格者不得用于生产c 接收每批不同标签和包装材料无须签收测试无论是接收或拒收须保留其记录d 用于不同药品含量剂型及成份数量的标签和标示材料分别贮存并持上适当牌证只限经核准人员接近贮存地区e 作废和陈旧的标签标示材料及其他包装材料应销毁f 排字印刷在不同药品或同一药品不同规格的品种上`使用排字印刷考虑在印刷期间和印刷后印刷品的设置切裁和管理等应制订包装和标签工作专门控制程序g 在药品印标签的生产线其上的或与其有关的印刷设备应受到临近保证所有印痕与本批产品记录中说明的印痕一致。
美国药品生产质量管理规范(cGMP)

GOOD MANUFACTURE PRACTICE美国药品生产质量管理规范(CGMP)二○○三年十二月目录210.1 cGMP法规的地位 (2)210.2 cGMP法规的适用性 (2)210。
3 定义 (2)总则 (4)211—A—211-B- 组织与人员 (4)211-C- 厂房和设施 (5)211-D—设备 (7)211-E—成份、药品容器和密封件的控制 (8)211-F—生产和加工控制 (10)211—G- 包装和标签控制 (11)211-H—贮存和销售 (13)211—实验室控制 (14)I—211—J- 记录和报告 (16)211—K- 退回的药品和回收处理 (20)210部分—人用及兽用药品的生产、加工、包装或贮存的CGMP210.1 cGMP法规的地位(a)在本部分及21CFR 211—226部分中陈述的法规是在药品生产、加工、包装或贮存中使用的现行生产质量管理规范及使用的设施或控制的最低标准,以保证该药品符合联邦食品、药品及化妆品法对安全性的要求,具有均一性和效价(或含量)并符合或代表其生产过程的质量及纯度等特征。
(b)凡是在药品生产、加工、包装或贮存过程中存在任何不符合本部分及21CFR 211—226部分中陈述的法规的药品,依据联邦食品、药品及化妆品法501 (a)(2)-(B),该药应被视为劣药,同时导致该事故发生的负责人应受相应的法规的制裁。
210.2 cGMP法规的适用性(a) 本部分及21CFR 211—226适用于普通药品,21CFR 600—680适用于人用生物制品,除非另有明确规定,否则上述两者之间应该是相互补充而不是相互取代。
如有上述两部分的法规不适用的药品,则可用特定的具体法规来替代。
210。
3 定义(a)在联邦食品、药品及化妆品法201部分中包含的定义和解释、说明适用于21CFR 211—226部分中的术语.(b)下面定义的术语适用于本部分及21CFR 211—226。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
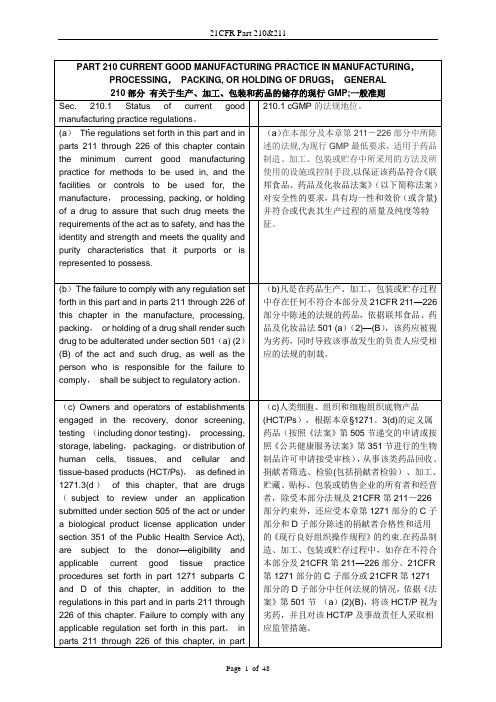
PART 210 CURRENT GOOD MANUFACTURING PRACTICE IN MANUFACTURING, PROCESSING, PACKING, ORHOLDING OF DRUGS; GENERAL210部分有关于生产、加工、包装和药品的储存的现行GMP;一般准则Sec. 210.1 Status of current goodmanufacturing practice regulations.210.1 cGMP的法规地位。
(a) The regulations set forth in this part and in parts 211 through 226 of this chapter contain the minimum current good manufacturing practice for methods to be used in, and the facilities or controls to be used for, the manufacture, processing, packing, or holding of a drug to assure that such drug meets the requirements of the act as to safety, and has the identity and strength and meets the quality and purity characteristics that it purports or is represented to possess. (a)在本部分及本章第211-226部分中所陈述的法规,为现行GMP最低要求,适用于药品制造、加工、包装或贮存中所采用的方法及所使用的设施或控制手段,以保证该药品符合《联邦食品、药品及化妆品法案》(以下简称法案)对安全性的要求,具有均一性和效价(或含量)并符合或代表其生产过程的质量及纯度等特征。
(b) The failure to comply with any regulation set forth in this part and in parts 211 through 226 of this chapter in the manufacture, processing, packing, or holding of a drug shall render such drug to be adulterated under section 501(a) (2)(B) of the act and such drug, as well as the person who is responsible for the failure to comply, shall be subject to regulatory action. (b)凡是在药品生产、加工、包装或贮存过程中存在任何不符合本部分及21CFR 211—226部分中陈述的法规的药品,依据联邦食品、药品及化妆品法501 (a)(2)-(B),该药应被视为劣药,同时导致该事故发生的负责人应受相应的法规的制裁。
(c) Owners and operators of establishments engaged in the recovery, donor screening, testing (including donor testing), processing, storage, labeling, packaging, or distribution of human cells, tissues, and cellular and tissue-based products (HCT/Ps), as defined in 1271.3(d) of this chapter, that are drugs (subject to review under an application submitted under section 505 of the act or under a biological product license application under section 351 of the Public Health Service Act), are subject to the donor-eligibility and applicable current good tissue practice procedures set forth in part 1271 subparts C and D of this chapter, in addition to the regulations in this part and in parts 211 through 226 of this chapter. Failure to comply with any applicable (c)人类细胞、组织和细胞组织底物产品(HCT/Ps),根据本章§1271.3(d)的定义属药品(按照《法案》第505节递交的申请或按照《公共健康服务法案》第351节进行的生物制品许可申请接受审核),从事该类药品回收、捐献者筛选、检验(包括捐献者检验)、加工、贮藏、贴标、包装或销售企业的所有者和经营者,除受本部分法规及21CFR 第211-226部分约束外,还应受本章第1271部分的C子部分和D子部分陈述的捐献者合格性和适用的《现行良好组织操作规程》的约束。
在药品制造、加工、包装或贮存过程中,如存在不符合本部分及21CFR第211—226部分、21CFR第1271部分的C子部分或21CFR 第1271部分的D子部分中任何法规的情况,依据《法案》第501节 (a)(2)(B),将该HCT/P 视为劣药,并且对该HCT/P及事故责任人采取相应监管措施。
regulation set forth in this part, in parts 211through 226 of this chapter, in part 1271subpart C of this chapter, or in part 1271subpart D of this chapter with respect to themanufacture, processing, packing or holdingof a drug, renders an HCT/P adulterated undersection 501(a)(2)(B) of the act. Such HCT/P,as well as the person who is responsible forthe failure to comply, is subject toregulatory action.Sec. 210.2 Applicability of current goodmanufacturing practice regulations.210.2 cGMP法规的适用性(a) The regulations in this part and in parts 211 through 226 of this chapter as they may pertain to a drug; in parts 600 through 680 of this chapter as they may pertain to a biological product for human use; and in part 1271 of this chapter as they are applicable to a human cell, tissue, or cellular or tissue-based product (HCT/P) that is a drug (subject to review under an application submitted under section 505 of the act or under a biological product license application under section 351 of the Public Health Service Act); shall be considered to supplement, not supersede, each other, unless the regulations explicitly provide otherwise. In the event of a conflict between applicable regulations in this part and in other parts of this chapter, the regulation specifically applicable to the drug product in question shall supersede the more general. (a)本部分及21CFR211-226适用于普通药品,21CFR 600-680适用于人用生物制品,21CFR 1271部分适用于人类的细胞、组织或是细胞组织底物产品(HCT/P) 类药品(按照《法案》第505节递交的申请或按照《公共健康服务法案》第351节进行的生物制品许可申请接受审核),它们之间应该是相互补充而不是相互取代,法规另有明确规定除外。
在适用本部分法规和21CFR其它部分法规发生冲突的情况下,特别法规应替代普通法规适用于所涉及的药品。
(b) If a person engages in only some operations subject to the regulations in this part, in parts 211 through 226 of this chapter, in parts 600 through 680 of this chapter, and in part 1271 of this chapter, and not in others, that person need only comply with those regulations applicable to the operations in which he or she is engaged. (b)如果一个人的具体操作仅涉及到21CFR中的本部分,211到226部分,600到680部分和1271部分,而不参与到其他部分中,这个人仅仅需要遵守他/她所涉及的相关操作的规范。