金属有机化学第十三章
有机化学第十三章

(3)单键的裂解 光解或电荷分解NH3或N2H4,也能生成没有取代 的乃春。
13.5 乃春的反应 乃春的反应也和卡宾的相似。乃春很容易发生插 入、加成、重排、氢提取、二聚和歧化反应。 (1)插入反应 乃春,特则是羧酰基乃春和磺酰基乃春,可以插 入C-H和其他的键,例如:
(2)对C=C键的加成 烷基乃春对双键环加成,形成氮杂环丙烷衍生 物。
(d)其它乃春 在-75°C,及四甲基乙烯中,用苯基锂处理氯 胺,确实产生了少量的四甲基氮杂环丙烷。
对甲苯磺酰基叠氮热分解,形成对甲苯磺酰基乃 春。
13.4 乃春的生成 (1)α-消去 例如,用碱处理苯磺酰羟胺,则生成乃春。
(2)叠氮化合物的分解 生成乃春最常用的方法是叠氮化合物的光分解或 热裂解。
(3)重排反应 烷基乃春的重排发生得很快,因而一般不发生上 述两种反应,烷基乃春重排,形成Schiff碱。
芳基乃春高温容易重排。
(4)氢提取 乃春可以从烷烃提取氢原子,形成游离基。
(5)二聚
(6)歧化反应 HN可以歧化,形成热力学稳定的氮乃春(nitrenes),R-N:是卡宾的氮类似物。它非常 活泼,以致在普通条件下是难以离析到的。在4K,在 模床中捕集,曾经离析到过烷基乃春;在77K,曾经 捕集过芳基乃春。HN:的基态是三线态;但是,乃春 也有两种形式单线态和三线态生成。
最简单的乃春是NH,可以把它看成是乃春的母 体,其他乃春则看成是它的衍生物。可用通式R-N表 示,式中的R用来代表H、F,Cl,Br,烷基、芳 基、酰基或磺酰基等。 13.2 乃春的结构 乃春的氮原于只有六个价电子,是缺电子的中性 物种。它与卡宾是等电子的,其结构与卡宾有些类 似。单重态氮烯具有亲电性,而三重态氮烯的行为则 象双自由基。
有机化学-第十三章
3)由于杂原子的电负性为 O>N>S,它们的给电子能力为 S>N>O,所以环上电子云密度的大小顺序为:
它们的离域能分别为117KJ·mol-1,88KJ·mol-1, 67kJ·mol-1 也说明了这点。如呋喃就表现出某些共轭二 烯的性质,可以进行双烯加成反应,有介于芳香族及不饱 和脂肪族化合物之间的某些特征。
3.工业制备 呋喃由呋喃甲醛在催化剂存在下脱去羰基而得:
4.应用 近年发现不少天然存在的呋喃或氢化呋喃衍生物,具
呋喃、噻吩和吡咯具有芳香性还可以从分子偶极矩看出。 在非芳香性杂环中只有诱导效应,而在相应的芳香杂环中 除诱导效应外,还有共轭效应,并且两者的方向相反。
μ=5.77x10-30C·m μ=6.33x10-30C·m μ=5.27x10-30C
四氢呋喃
四氢噻吩
四氢吡咯
在呋喃、噻吩、吡咯杂环中,由于杂原子不同,显示的芳 香性也不完全一致。键长的平均化程度也不一样。实测的
键长数据如下:
由此可见:
1)五元杂环分子中的键长有一定的平均化,但不像苯那 样完全平均化,因此芳香性较苯环差,有某种程度的不饱 和化合物的性质和环的不稳定性;
2)杂原子有给电子性,环上电子云密度比苯环上的高, 因此比苯更容易发生亲电取代反应,取代主要发生在 α 位,其活性相当于苯环上连接 -OH、-SH、-NH2;
2)当环上含有两个或两个以上的杂原子时,应使杂原子所 在的位次数字最小;当环上有不同的杂原子时,按 O>S->N 的次序开始编号。
3)当环上连有不同的取代基时,习惯上根据顺序规则及最 低系列原则编号。
3.命名 取代基位号及名称加上母环的名称即为杂环化合物的
名称,例如:
课堂练习
有机化学第十三章有机含氮化合物

CH3 N(C2H5)2 N,N- dimethyl-3-methyl-2-pentanaime
CH3CH2CH CHCH3
N,N-二乙基-3-甲基-2-戊胺
CF3 NH2
CF3 2,5-bis(trifluoromethyl)benzenamine
2,5-双(三氟甲基)苯胺
NH3HSO4 H2O
NHSO 3H 180℃
NH2 SO3H
NH3 SO2O
(四)胺类化合物的制备方法:
1 、氨的烷基化(卤代烷的取代,SN2 机理)
RX
NH3(过量)
R NH2 +
RX
NH4X
R
R
R NH R + R N R + R N R X
R
• 伴有多取代产物,分离可能有困难
• 仲卤代物和叔卤代物伴有消除产物
3、结构
O N O(由一个N=O和一个N→O配位键组成)
物理测试表明,两个N—O键键长相等,这说明硝基为一P-π 共轭体系(N原子是以sp2杂化成键的,其结构表示如下:
O
RN O
O RN
O
O RNOBiblioteka (二)硝基化合物的物理性质
脂肪族硝基化合物是无色有香味的液体。芳香 族硝基化合物多为淡黄色固体,有杏仁儿气味 并有毒。硝基化合物比重大于一,硝基越多比 重越大;不溶于水,溶于有机溶剂;分子的极 性较大,沸点较高。多硝基化合物受热时以分 解爆炸。
R NH2 +
CO2
6、Gabriel 伯胺合成法
O NH
O 邻苯二甲酰亚胺
KOH
or K2CO3
* 与强碱作用
1、胺的碱性和胺盐的生成
《有机化学》第13章 杂环化合物和生物碱

4-甲基嘧啶
4-甲基噻唑
⑶ 连有取代基的杂环化合物命名时,也可将杂环作为取代基,以侧链为母体来命名。
4-嘧啶磺酸
β-吲哚乙酸(3-吲哚乙酸)
2-苯并咪唑甲酸乙酯
⑷ 为区别杂环化合物的互变异构体,需标明杂环上与杂原子相连的氢原子所在的位 置,并在名称前面加上标位的阿拉伯数字和大写H的斜体字。
2023/6/13
⑴ 卤代反应
在室温条件下,吡咯、呋喃和噻吩能与氯或溴发生激烈反应,得到多卤代物。将反应 物用溶剂稀释并在低温下进行反应时,可以得到一氯代物或一溴代物。碘化反应需要 在催化剂存在下进行。例如:
2023/6/13
6
(2)硝化反应
在低温条件下,吡咯、呋喃和噻吩能与比较缓和的硝化剂硝酸乙酰酯(CH3COONO2) 发生硝化反应,主要生成α-硝基化合物。例如:
3. 颜色反应
生物碱能与一些试剂发生颜色反应,比如钒酸铵的浓硫酸溶液、浓硝酸、浓硫酸、 甲醛、氨水等,利用此性质可鉴别生物碱。比如莨菪碱遇1%钒酸铵的浓硫酸溶液显 红色,可待因遇甲醛-浓硫酸试剂显紫红色等。
二、重要的生物碱 1. 烟碱 又叫尼古丁,主要以苹果酸盐及柠檬酸盐的形式存在于烟草中。其结构式
2023/6/13
13
血红素是卟啉环与Fe2+形成的配合物;叶绿素是卟啉环与Mg2+形成的配合物,它们的 结构式如下:
血红素在体内与蛋白质结合形成血红蛋白,存在于红细胞中,是人和其他哺乳动物 体内运输氧气的物质。叶绿素是植物进行光合作用不可缺少的物质。
2023/6/13
14
二、呋喃衍生物
呋喃甲醛是最常见的呋喃衍生物,又称为糠醛,它是一种无色液体,沸点为161.7℃, 在空气中易氧化变黑,是一种良好的溶剂。 糠醛是合成药物的重要原料,通过硝化可制得一系列呋喃类抗菌药物,如治疗泌尿 系统感染的药物呋喃坦丁、治疗血吸虫病的药物呋喃丙胺等。
有机化学第十三章 取代羧酸

稀 OH -
CH3COCHC 2H5 CH3
1. 合成甲基酮 经乙酰乙酸乙酯合成:
引入基团
引入基团
CH3 CH3CO CHCH 2CH 3
CH3CO CH 2CH 3
分析:(1) 产物为甲基酮,合成时一定要经过酮式分解。 (2) 将TM的结构与丙酮进行比较,确定引入基团。 (3) 最后确定合成路线。
-OH、-COOH均可形成氢键。 二、 物理性质 1、 比羧酸更易溶于水
2 、 熔点比羧酸高 三 、化学性质
1、 酸性: -OH吸电子,酸性增强,且-位大于-位
CH3-CH2-COOH Ka=1.34×10-5
CH2 CH2 COOH Ka=3.10×10-5 OH
CH3 CH OH COOH Ka=1.38×10-4
HO
HO
COOCH 3
尼泊金M
COOC 2H5 尼泊金A
HO
COOC 3H7
尼泊索 Nipasol
COOH
没食子酸
HO
OH
OH 没食子酸又名五倍子酸,3,4,5-三羟基苯甲酸,无色结晶, 以游离形式存在于茶叶中,以鞣质(丹宁)存在于五倍子等 植物中。是自然界中广泛存在的一种有机酸。
没食子酸用途:照像中用作显影剂,遇三氯化铁显蓝 黑色,作蓝黑墨水。
O C COOH
CO2
RCHO O
CO2
R
C
CH 3
四 醇酸的制备 1 α-羟基酸 羟基腈水解( ―醛的化学性质”)
O R-C-R(H) + HCN
2 β-羟基酸(酯)
OH R-C-R(H) CN
H3O
+
OH α R-C-R(H) COOH
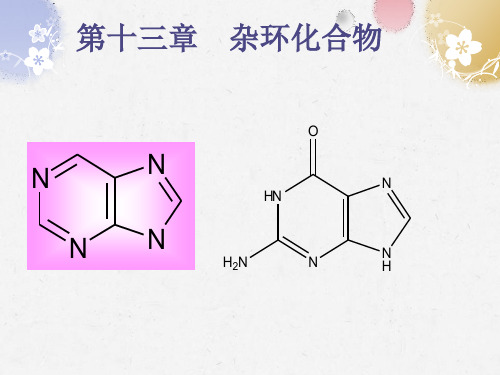
有机化学第十三章 羧酸及其衍生物
2) 与羰基相连的基团(Y) 空间体积; 3) 与羰基相连的基团(Y)
离去能力; 4) 反应物稳定化程度。
离去基团的离去能力: X- > RCOO- > RO > H2N
O R C Y
OR C Y+
羧酸衍生物亲核取代反应活性顺序:
O O O O > RCNH2 ~ ~ RCN
RCX > RCOCR >
RCH2Cl3 + H2O RCH2CN + H2O RCH2COOR' + H2O ......
RCH2COOH
13.3 羧酸的物理性质(P309) 13.4 羧酸的化学性质(P311) 1、酸性和成盐
sp
2
自学
R C
O OH R C
O
O OH
O C O
R C O-
两个碳氧键键长不同
四电子三中心的分子轨道 两个碳氧键键长等同。
O HO HC HOH2C OH
O
CH3
OH
Erythromycin A (红霉素) A
Vitamin C
• 酰胺
CH3 O O HCN-CH3 CH3
N,3-二甲基戊酰胺 N,N-二甲基甲酰胺
COOH
CH3CH2CHCH2CNHCH3
NHCOCH3
4-乙酰氨基-1萘羧酸
氮原子上有取代基, 在取代基名称前加N标出。
O NH R O R-C-NH N O S CH3 CH3 COOH
R=C6H5CH2(penicillin G)
γ -内酰胺
青霉素 ( penicillin )
物理性质(自学)
13.9 羧酸衍生物的物理性质
有机化学第十三章羧酸衍生物总结

反应活性较差
叔醇
吡啶
O C OC(CH3)3
不仅起催化作用 还可吸收产生的HCl
2) 酯交换:由低级醇酯制高级醇酯
= H+
CH2 CHCOOCH3 + CH3(CH2)2CH2OH
b.p: 80.5℃ 低沸点酯
= CH2 CHCOO(CH2)3CH3 + CH3OH
b.p: 145℃
b.p: 64.7℃
R C =O > X
=
R C =O
O RC
O
> R C =O > R C =O
OR′
NH2
第二步——取决于离去基团的离去能力。
基团的离去能力:
! 试解释之
X > RCOO > RO > NH2
碱性越小离去能力越强。
碱性强弱:
X < RCOO < RO < NH2
结论:
羧酸衍生物的反应活性顺序为
R C =O > X
== =
== =
O C O C4H9_ n
C OH
O
邻苯二甲酸单正丁酯
O CH3 C O CH2 CH3 C O CH2
O
乙二醇二乙酸酯
CH2OCOR CHOCOR'
甘油三酯:脂肪和油
CH2OCOR''
O
H3C
CH3
H3C
HO H3CH2CH
OH O
O
CH3
CH3 OH HO O
O O
N(CH3)2
青霉素 ( penicillin )
酸酐:两个羧酸名加“酐”字。
O
OO
OO
COC
有机化学第十三章 取代羧酸
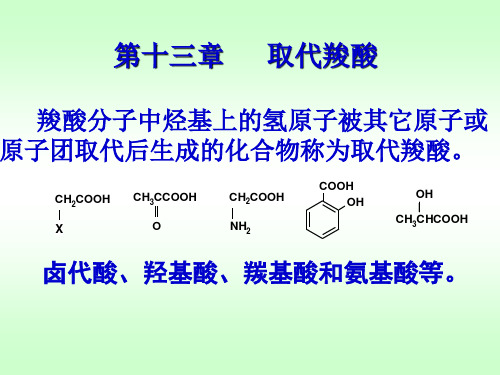
C HO 浓NaOH CH2OH COOH ’ COOH C anniz z aro 反 应 COOH + COOH
三
丙酮酸: 最简单的α -酮酸
CH3
O C COOH
CH3 CH3
O 浓 H2SO4 CO + CH3 COOH(脱羰) C COOH O 稀 H2SO4 CO + CH CHO (脱羧) 2 3 C COOH
OH O C6H5C=CHCCH 3 90.0%
2 乙酰乙酸乙酯的分解反应 稀OH-
O O CH3C-CH 2-COC 2H5
浓OH-
O CH3CCH3
酮式分解
O CH3C-OH + 其余 酸式分解
3 α-H的活性:被取代
O O CH3C-CH2-COC 2H5
RONa
O Na+ O CH3C-CH--COC 2H5
稀 OH -
CH3COCHC 2H5 CH3
1. 合成甲基酮 经乙酰乙酸乙酯合成:
引入基团
引入基团
CH3 CH3CO CHCH 2CH 3
CH3CO CH 2CH 3
分析:(1) 产物为甲基酮,合成时一定要经过酮式分解。 (2) 将TM的结构与丙酮进行比较,确定引入基团。 (3) 最后确定合成路线。
O C COOH
CO2
RCHO O
CO2
R
C
CH 3
四 醇酸的制备 1 α-羟基酸 羟基腈水解( “醛的化学性质”)
O R-C-R(H) + HCN
2 β-羟基酸(酯)
OH R-C-R(H) CN
H3O
+
OH α R-C-R(H) COOH
有机化学:13 取代羧酸

2. 乙酰乙酸乙酯的互变异构
两种或以上异构体互相转变,并以动态平衡而 存在——互变异构现象(tautomerism)
O O CH3 C H 7% O CH C OC2H5
CH3CCH2COOC2H5 93%
O
COOH O OH
γ -丁内酯(1,4-丁内酯)
38
+ H2O
3.脱水反应
γ -,δ -醇酸:分子内脱水 内酯
O COOH OH O + H2O
Δ
δ -戊内酯(1,5-戊内酯)
39
3.脱水反应
γ-内酯是稳定的中性化合物 与热碱溶液作用能水解
O COONa O NaOH / H2O OH
COOH
45
(一)制备
其他酚酸:
OH +CO 2 OH NaHCO 3 OH COOH OH
46
(二)性 质
OH COOH OH 200~220℃ OH
酚酸为结晶固体,具 有酚和芳酸的性质 羧基处于羟基邻对位 时,受热易脱酸:
COOH 200℃ HO OH OH
COOH
+CO 2 HO OH
56
(二)乙酰乙酸乙酯的酸性和互变异 构
α-H的活泼性 思考:
O O CCH3
CH3C CH2
的酸性大小?
O C2H5OC CH2
O COC2H5
57
2. 乙酰乙酸乙酯的互变异构
O O CH3 C CH2 C OC 2H5
+2,4-二硝基苯肼橙色苯腙 ——羰基 +Br2-CCl4 褪色 ——不饱和键 +Na H2 ↑ ——活泼氢 +CH3COCl 酯 ——醇羟基 +FeCl3水溶液 紫红色——烯醇式结构
有机化学第13章 羧酸衍生物

35
另一方面,乙酰胺的水溶液能与氧化汞作用生成稳定的汞盐。
酰胺与金属钠在乙醚溶液中作用,也能生成钠盐,但它遇水即 分解。这些说明酰胺具有弱酸性。
O C NH C O
邻苯二甲酰亚胺
36
2.酰胺脱水
酰胺与强脱水剂共热或高温加热,则分子内脱水生成腈,这
是合成腈最常用的方法之一。常用的脱水剂有五氧化二磷和亚硫
10
IR:
11
酰卤的C=O伸缩振动在1815~1785cm-1区域有强吸收,其C—X
的面内弯曲振动在645 cm-1附近。
12
酸 酐的 C=O伸 缩 振动 吸 收 与其它 羰 基 化合物 明 显不 同 , 在 1850~1800cm-1和1780~1740 cm-1区域内有两个C=O伸缩振动的强 吸收峰。线型酸酐的高频峰强于低频峰,而环状酸酐则相反。酸酐
26
酰基化试剂中离去基团离去的难易,取决于离去基团(L)的碱 性。碱性愈弱,愈易离去。离去基团L的碱性由弱到强的次序是:
C1—<RCOO—<RO—<NH2—。因此氯原于是最容易离去的基团,
而氨基则是最难离去的基团。即酰氯是最活泼的酰基化试剂,而酰 胺是最弱者。 离去基团碱性的强弱,可从其共轭酸的酸性强弱得知(强酸的 共轭碱是弱碱):
酯的醇解亦称酯交换反应。例如:
21
腈的醇溶液和浓硫酸或盐酸共热,则发生醇解反应得到酯。 例如:
22
3.氨解
酰氯、酸酐和酯与氨或胺作用,都可以生成酰胺。例如:
23
N-未取代的酰胺与胺反应生成N-取代酰胺。例如:
以上这些反应对羧酸衍生物是发生了水解、醇解或氨解;但对 水、醇或氨则是发生了酰基化反应。酰氯、酸酐和酯都是酰基化试 剂,酰胺的酰化能力极弱,一般不用作酰基化试剂。
有机化学学习笔记 第十三章羧酸

CH2CH2COOH CH2 (CH3CO)2O CH2CH2COOH
O + CO2 + H 2O
如何将环己酮转化为环戊酮?
O HNO3 CH2CH2COOH CH2CH2COOH (CH3CO)2O O
3.羧酸的脱羧卤代反应
A. Hunsdiecker反应
O R-C-OH Ag2O
O R-C-O Ag O
O CH3 CH3C-C H CH3
COOH CH2 COOH
CH3COOH + CO2
O CH2COOH
n=2,3
CH2COOH (CH3CO)2O
O + H 2O O
CH2COOH CH2 (CH3CO)2O CH2COOH
O O + H2O O
n=4,5
O CH2CH2COOH CH2CH2COOH (CH3CO)2O + CO2 + H2O
BrCH2COOH 2.90
0.64
ICH2COOH 3.18
FCH2COOH ClCH2COOH 2.66 2.88
羧酸 pKa
羧酸 pKa
HCOOH CH3COOH CH3CH2COOH (CH3)2CHCOOH (CH3)3CCOOH 3.77 4.76 4.87 4.86
Cl CH2CH2CH2COOH 4.70
5.05
n-C3H7COOH 4.80
Cl Cl CH3CH2CHCOOH CH3CHCH2COOH 2.82 4.41
羧酸 pKa
CH=CCH2COOH 3.30
CH2COOH 4.31
CH2=CHCH2COOH 4.35
n-C3H7COOH 4.82
有机化学 第十三章__羧酸1

R CH 2OH
①干醚 ② H2O
H 2C
CH(CH 2)4COOH
+
LiAlH4
H 2C
CH(CH 2)4CH 2OH 83%
合成上应用 —— 制备伯醇
O R C OH 2. H2O
1. LiAlH4 酯化 R O C OEt 2. H2O
1. LiAlH4
R
CH2OH
直接还原羧酸: LiAlH4用量多, 反应开始剧烈, 后较慢,
先酯化再还原: 反应较易进行,产率较好, 反应条件相对比较温和, LiAlH4用量较少
合成上由羧酸制备 伯醇,宜先酯化再 还原
补充内容:能被LiAlH4还原的化合物及其产物类型
O R C O R C O H O C R R R O C OR' O C OH R O C Cl 1. LiAlH4 R 2. H2O CH2OH
CHN H(a -aC OOO N→ aC +H N4 a+ O3 ) 3C2
O O CH3 C CH2 COOH COOH C2H5 COOH CH3CH COOH CH3 C O CH3
+
CO2
O
C2H5
+
CO2
CH3CH2COOH
+
CO2
羧酸与金属有机试剂的反应
与RMgX反应
O R
O OH R
R
R
OH
Ⅰ. 酰氧断裂
O
Ⅱ. 烷氧断裂
OH
OH
Ⅰ: R C OH
H2O OH
H
+
R C OH
HOR
R C OH HOR
R C OH H OR
有机化学第十三章羧酸衍生物

C3H7 C OH H
这是用二元酸酯合成大环化合物很好的方 法。
2019/9/30
CO3OCH Na
(C 2 )8H CO3O二 C 甲 苯 H
HAc
CO
(C 2 )8HCO
3、酸酐的还原 酸酐的活性不如酰卤, 但酸酐可以被LiAlH4 、NaBH4还原成醇。
4、酰胺的还原 酰胺在LiAlH4 作用下 还原成伯胺。
L
(1)与 酰 基 相 连 的 原 子 的 电 负 性 都 比 碳 大 , 故 有I效 应 (2)L和 碳 相 连 的 原 子 上 有 未 共 用 电 子 对 , 故 具 有 +C (3) 当 +C> I时 , 反 应 活 性 将 降 低
Pπ共 20轭 19/9体 /30系
当 +C< I时 , 反 应 活 性 将 增 大
酯在酸催化下进行的水解为可逆水解:
H +
C H 3C O O C 2H 5 +H 2O
C H 3C O O H+C 2H 5O H
酯在碱催化下进行的水解为不可逆水解:
C H 3C O O C 2H 5 +H 2ON aO HC H 3C O O N a+C 2H 5O H
因此,在碱性下水解反应更为彻底,其 碱性水解称为皂化,即为工业上制造肥皂的 原理。
R C OH R
从产物的结构看,格氏试剂与酯作用是 合成具有两个相同烃基的叔醇的最有效方法。 例如:
COCH3
1)乙醚 +2CH3M gX2)3O H +
CH3 C OH CH3
2019/9/30
当使用HCOOR与RMgX作用,可以制备 结构对称的仲醇。例如
第十三章 EDOT杂环类似物和在噻吩上具有取代基的EDOT衍生物
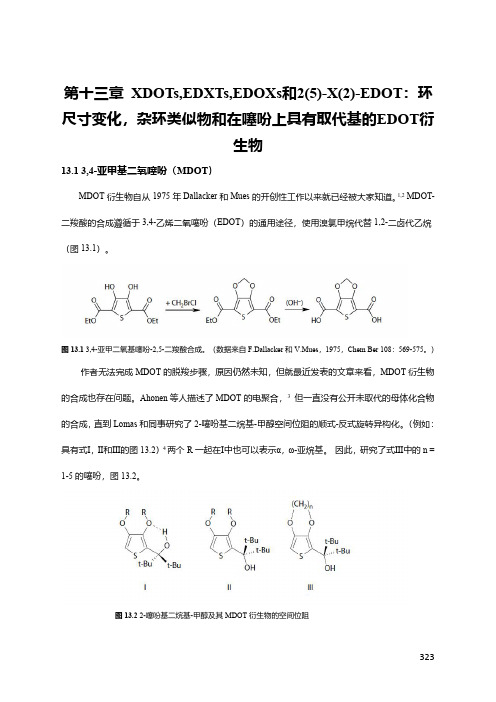
323第十三章XDOTs,EDXTs,EDOXs 和2(5)-X(2)-EDOT :环尺寸变化,杂环类似物和在噻吩上具有取代基的EDOT 衍生物13.13,4-亚甲基二氧噻吩(MDOT )MDOT 衍生物自从1975年Dallacker 和Mues 的开创性工作以来就已经被大家知道。
1,2MDOT-二羧酸的合成遵循于3,4-乙烯二氧噻吩(EDOT )的通用途径,使用溴氯甲烷代替1,2-二卤代乙烷(图13.1)。
图13.13,4-亚甲二氧基噻吩-2,5-二羧酸合成。
(数据来自F.Dallacker 和V.Mues ,1975,Chem Ber 108:569-575。
)作者无法完成MDOT 的脱羧步骤,原因仍然未知,但就最近发表的文章来看,MDOT 衍生物的合成也存在问题。
Ahonen 等人描述了MDOT 的电聚合,3但一直没有公开未取代的母体化合物的合成,直到Lomas 和同事研究了2-噻吩基二烷基-甲醇空间位阻的顺式-反式旋转异构化。
(例如:具有式Ⅰ,Ⅱ和Ⅲ的图13.2)4两个R 一起在Ⅰ中也可以表示α,ω-亚烷基。
因此,研究了式Ⅲ中的n =1-5的噻吩,图13.2。
图13.22-噻吩基二烷基-甲醇及其MDOT 衍生物的空间位阻如Lomas等人所指出的,MDOT可期望作为一种非常有用的单体聚合导电聚噻吩。
逻辑上,通过从EDOT,3,4-亚丙基二氧噻吩(ProDoT)和3,4-亚乙基二氧噻吩(BuDOT)已知的特性的简单线性延续,来自MDOT的聚合物应该表现出比PEDOT更好的电性能。
换句话说,“EDOT位于3,4-亚甲基二氧噻吩和其它3,4-亚烷基二氧基,3-烷氧基和3,4-二烷氧基噻吩之间的连续体上。
”4但这个连续体最优不是结束在MDOT,而是EDOT。
Lomas的上述引用是基于图13.2中描述的噻吩甲醇衍生物的旋转异构行为。
4因为空间位置的关系,由于更长的O-H距离,MDOT衍生物中产生的氢键强度最低,所以MDOT聚合物中相邻单体部分之间的空间位阻低。
有机化学-第十三章

3.命名
取代基位号及名称加上母环的名称即为杂环化合物的 名称,例如:
课堂练习
1、命名下列杂环化合物: (1)
Br
(2)
CH3
N
O
NO2
2 – 硝基 – 4 –溴呋喃
3 – 甲基吡啶
杂环化合物的结构与芳香性
一、五元单杂环化合物的结构和芳香性
五元单杂环如呋喃、噻吩、吡咯,在结构上,都符 合 Huckel 的关于芳香性的规则,即环上原子共平面,彼 此以 σ 键相连接,四个碳原子各有一个电子在 p 轨道 上,杂原子有两个电子在 p 轨道上,这些 p 轨道都垂直 于σ 键所在的平面,相互重叠形成大 π 键——闭和的 共轭体系,π 电子数目为4n+2。结构如图所示:
亲电取代反应主要发生在 α 位上
二、定位规律
对五元杂环的亲电取代反应环上取代基有一定的定位 作用,以噻吩为例,说明如下: (1)当 α 位上有邻对位定位基(x)时,亲电试剂(E)主 要进入 5 位,3 位次之:
(2)当 α 位上有间定位基(Y)时,则 E 主要进入 4 位,少量进入 5 位:
近年发现不少天然存在的呋喃或氢化呋喃衍生物,具 有明显的生物活性或药用价值。例如,从重斑病感染的薯 类植物块根中分离的苦味成为含多种呋喃衍生物,如3-呋 喃甲酸,巴他酸(bacatic acid),番薯酮等
二、α-呋喃甲醛
1.来源与制备 呋喃甲醛最初是从米糠中得来,故俗称糠醛,因为这 些农副产品中都含有戊聚糖,在稀酸作用下水解成戊醛糖 ,再进一步脱水环化,得到糠醛:
4)综上所述,它们的芳香性由强到弱的顺序为:
二、六元单杂环化合物的结构和芳香性
吡啶具有六元单杂环的典型结构和苯的结构很相似, 是苯中的一个碳原子被氮原子代替,氮原子以 sp2 杂化 轨道和两个相邻碳原子的 sp2 杂化轨道形成两个 σ 键 。环上每个原子均有一个 p 轨道垂直于环的平面,组成 闭合的6电子大 π 轨道,因此,吡啶环也有芳香性。吡 啶的结构如图所示:
有机化学第13章 含氮化合物

CH2CN H2
Ni
6.霍夫曼降解反应
CH2CH2NH2
NaOCl,OHRCONH2
或NaOBr,OH-
RNH2
CH3(CH2)4CONH2 Br2+NaOH
CH3(CH2)4NH2
三、胺的物理性质
低级和中级脂肪胺为无色气体或液体,高级脂肪胺为固体, 芳香胺为高沸点的液体或固体。低级胺具有氨的气味或鱼腥味, 高级胺没有气味,芳香胺有特殊气味,并有较大的毒性。
根据分子中烃基的结构,可把胺分为脂肪胺和芳香胺 。例如:
铵盐(NH4)+X-分子中的四个氢原子被四个烃基取代 后的化合物,成为季铵盐,如[N(CH3)4+]I-。
根据分子中氨基的数目,可把胺分为一元胺、二元 胺和多元胺等。例如:
2.胺的命名法 构造简单的胺一般用衍生命名法命名,此时把氨看
作母体,烃基看作取代基。命名时省去“基”字。
2.硝基化合物的命名 硝基化合物命名与卤代烃相同,即以烃为母体,硝基为
取代基。如:CH3NO2(硝基甲烷)、CH3CH2CH2NO2 (硝基丙烷)、硝基苯等。
CH3CHCH3
NO 2
2-硝基丙烷
O2N
OH NO 2
NO 2
HOOC
NO 2
对硝基苯甲酸
O2N
CH 3 NO 2
NO 2
O2N
NO 2
NO 2
第二节 胺
一、胺的分类和命名 1.胺的分类 氨分子中的一个或几个氢原子被烃基取代物称为胺。氨
中一个、两个或三个氢原子被烃基取代生成的化合物分别称 为伯胺、仲胺和叔胺。也被称为一级胺、二级胺和三级胺。
注意: 伯、仲、叔胺的含义和醇、卤代烃等的伯、仲、叔含义不同
有机化学 第十三章 羧酸衍生物

羧酸衍生物
O
OO
O
O
R CXR CO C RR CO RR ' CN 2H ( RCN)
O
上述化合物中均含有 R C 因此它们的化学性质相似:
① 均可以核亲核试剂发生亲核加成消去反应; ② 均可由羧酸制备; ③ 水解均可以得到羧酸。 一、羧酸衍生物结构的比较:
1、酰胺:
O
CN O
H C NH 2 CH 3 NH 2
1.376
1.474 Oδ -
H C NH 2
C
δ+
NH 2
① 羰基碳正电荷降低,亲核加成困难;
② N原子电荷降低,碱性减弱,显弱酸性。
2、酯:
O
H C O CH 3 CH 3 OH
CO
1.334
1.430
OC O
由于O的电负性大于N,因此,酯中羰基碳 的正电荷应大于酰胺中羰基碳的正电荷。
酯 +酯
β -酮 酸 酯
酯 + 甲酸酯
α -甲 酰 酯
酯 + 苯甲酸酯
α -苯 甲 酰 酯
酯 + O C( OE t) 2 酯 + 草酸酯
取代丙二酸酯 β -环 二 酮 酯
酯+ 酮
β -二 酮
二元酸酯
β -环 酮 酯
七、达参缩合:
O
α-卤代酸酯+ C O
α ,β -环氧酯 O E tO -
O + C Cl2HC Ot E
取代基—I效应:
O
X2>R C O >RO> N2 H
羰基的正电荷:
O
OO
O
O
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
一. Typical Lanthanide Complexes
Cl Ln Ln Cl Ln
N
S Ln S
a
rK o
C p N a + L n I2
H Ln Ln H
N aH
Cl Ln Cl Ln
0
S iH 2 Ph
Livinghouse, T. ; et al, Organometallics 2004, 23, 12-14.
C p * 2L n R ' R 3 S iH H R" S iR 3 "C p * 2 L n H " R" -R 'S iR 3
R 3 S iH R"
H L n C p *2
Lanthanide Complexes Catalyzed Hydroamination/Cyclization Reactions of aminoalkynes
R NH 2 c a t. L Y -N (T M S ) 2
n N
R
n = 1, 2
Et Et
Pr N or
i
L =
Et
N N
Et
Cy Pri Cy Cy Pri Cy Pri Cy
H2N NO2
H 2N NO2
r.t. / 12 110 / 36 110 / 36 110 / 36
NH
2
H 2N O 2N
110 / 12 110 / 12
22
23
2
2
Pri
Cy
NH2
110 / 24
110 / 24
25
26
81 (84)b
85
13 14 15 16 17 18 19 20 21 2 2 2 2 2 2 2 2 2 Pri
H 2N Cl
r.t. / 12 r.t. / 12 r.t. / 12
H2N Br
16 17 18 19 20 20 20 23 24
93 92 92 (88)c 93 (95)b 80 (82)b No reaction a,b,c No reaction a,b,c 95 97
O O n -C 5 H 1 1 O P h S iH 3 , 5 0 0 C 24 h, 85% H S iH 2 P h C p * 2 Y M e (T H F ) O n -C 5 H 1 1
L a[ N (T MS) 2 ] 3 3 m o l% c at. P h S iH 3 60 C , 9h
二. Reactivity
1. Catalytic Hydrogenation
5 % C p * 2 Y M e (T H F ) 1 a tm H 2 , b e n z e n e rt, 1 h , 7 2 %
H 0 .7 % [M e 2 S i(C p " )(C 5 H 3 R * )]S m C H (T M S ) 2 1 a tm . H 2 , h e p ta n e , rt, < 1 h , 9 5 % , 7 1 % e e
3. Lanthanide Complexes Catalyzed
Hydrophosphination/Cyclization Reactions
H 2P n Ph n = 1, 2 C a t. H P Ph n
C a t.
Si N
Sm
N (S iMe 3 ) 2
O r [(M e 3 S i) 2 N ] 3 L n
Organometallics, 2008, ASAP Article,
Lanthanide Phosphinidene Complex
J. AM. CHEM. SOC. 2008, 130, 2408-2409
L n I 2 (T H F ) x Ln = Y b, Eu, Sm
L n I 2 (T H F ) x a n d r e la te d c o m p le x e s L n = T m , D y, N d
选择性 100:0 100:0 100:0 11:1 3.2:1 1.9:1 31:1 1:2 1:2
3 % C p * 2 Y C H (T M S ) 2 P h S iH 3 , b e n z e n e , rt 92 h, 91% 3 % C p * 2 Y C H (T M S ) 2 P h S iH 3 ,to lu e n e , rt 18 h, 97% 3 % C p * 2 Y C H (T M S ) 2 P h S iH 3 , b e n z e n e , rt 9 6 h , 6 1 % , 2 :1 d s S iH 2 P h S iH 2 P h
H 2N
r.t. / 12 r.t. / 12
H 2N
r.t. / 12 r.t. / 12
NH2
r.t. / 12 r.t. / 12
OCH3
r.t. / 12 r.t. / 12 r.t. / 12
Catalytic Addition of the Amines to the Carbodiimides
Marks, T. J. et al, J. Am. Chem. Soc. 2000, 122, 1824. Marks, T. J. et al, Organometallics 2003, 22, 4630
C p * 2 L n C H (T M S ) 2 + H 2 P
n
C H 2 (T M S ) 2 * PH C p*2L n PH
C a ta ly tic C y c le fo r H y d ro s ily la tio n re a c tio n
5. Catalytic Hydroboration Reaction
C p * 2 L n C H (T M S ) 2
O B O O B O H R H
O B O C H (T M S ) 2
R1 N R
R2 R N H N
RN
C
NR
R 1R 2N H
R2 = H 1 ,3 - H s h ift
R1 N R N H N H R
C a ta ly s t = [ 5 -(C H 2 ) 2 (C 9 H 6 ) 2 ]L n N (S iM e 3 ) 2 ( L n = Y (1 ); S m (2 ); Y b (3 ) )
H 2P
n C p*2L n PH
C p*2L n
*
PH
E = C H (T M S )2
4. Catalytic Hydrosilylation
5 % c a ta ly st n -C 8 H 1 7 P h H 2S i P h S iH 3 , rt, 1 -2 4 h , 7 5 % , 2 8 :1 n -C 8 H 1 7 + n -C 8 H 1 7 S iH 2 P h
S iH 2 P h
n -C 6 H 1 3 + 5 % (C 5 M e 4 i-P r) 2 Y C H (T M S ) 2 n -C 6 H 1 3 P h S iH 3 , 5 0 0 C 1 2 h , 7 8 % , 4 :1 H H P hH 2S i n -C 6 H 1 3 S iH 2 P h
Marks, T. J. et al, Organometallics 1999, 18, 2568
H C a t. NH 2 n -C 5 H 11 H n -C 5 H 11
H
N
H CH 3
C a t. =
Si N
Sm
N (S iMe 3 ) 2
Marks, T. J. ; et al, J. Am. Chem. Soc. 1999, 121, 3633.
Catalytic Addition of the Amine to the Carbodiimides
entry 1 2 3 4 5 6 7 8 9 10 11 12 Cat. 2 2 2 2 2 2 2 2 2 2 2 2 R Pri Cy Pri Cy Pri Cy Pri Cy Pri Cy Pri Cy
N Pr
i
Livinghouse, T. et al, Adv. Synth. Catal. 2006, 348, 701-704.
C p*2L nE H 2N n HE
*
NH
C p * 2L n
NH
H 2N
n C p * 2L n NH
C p * 2L n
*
NH
E = H , C H (T M S ) 2 , N (T M S ) 2 C a ta ly tic C y c le fo r H y d ro a m in a tio n
"C p*2L nH "
-C p * 2 L n H 脱 氢 H R n 氢化环化的催化循环
2. Lanthanide Complexes Catalyzed Hydroamination/ Cyclization Reactions
H N
C a t.
H 2N
C a t. ) 2 L n = S m , N d
Catalytic Activity of the Cp-Free Lanthanide Amides on the Addition of Amines to Carbodiimides
R2 HN R2 c a t. R3 NH2 R1 N C N R1 c a t.
??