CNBr活化琼脂糖填料明书(GE)
快速高效偶联亲和层析介质试剂盒说明书

快速高效偶联亲和层析介质试剂盒说明书【产品名称】通用名称:CDI预活化琼脂糖凝胶4B试剂盒。
商品名称:快速高效偶联试剂盒。
【包装规格】20mL活化柱+500mL缓冲液/盒,附送亲和层析用空柱一支。
【预期用途】快速高效偶联试剂盒适用于亲和层析填料的快速制备以及固相吸附、固定化酶等用途。
【主要组成成份】●预活化琼脂糖凝胶4B●缓冲液【原理】本试剂盒系用优质琼脂糖凝胶 4B活化后存放于保护液中和适合的偶联缓冲液制成,用活性咪唑碳酸酯基团偶联需要的配体。
使用时,保护液被洗去后就可使欲偶联的配体的氨基和活性咪唑碳酸酯反应,脱去咪唑基,使得配体的氨基通过共价键牢固的链接在基质上,即可方便得到高偶联效率的亲和层析填料。
【存储条件和有效期】存储条件:原包装应储存于4-8 C。
密封保存在阴凉干燥处切忌冷冻。
有效期:一年。
【适用仪器】本产品无需特殊仪器辅助使用,只需要在操作时使用抽滤装置帮助抽去洗涤液,以免洗涤时间过长造成偶联效果不理想。
附送的亲和层析空柱可安装适合的接口用于自动纯化仪。
【配体要求】配体溶液的质量至关重要。
配体需溶解于偶联缓冲液中并使得浓度大于15mg/mL,并且浓度越大,偶联效果越好,如已经溶解在其他缓冲液中,可用偶联缓冲液透析平衡。
【偶联方法】方法一:1、预先准备好溶解在偶联缓冲液中的配体(可用直接溶解或透析方法制备),浓度>15mg/ml或者更高,浓度越高偶联效果越好。
2、将活化琼脂糖摇匀,倒入抽滤漏斗,加5倍体积的去离子水,抽气至快干时拔掉抽气管,注意不可抽干,此步速度宜快。
3、用配体溶液悬浮抽滤漏斗里的琼脂糖固体,摇床缓慢振荡(不可使用磁力搅拌器),室温3小时或4摄氏度过夜,然后洗净即可。
洗出来的配体可回收使用。
此方法适用于比较昂贵的配体。
方法二:1、用干燥枪头吸去保护剂。
制备配体在溶液B中的溶液(可用直接溶解或透析方法制备),浓度>15mg/ml或者更高,浓度越高偶联效果越好。
CNBr-4B活化填料使用说明书(译文)

CNBr活化柱说明书溴化氰活化琼脂糖™-4B是一种预活化并固定配体来结合氨基的介质。
它提供了一个非常方便的固定溴化氰配体的方法。
这个偶联反应是自发的、快速和容易实现的。
它的应用领域涵盖了固定化蛋白质、多肽和核酸等。
注:线性流速=体积流速(L/h)/ 层析柱横截面积(cm2)这里所给出的范围是基于我们的立论知识和经验估计的。
请注意以下几点:pH值稳定、长期保存的pH值区间指的是凝胶在很长一段时间内可以保持稳定,没有在其随后的层析分离时出现不利的影响。
pH值稳定、短期保存的pH值区间是相对于再生、在线清洗和维护程序来说的。
准备阶段溴化氰活化琼脂糖™-4 B是以添加剂的形式冻干处理得到的。
这些添加剂在填料螯合所需的配体前必须在低pH(pH 2 – 3)条件下进行清洗。
活化后的活性基团也应该使用低pH 值(pH值2 – 3)保存,而在高pH值则会出现降解。
称取所需量的冻干粉(1 g冻干粉可获得3.5ml终体积的介质),使用1mM HCl悬浮。
介质会立刻膨胀,再用1mM HCl在玻璃过滤器中进行清洗15min。
每克冻干粉添加约200ml 1mM 的HCl溶胀,实际按几等分的量来添加。
螯合配体常规的配体螯合过程1.在螯合前,使用0.1 M NaHCO3、0.5 M NaCl pH 8.3的螯合缓冲液来溶解配体,每克冻干粉使用5ml螯合缓冲液溶解。
一般,每毫升的填料介质可以结合5-10mg的蛋白配体。
对于较小的配体,一般每毫升填料介质添加1-10μM的配体。
2.称出所需量的溴化氰活化琼脂糖-4B填料。
如上所述,用1mM HCl来溶胀和清洗冻干粉。
3. 在一个密封的容器里,将溶解的配体与待螯合的介质混合。
在室温下反复旋转混合约1 h或在4℃下过夜混合,其他温和的搅拌方法也可以使用的。
4.使用至少5倍填料体积的螯合缓冲液来洗去多余的配体。
5.排除任何剩余的活性配体,将填料转移到0.1M Tris-HCl缓冲液pH 8.0或1M 乙醇胺,pH值8.0溶液中,保持2h。
亲和纯化糖蛋白常用填料

亲和纯化糖蛋白常用填料好啦,今天咱们聊一聊“亲和纯化糖蛋白常用填料”这个话题,虽然听起来像是科研实验室里的难题,但其实它并不难懂,咱们一起轻松愉快地聊聊。
你看,糖蛋白这种东西,其实是咱们人体里非常重要的一类分子,它们参与了很多生物过程,比如免疫反应、信号传递这些,跟咱们的健康息息相关。
所以,如何提纯这些糖蛋白,变得超级重要了。
而要从复杂的生物混合物中把糖蛋白分离出来,亲和纯化就成了一个好帮手。
你可以想象,亲和纯化就像是一个超级有眼力的“猎人”,它能一眼就识别出糖蛋白,然后把它们从“人海”中抓出来。
它有一个特别厉害的地方,就是“专一性”极强,能“锁定”目标蛋白,而不管其他杂七杂八的东西。
好了,说回填料。
其实这就是亲和纯化过程中的“筛子”或“钓饵”。
填料在亲和纯化中起着决定性作用,它们能帮助你将糖蛋白准确地“捕获”住。
你想啊,如果“钓饵”不对,怎么能钓到想要的“大鱼”呢?所以,亲和纯化糖蛋白常用的填料就是一系列能和糖蛋白亲和结合的物质。
常见的填料有很多种,不同的填料有不同的“特点”,就像不同的工具,各有所长。
亲和纯化的填料中最常见的一种,就是用“凝血酶”这种物质做填料了。
你想象一下,凝血酶就像是糖蛋白的好朋友,能够准确地和目标蛋白结合,把它“锁”住不放。
而这个过程中,其他杂质什么的就像是被“甩掉”的,剩下的就是纯净的糖蛋白。
这种填料的好处就是,它能够在非常温和的条件下进行分离和纯化,不会损伤糖蛋白本身。
你想,谁不想在不伤害“珍宝”的情况下,得到它呢?这种方法既能保护糖蛋白,又能提高产率,简直是科研人员的心头好。
有些填料是专门针对糖分子设计的。
这种就像是糖蛋白的“糖尿病治疗师”,专门去吸附那些含糖的蛋白。
通过设计一些糖基配体,它们能够巧妙地捕捉到糖蛋白,进行分离。
你能想象吗,就像是找到了糖蛋白的“专属收信人”,给它送去了“情书”。
这种填料不仅效率高,而且相对简单,不需要过多复杂的操作,适合大规模的应用。
CNBr活化琼脂糖填料明书(GE)

GE HealthcareInstructions 71-7086-00 AF Affinity mediaCNBr-activated Sepharose™ 4B CNBr-activated Sepharose 4B is a pre-activated medium for immobilization of ligands containing primary amines. It provides a very convenient way to immobilize ligands by the cyanogen bromide method. The coupling reaction is spontaneous, rapid and easy to carry out.The application area covers immobilization of proteins, peptides and nucleic acids.Table 1. Medium characteristics.Bead structure: 4% agaroseSpacer: NoneCoupling capacity: 25–60 mg α-chymotrypsinogen/ml drainedmediumBead size range: 45–165 μmAverage bead size: 90 μmMax linear flow rate*: 75 cm/h at 25°C, HR 16/10 column, 5 cm bed height pH stability**:Long term: 3–11Short term: 2–11Chemical stability***: Stable to all commonly used aqueous solutions.Can be used with non-ionic detergents, denaturingsolvents, e.g. 8 M urea and 6 M guanidinehydrochloride. Stable in organic solvents, such as50% dimethylformamide and 50% dioxane. Autoclavable: Not recommended* Linear flow rate = volumetric flow rate (cm3/h) column cross-sectional area (cm2)** The ranges given are estimates based on our knowledge and experience. Please note the following:pH stability, long term refers to the pH interval where the gel is stable over a longperiod of time without adverse effects on its subsequent chromatographic perfor-mance.pH stability, short term refers to the pH interval for regeneration, cleaning-in-place and sanitization procedures.*** Data refer to the coupled product, provided that the ligand can withstand the pH or chemical environment.p.Contents1. Preparing the medium 42. Coupling the ligand 43. Factors effecting the coupling efficiency 54. Packing Sepharose 4B 75. Binding 96. Elution 97. Regeneration 108. Storage 109. Further information 1010. Ordering information 11p.1. Preparing the mediumCNBr-activated Sepharose 4B is supplied lyophilized in the presence ofadditives. These additives must be washed away at low pH (pH 3) beforecoupling the desired ligand. The use of low pH (pH 3) preserves the activity ofthe reactive groups, which otherwise hydrolyze at high pH.Weigh out the required amount of powder (1 g lyophilized powder givesabout 3.5 ml final volume of medium) and suspend it in 1 mM HCl. Themedium swells immediately and should now be washed for 15 minutes with1 mM HCl on a sintered glass filter (porosity G3). Use approximately200 ml 1 mM HCl per gram freeze-dried powder, added in several aliquots. 2. Coupling the ligandGeneral ligand coupling procedure1. Dissolve the ligand to be coupled in coupling buffer, 0.1 M NaHCO3 pH 8.3 containing 0.5 M NaCl. Use about 5 ml coupling solution/glyophilized powder.About 5–10 mg protein per ml medium is recommended. For smallerligands add 1–10 μmoles per ml medium.2. Add the coupling solution containing the ligand with the preparedmedium suspension in a stoppered vessel.3. Rotate the mixture end-overend for 1 h at room temperature orovernight at 4 °C. Other gentle stirring methods may be employed.Do not use a magnetic stirrers as these can disrupt the Sepharosebeads.4. Wash away excess ligand with at least 5 medium (gel) volumes ofcoupling buffer.5. Block any remaining active groups. Transfer the medium to 0.1 M Tris-HCl buffer, pH 8.0 or 1 M ethanolamine, pH 8.0. Let it stand for 2 hours.p.6. Wash the medium with at least three cycles of alternating pH. Wash withat least 5 medium volumes of each buffer.Each cycle should consist of a wash with 0.1 M acetic acid/sodiumacetate, pH 4.0 containing 0.5 M NaCl followed by a wash with0.1 M Tris-HCl, pH 8 containing 0.5 M NaCl.3. Factors effecting the coupling efficiencypHThe coupling reaction proceeds most efficiently in the pH range 8–10 where the amino groups on the ligand are predominantly in the unprotonated form. A buffer at pH 8.3 is most frequently used for coupling proteins. Coupling at low pH is less efficient but may be advantegeous if the ligand looses biological activity when it is fixed firmly by multi-point attachmentor if steric hindrance between binding sites occurs when a large amount of high molecular weight ligand is immobilized. A buffer of approximately pH 6is used for coupling at low pH.Coupling solutionCoupling should be performed in bicarbonate or borate buffers. Tris andother buffer salts containing amino groups should not be used since thesewill couple to the medium.Organic solvents may be needed to dissolve the ligand. Dimethylformamide and dioxane may be used up to 50% of the final mixture. The same concentration of organic solvents should be included in the coupling buffer. Always adjust the pH after dissolving the ligand, since organic solvent usually lowers pH.p.SaltTo minimize protein-protein adsorption and the formation of proteinaggregates, it is recommended to have a high salt content, 0.5 M NaCl, in the coupling buffer.TemperatureCoupling is completed within 2 hours at room temperature, 20–25 °C. If cold room temperatures are necessary, coupling can be carried out overnight. Ligand concentrationA very high ligand concentration can have adverse effects on affinitychromatography. Firstly, the binding efficiency of the adsorbent maybe reduced due to steric hindrance between the active sites. Secondly,substances are more strongly bound to the immobilized ligand and this may result in difficult elution. Thirdly, the extent of non-specific binding increases at high ligand concentrations.For an efficient adsorbent, 1–10 μmoles ligand per ml medium isrecommended. For protein ligands, 5–10 mg protein per ml medium isrecommended.Controlling the coupling efficiencySometimes it may be necessary to reduce the number of coupling groups on the matrix to preserve the structure of the binding site in a labile molecule, or to facilitate elution when high binding constants make elution difficult or when steric effects reduce the binding efficiency of a large ligand.Reduced coupling activity may be achieved by controlled hydrolysis ofthe activated medium prior to coupling, or by coupling at a lower pH. Pre-hydrolysis reduces the number of active groups available for couplingand reduces the number of points of attachment between the protein and matrix as well as the amount of protein coupled. In this way a higher binding activity of the product is obtained. At pH 3, coupling activity is lost onlyslowly, whereas at pH 8.3 activity is lost fairly rapidly. A large molecule is coupled at only about half as many points after 4 h pre-hydrolysis at pH 8.3 as it is before the number of active groups was reduced.p. 6Blocking excess remaining groupsRemaining active groups on the medium should be deactivated or blocked after the coupling. These can be hydrolyzed in a mildly alkaline pH (2 hoursat room temperature or 16 h at 4 °C).Alternatively, these can also be blocked by adding an excess of a small primary amine (e.g. Tris-HCl, ethanolamine, glycine) at approximatelypH 8 (2 hours at room temperature or 16 h at 4 °C).These blocking agents introduce a small number of charged groups intothe medium. The effect of these charged groups is overcome by the useof a relatively high salt concentration (0.5 M NaCl) in the buffer for affinity chromatography.Washing the adsorbentTo remove excess of uncoupled ligand after coupling, the adsorbentis washed alternatively with high and low pH buffer solutions at leastthree times. Acetate buffer (0.1 M, pH 4) and coupling buffer (pH 8.3) each containing 0.5 M NaCl are suitable. This procedure ensures that no freeligand remains ionically bound to the immobilized ligand.4. Packing Sepharose 4BPrepare a slurry with binding buffer, see below, in a ratio of 75% settled medium to 25% buffer. The binding buffer should not contain agents which significantly increase the viscosity. The column may be equilibrated with viscous buffers at reduced flow rates after packing is completed.1. Equilibrate all material to the temperature at which the chromatographywill be performed.2. De-gas the medium slurry.3. Eliminate air from the column dead spaces by flushing the end pieceswith buffer. Make sure no air has been trapped under the column net.Close the column outlet with a few centimeters of buffer remaining in the column.p. 74. Pour the slurry into the column in one continuous motion. Pouring theslurry down a glass rod held against the wall of the column will minimize the introduction of air bubbles.5. Immediately fill the remainder of the column with buffer, mount thecolumn top piece onto the column and connect the column to a pump.6. Open the bottom outlet of the column and set the pump to run at thedesired flow rate. This should be at least 133% of the flow rate to beused during subsequent chromatographic procedures. However, themaximum flow rate, see Table 1, is typically employed during packing. Note: If you have packed at the maximum linear flow rate, do not exceed 75% of this in subsequent chromatographic procedures.7. Maintain the packing flow rate for 3 bed volumes after a constant bedheight is reached.Using an adapterAdapters should be fitted as follows:1. After the medium has been packed as described above, close thecolumn outlet and remove the top piece from the column. Carefully fillthe rest of the column with buffer to form an upward meniscus at thetop.2. Insert the adapter at an angle into the column, ensuring that no air istrapped under the net.3. Make all tubing connections at this stage. There must be a bubble-freeliquid connection between the column and the pump.4. Slide the plunger slowly down the column so that the air above thenet and in the capillary tubings is displaced by eluent. Valves on theinlet side of the column should be turned in all directions during thisprocedure to ensure that air is removed.5. Lock the adapter in position on the medium surface, open the columnoutlet and start the eluent flow. Pass eluent through the column at thepacking flow rate until the medium bed is stable. Re-position the adapter on the medium surface as necessary.The column is now packed and equilibrated and ready for use.p. 85. BindingConditions for binding depend on which ligand is used. Literature references and textbooks may give good guidelines.The adsorption will depend upon parameters such as sample concentration, flow rate, pH, buffer composition and temperature. General guidelines for adsorption are:• Sample pH should be the same as that of the binding buffer. Filter the sample through a 0.22 μm or 0.45 μm filter to prolong the working life of the medium.• After the sample has been loaded, wash the medium with binding buffer until the base line is stable.6. ElutionConditions for elution of bound substances depend on which ligand is used. Literature references and textbooks may give good guidelines.General guidelines are described below.• pH change: A change in pH alters the degree of ionization of charged groups at the binding sites. Elution is generally affected by a decrease in pH. The chemical stability of the matrix, ligand and adsorbed substances determines the limits of pH which may be used.•Ionic strength: A buffer with increased ionic strength is used. Elution with a continuous or step-wise gradient may be used. A gradient ofincreasing salt concentration can be used to separate substances bound to the adsorbent. NaCl is most frequently used and enzymes usuallyelute at a concentration of 1 M NaCl or less. If the interaction has a very high affinity, a chaotropic salt may be required.• Competitive elution: Competitive eluents are often used to selectively elute substances from a group specific adsorbent and also when theaffinities are relatively low. Selectively retained substances are usuallydisplaced at low concentrations of eluting agents, often less then 10 mM.Either continuous or step-wise gradients may be used.p.• Reduced polarity: Conditions which lower the polarity of the eluent to promote elution may be used if they do not inactivate elutedsubstances. Dioxane (up to 10%) or ethylene glycol (up to 50%) may beused.• Deforming eluents: If the elution methods described above fail to affect elution, deforming agents, such as chaotropic salts, guanidine-HCl orurea, which alter the structure of the proteins can be used.7. RegenerationConditions for regeneration depend on which ligand has been coupled.Literature references and textbooks may give good guideines.A general regeneration method is described below:An affinity medium may be regenerated for re-use by washing the medium with 2–3 column volumes of alternating high pH (0.1 M Tris-HCl, 0.5 M NaCl, pH 8.5) and low pH (0.1 M sodium acetate, 0.5 M NaCl, pH 4.5) buffers. This cycle should be repeated 3 times followed by re-equilibration in binding buffer.8. StorageLyophilized CNBr-activated Sepharose 4B should be stored below 8 °C.Swollen coupled medium should be stored at 4–8 °C in presence of abacteriostatic agent, e.g. 20% ethanol.9. Further informationCheck /protein-purification for more information.Useful information is also available in the Affinity ChromatographyHandbook, se ordering information.p. 1010. Ordering informationProduct Pack size Code No. CBNr-activated Sepharose 4B 15 g 17-0430-01250 g 17-0430-02 LiteratureAffinity Chromatography Handbook, 1 18-1022-29 Principles and MethodsAffinity Columns and Media, 1 18-1121-86 Product Profilep. 11GE, imagination at work and GE monogram are trademarks of General Electric Company.Drop Design and Sepharose are trademarks of GE Healthcare companies. © 1996-2009 General Electric Company – All rights reserved. Previously published Aug. 1996.All goods and services are sold subject to the terms and conditions of sale of the company within GE Healthcare which supplies them. A copy of these terms and conditions is available on request. Contact your local GE Healthcare representative for the most current information.GE Healthcare Europe GmbH Munzinger Strasse 5D-79111 Freiburg GermanyGE Healthcare UK Limited Amersham Place Little ChalfontBuckinghamshire, HP7 9NA UKGE Healthcare Bio-Sciences Corp.800 Centennial Avenue P.O. Box 1327Piscataway, NJ 08855-1327USAGE HealthcareJapan Corporation Sanken Bldg.3-25-1, HyakuninchoShinjuku-ku, Tokyo 169-0073JapanFor local offoce contact information, visit /contact GE Healthcare Bio-Sciences AB Björkgatan 30751 84 Uppsala Sweden/protein-purification71-7086-00 AF 12/2009imagination at work。
琼脂糖凝胶纯化回收试剂盒操作方法及步骤说明书

5. 快速、方便,不需要使用有毒的苯酚、氯仿等试剂,也不需要 乙醇沉淀。
注意事项
1. 所有的离心步骤均在室温完成,使用转速可以达到13,000rpm的 传统台式离心机,如Eppendorf 5415C 或者类似离心机。
2. 溶胶液中含有刺激性化合物,操作时要戴乳胶手套,避免沾染 皮肤,眼睛和衣服。若沾染皮肤、眼睛时,要立即用大量清水 或者生理盐水冲洗。
1. 在长波紫外灯下,用干净刀片将所需回收的DNA条带切下,尽 量切除不含DNA的凝胶,得到凝胶体积越小越好。
2. 将切下的含有DNA条带凝胶放入1.5ml离心管,称重。 先称一个空1.5ml离心管重量,然后放入凝胶块后再称一次,两次 重量相减,得到凝胶的重量。
3. 加3倍体积溶胶液DD。 如果凝胶重为100mg,其体积可视为100μl,则加入300μl溶胶液。 如果凝胶浓度大于2%,应加入6倍体积溶胶液。 4. 56℃水浴放置10分钟(或直至胶完全溶解)。每2-3分钟涡旋震
3. 回收纯化的DNA片段一般在100bp到40kb之间,过长、过短片段 的回收效率迅速降低。
4. 回收DNA的量和起始DNA的量、洗脱体积、DNA片断大小有 关。一般1-15μg, 100bp-5kb的DNA片段,回收率可高达85%。
5. 切胶回收时,紫外灯观察对DNA片段有损坏作用,应该尽可能 使用能量低的长波紫外线,并且尽可能的缩短紫外线下处理的 时间。
试剂盒组成保存50次dr0101100次dr0102200次dr0103平衡液室温5ml10ml20ml溶胶液dd室温50ml50ml2200ml漂洗液wb室温15ml25ml50ml第一次使用前按说明加指定量乙醇洗脱缓冲液eb室温10ml15ml15ml吸附柱ec室温50个100个200个收集管2ml室温50个100个200个本试剂盒在室温储存12个月不影响使用效果
填料的选择

最适用层析技术: 凝胶过滤/离子交换 / 疏水 层析/反相层析
分辩率
速度 回收率
载量
14 / GE /
精细纯化: 凝胶过滤填料
Superdex™ Peptide
Mr 100 - 7 000
Superdex 75
Mr 3 000 - 70 000
Superdex 200
Mr 10 000 - 600 000
Gel type Bead size Fractionation range Globular proteins Fractionation range Dextrans Exclusion limit DNA 1000-100000 5000-250000 10000-1500000 20000-8000000 ND ND 1000-80000 2000-400000 10000-2000000 40000-20000000 ND 118 118 271 1078 Sephacryl S-100 HR 25-75um Sephacryl S-200 HR 25-75um Sephacryl S-300 HR 25-75um Sephacryl S-400 HR 25-75um Sephacryl S-500 HR 25-75um
精细纯化:反相层析填料
Sephasil™ µ RPC
硅胶 C4, C8, C18
5 and 12 µ m 100 Å: 肽类 300 Å : 蛋白质
SOURCE™ RPC
聚苯乙烯/二乙烯基苯
硅胶 C2/C18 3µ m
120 Å: 肽类
pH 1 - 12 (14)
5, 15 and 30 µ m 肽类
19 / GE /
凝胶过滤介质
环氧活化琼脂糖说明书

环氧活化琼脂糖凝胶介质产品说明书1. 产品简介环氧活化琼脂糖凝胶介质可用于各种含有氨基、巯基或羟基的亲和配基(蛋白质、多肽、氨基酸或糖等)的偶联,应用十分广泛。
该活化偶联介质以琼脂糖凝胶为基质,采用环氧活化的方法激活琼脂糖上的羟基,活化后即可直接用于各种配基的偶联。
本中心开发的环氧活化偶联介质具有使用方便、偶联条件温和、偶联效率高和应用范围广(可偶联氨基、巯基和羟基)的特点。
2. 产品特性3. 应用环氧活化琼脂糖凝胶介质可用于各种含有氨基、巯基或羟基的亲和配基(蛋白质、多肽、糖等)的偶联。
具体的操作步骤如下(以10 ml活化胶偶联BSA为例):清洗:用10-20倍胶体积的去离子水冲洗保存于50%DMSO的环氧活化胶,除去DMSO,抽干;偶联:称取200mg BSA,溶解于10ml 0.1M碳酸钠缓冲液(pH 9.0),加入到抽干的活化胶中,在摇床上振荡,37℃偶联12-24h(反应完毕后将料液转移到砂芯漏斗中抽干,收集料液用于未偶联BSA的分析和偶联配基密度的计算,活化胶水洗后抽干);封闭:将10ml抽干的偶联BSA的胶加入到100 ml三角瓶中,加入30 ml 1 mol/L 乙醇胺溶液。
反应温度恒定在37 ℃,搅拌速度120 rpm,反应时间4h。
反应停止后将料液转移到砂芯漏斗中抽干,用去离子水清洗。
清洗:偶联BSA的介质依次用5倍的去离子水、0.1 M含0.5 MNaCl的乙酸-乙酸钠缓冲液(pH 4.0)、去离子水、0.1M含0.5 MNaCl的硼酸-四硼酸钠缓冲液(pH 8.0)和去离子水充分洗涤后,抽干。
保存:保存于4℃、20%乙醇溶液中。
备注:其他配基的偶联方法与BSA类似,但反应条件(pH、温度、时间、配基加入量)需要根据配基的特性和应用的需求进行适当调整。
注意事项:偶联缓冲液可用碳酸盐、硼酸盐和磷酸盐缓冲液,但绝对不能使用含有氨基的Tris、甘氨酸缓冲液。
4. 其他服务(1)提供介质筛选及工艺开发的咨询。
Heparin层析填料说明书

Heparin Sepharose 6 Fast Flow原理肝素是一种含硫酸酯的酸性多糖,将它偶联到交联及活化的琼脂糖凝胶上,该填料具有很高的物理化学稳定性。
肝素能和抗凝血因子Ⅲ、凝血因子、蛋白合成因子、脂蛋白、干扰素、核酸结合蛋白、限制内切酶、凝血酶及类凝血酶等生物大分子结合,所以肝素琼脂糖凝胶可以用于这类物质的纯化。
*图为含有交互转换的抗坏血酸的肝磷脂多糖的结构(A)和D-葡萄糖残基(B)分离操作结合缓冲液:20mM Tris-HCl, pH 8.0或者10mM 磷酸钠, pH7.0洗脱缓冲液:20mM Tris-HCl, 1~2M NaCl, pH 8.0或者10mM 磷酸钠,1~2M NaCl, pH7.01,用10倍柱体积的结合缓冲液平衡柱子。
2,上样。
3,用5-10倍柱体积的结合缓冲液平衡分离柱,直到基线,即所有未结合物质都被冲洗出柱子。
紫外吸光A280nm处监测。
4,用5-10倍柱体积的洗脱缓冲液进行洗脱。
使用连续的或者阶梯式的梯度洗脱,洗脱缓冲液的浓度从0%-100%。
使用注意1,通过改变缓冲液的pH值或者离子强度来修饰肝磷脂的选择性。
洗脱时使用连续的或者阶梯式的洗脱方式,用NaCl,KCl或者硫酸铵溶液,浓度可以高达1.5~2M。
2,对于凝血因子而言,肝磷脂作为亲和配基,在结合缓冲液中含有一个低浓度的0.1M的NaCl是合适的。
3,如果增加盐离子浓度的梯度产生一个令人不满意的结果,使用肝磷脂(1~5mg/ml)在洗脱缓冲液中作为一个竞争性试剂。
净化1,用0.5个柱体积的2M的NaCl冲洗10分钟去除离子键结合蛋白。
2,通过用4倍柱体积的0.1M NaOH溶液冲洗柱子1~2小时去除沉淀物或变性蛋白或用2倍的柱体积的6M的盐酸胍冲洗柱子30~60分钟,或者用2倍柱体积的6M的尿素冲洗30~60分钟。
3,用4倍柱体积的0.1%~0.5%的TritonX-100冲洗1~2小时,去除疏水键结合的蛋白质。
环氧氯丙烷法活化琼脂糖凝胶及其动力学分析_甄宇红

第27卷第4期2005年8月大连医科大学学报Journal of Dalian Medical UniversityVol.27No.4Aug.2005环氧氯丙烷法活化琼脂糖凝胶及其动力学分析甄宇红1,杨青2,张宝1(1.大连医科大学药学院,辽宁大连116027; 2.大连理工大学环境与生命学院生物科学与生物工程系,辽宁大连116023)摘要:[目的]探索环氧氯丙烷法活化琼脂糖凝胶的最佳反应条件,建立活化反应动力学模型。
[方法]研究了活化反应中氢氧化钠、环氧氯丙烷和硼氢化钠的浓度以及反应温度、时间和溶剂对活化反应的影响。
通过测定活化反应中环氧基的浓度,计算反应的活化度,以确定环氧氯丙烷活化琼脂糖凝胶的最佳反应条件。
对影响活化反应的主要因素进行分析,建立活化反应动力学模型。
[结果]环氧氯丙烷法活化琼脂糖凝胶的最佳反应条件为:5g琼脂糖、7.5mL0.8mol/L氢氧化钠、2 mL环氧氯丙烷以及10mg硼氢化钠于25e反应8h。
根据活化反应动力学模型推导出了反应过程的宏观动力学公式。
[结论]对影响活化反应的主要因素进行了分析,确定了环氧氯丙烷活化琼脂糖凝胶的反应条件。
将琼脂糖凝胶活化实验结果与动力学公式计算结果相比较,发现二者基本一致。
关键词:环氧氯丙烷;活化;琼脂糖凝胶;动力学模型中图分类号:Q503文献标识码:A文章编号:1671-7295(2005)04-0268-05载体的活化技术在色谱介质制备,融合蛋白质的固相酶切割[1]以及重组蛋白质的固相重折叠[2]等生物工程及相关领域中的应用越来越广泛。
目前,实验室内对载体进行活化,用得最多的是溴化氰(CNBr)法。
该法简便、快速、活化密度高。
但CNBr 是剧毒品,而且CNBr活化的介质在偶联配基后常有泄漏现象,因此,Clonis[3]提出了3种有可能取代溴化氰并适用于工业化生产的活化方法,即二溴丙醇法,双环氧化合物法和环氧氯丙烷法。
本文研究了环氧氯丙烷活化琼脂糖凝胶的反应条件及反应过程,对影响活化反应的主要因素进行了分析,确定了最佳反应条件并建立了活化反应动力学模型,推导出反应过程的宏观动力学公式。
蓝晓生物科技CNBr Activated Seplife FF介质说明书

CNBr Activated Seplife FF说明书1.产品介绍CNBr Activated Seplife FF琼脂糖层析介质是蓝晓科技自主研发的一种新型活化中间体填料,它是通过溴化氰和琼脂糖上面的羟基反应生成活化的氰酸酯基团。
蛋白质、多肽、氨基酸等可以偶联到溴化氰活化琼脂糖凝胶上。
具有以下优点:偶联条件温和,效率高;可偶联含有氨基的配体,应用范围广。
2.性能介绍产品牌号CNBr Activated Seplife FF外观白色球状冻干粉,无臭无味基质Seplife4FF配基溴化氰形状球形最大线性流量75cm/h(25℃),直径1.6cm色谱柱,5cm柱床高粒径(μm)45~165偶联能力20~60mg α-胰凝乳蛋白酶原/ml凝胶pH稳定性2~11在以下溶液中稳定:6M盐酸胍;8M尿素;非离子型表面活性剂;化学稳定性一般有机溶剂:50%二甲基甲酰胺、50%二氧六环应用活化中间体,用于偶联配基3.使用方法下面简要介绍介质偶联配体的使用过程。
具体实验条件取决于配基,应根据具体配基选择偶联、洗脱和再生条件。
3.1干粉的溶胀称取一定量的CNBr Activated Seplife FF冻干粉放置在1mmol/L HCl中悬浮溶胀,放置大约30分钟后抽滤,用约15倍胶体积的 1mmol/L HCl溶液冲洗干净,用偶联缓冲液洗涤,抽干。
3.2 配基的偶联一般配体偶联的过程如下:(1)将所需要偶联的配基溶解到含有0.5mol/L NaCl 的0.1mol/LNaHCO3 (pH8.3)缓冲溶液中,加入溶胀好的CNBr activated seplife®4B。
(2)在室温中搅拌反应2~4小时或者在4℃下反应过夜。
(3)反应完全后,先用5倍反应体积的偶联缓冲液洗涤掉其中多余的配体,然后用10倍体积的水洗涤。
(4)将反应完洗涤过的凝胶悬浮到水中,加入乙醇胺(1mol/L)或者pH8.0 T ris-HCl缓冲液,在常温最少搅拌反应2小时,以消除残余活性基团。
琼脂糖微球的制备及活化条件研究

摘要一般说来,琼脂糖磁性微球活化度越高,能连接的亲和配位体就越多,用其吸附纯化蛋白质及固定化酶效率就越高。
本实验研究了用环氧氯丙烷活化琼脂糖磁性微球的最优条件。
实验先用反相悬浮包埋法制备出琼脂糖磁性微球,微球因内部含有磁性四氧化三铁的超细粉末而具有磁响应性。
以微球表面的环氧基密度作为活化度的度量,考察了NaOH浓度、环氧氯丙烷体积分数、NaBH4的浓度、活化温度和活化时间等因素对琼脂糖磁性微球活化反应活化度的影响,得到了最佳活化条件为:NaOH溶液浓度为0.9mol/L,环氧氯丙烷体积分数为40%,NaBH4的浓度为0.5g/L,活化温度为40℃,活化时间为4h。
用环氧氯丙烷活化的载体稳定性好,且可提供相当于3个碳原子间距的空间臂。
本文还研究过相似活化步骤和条件下,改用1,4-丁二醇二缩水甘油醚做活化剂,但未能成功活化琼脂糖磁性微球,该研究尚待进一步开展。
关键词:琼脂糖磁性微球活化AbstractGenerally speaking, the higher the activation degree of agarose magnetic microspheres is, the more affinity ligands can be grafted. So that the adsorptive capacity of microspheres for protein purification and the efficiency of immobilized enzyme can be higher. This paper investigated optimal conditions for the activation of agarose magnetic microspheres, using epichlorohydrin as activating agent. Firstly, agarose magnetic microspheres were prepared by reverse suspended embedding method. The microspheres possessed magnetic responsibility because of the interior Fe3O4ultrafine powder. The effects of sodium hydroxide concentration,epichlorohydrin volume fraction,NaBH4 concentration,activation temperature and activation time on activation degree were investigated,according to the density of surface epoxy group .The optimal conditions are obtained as followed:sodium hydroxide concentration,epichlorohydrin volume fraction,NaBH4 concentration ,activation temperature and activation time is 0.9 mol/L, 40%, 0.5 g/L, 40 ℃, 4 h, respectively. The carrier activated by epichlorohydrin can be of good stability and has 3 carbon atom space arm. This paper also studied the activation of agarose magnetic microspheres using 1, 4-butyl glycol two glycidyl ether as activating agent. The experiments in which agarose magnetic microspheres were failed to be activated were operated under similar methods and conditions. The relevant researches are yet to be further developed.Keywords:Agarose; magnetic microspheres; activation目录摘要 (I)ABSTRACT ........................................................................................................... I I 目录. (IV)第一章文献综述 (1)1.1固定化酶技术的研究意义 (1)1.2固定化酶载体基质材料 (2)1.2.1固定化酶载体基质材料性质要求 (2)1.2.2固定化酶载体材料研究进展 (2)1.3固定化酶载体的表面修饰 (3)1.3.1固定化酶载体表面修饰原则 (3)1.3.2固定化酶载体表面修饰环氧基 (4)1.4固定化酶载体的间隔臂 (6)1.5固定化酶载体的配位体 (6)1.6展望 (7)1.7本论文研究内容及意义 (7)第二章琼脂糖微球的制备 (9)2.1实验试剂及仪器 (9)2.1.1实验试剂 (9)2.1.2实验仪器 (9)2.2实验方法及步骤 (10)2.3实验结果及讨论 (10)第三章琼脂糖磁性微球活化条件研究 (12)3.1微球活化方法论证 (12)3.2环氧基分析方法论证 (12)3.3实验试剂及仪器 (13)3.3.1实验试剂 (13)3.3.2实验仪器 (14)3.4实验方法及步骤 (14)3.4.1琼脂糖磁性微球的活化 (14)3.4.2琼脂糖磁性微球环氧基修饰密度的分析 (15)3.5实验结果及分析 (15)3.5.1活化后琼脂糖磁性微球的性质 (15)3.5.2琼脂糖磁性微球活化条件探究结果分析 (16)3.5.2.1氢氧化钠溶液浓度的影响 (16)3.5.2.2环氧氯丙烷体积分数的影响 (17)3.5.2.3硼氢化钠的浓度的影响 (18)3.5.2.4活化温度的影响 (18)3.5.2.5活化时间的影响 (19)第五章全文总结及展望 (21)5.1全文总结 (21)5.2展望 (21)参考文献 (23)致谢...................................................................................... 错误!未定义书签。
第7章 亲和层析

通用性配体一般是指特异性不是很强,能和某一类的蛋 白质等生物大分子结合的配体,如各种凝集素(lectine) 可以结合各种糖蛋白,核酸可以结合RNA、结合RNA的蛋白质 等。通用性配体对生物大分子的专一性虽然不如特异性配体, 但通过选择合适的洗脱条件也可以得到很高的分辨率。而且 这些配体还具有结构稳定、偶联率高、吸附容量高、易于洗 脱、价格便宜等优点,所以在实验中得到了广泛的应用。
4) 配体自身应具有较好的稳定性,在实验中能够 耐受偶联以及洗脱时可能的的较剧烈的条件,可以 多次重复使用。
根据配体对待分离物质的亲和性的不同,可以将其分为 两类:特异性配体(specific ligand)和通用性配体 (general ligand)。 特异性配体一般是指只与单一或很少种类的蛋白质等生 物大分子结合的配体。如生物素和亲和素、抗原和抗体、酶 和它的抑制剂、激素-受体等,它们结合都具有很高的特异 性,用这些物质作为配体都属于特异性配体。配体的特异性 是保证亲和层析高分辨率的重要因素,但寻找特异性配体一 般是比较困难的,尤其对于一些性质不很了解的生物大分子, 要找到合适的特异性配体通常需要大量的实验。
(4)凝集素 凝集素(lectin)是与糖特异性结合的蛋白质(酶和抗体除外)的 总称,大部分凝集素为多聚体,含有两个以上的糖结合部位,不同的 凝集素与糖结合的特异性不同。例如,常用做亲和配基的伴刀豆球蛋 白A(concanavalin A,con A)与葡聚糖和甘露糖的亲和结合作用较强, 而麦芽糖凝集素(wheat germ agglutinin,WGA)与N-乙酰葡糖胺 (N-acetyl-glucosamine)的亲和结合作用较强。 Con A可用做糖蛋白、多糖、糖脂等含糖生物大分子以及全细胞、 细胞膜片断和细胞表面受体蛋白的亲和配基。pH<5.6时con A为二聚 体,分子量为52KD;pH>5.6时为四聚体,分子量为102KD,每个亚基 (subunit)之间通过二硫键结合。因此,在利用con A为配基的亲和 层析操作中,操作条件(pH,溶液组成)应当适宜,不能使con A的亚 基发生解离。
多克隆抗体纯化
多克隆抗体纯化一、简介抗原分子通常具有多个抗原决定簇,免疫动物后可刺激具有相应抗原受体的B细胞发生免疫应答,产生多种抗体。
由不同B细胞克隆产生的抗体称多克隆抗体;免疫血清实际上是含有多种抗体的混合物;具有不均一性。
特异性差,易导致超敏反应。
抗原亲和纯化一般用在多抗的纯化上,这种纯化方式去掉了血清中那些非特异性结合的抗体分子,得到的抗体分子基本上都是能特异性与抗原结合的。
抗原亲和纯化需要先将抗原偶联到柱料上,然后通过亲和层析的方式去除非特异性抗体及杂蛋白,得到特异性抗体。
通常采用的柱料为溴化氢预处理和N-羟基琥珀酰亚胺预处理琼脂糖凝胶柱料,前者适合偶联大分子,后者适合偶联小分子物质,在实际操作中还是需要根据情况进行选择。
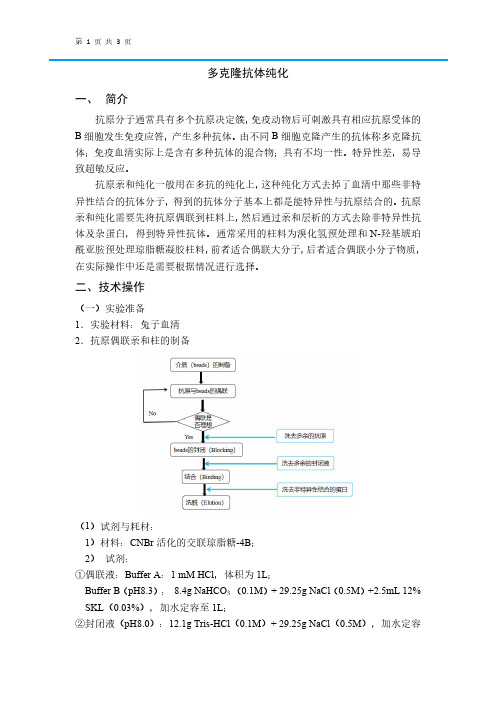
二、技术操作(一)实验准备1.实验材料:兔子血清2.抗原偶联亲和柱的制备(1)试剂与耗材:1)材料:CNBr活化的交联琼脂糖-4B;2)试剂:①偶联液:Buffer A:1mM HCl,体积为1L;Buffer B(pH8.3):8.4g NaHCO3(0.1M)+29.25g NaCl(0.5M)+2.5mL12% SKL(0.03%),加水定容至1L;②封闭液(pH8.0):12.1g Tris-HCl(0.1M)+29.25g NaCl(0.5M),加水定容至1L;③清洗液(pH4.0):29.25g NaCl(0.5M)+11.5mL冰醋酸,加水定容至1L;④亲和纯化试剂:1xPBS缓冲溶液(pH7.4),0.1M Gly-HCl(pH2.7);⑤其他试剂:硫酸铵,考马斯亮蓝G-2503)仪器与设备:Thermo台式离心机miro17R酶标仪(2)实验步骤①清洗:称取0.2g的CNBr活化交联琼脂糖-4B到柱子中,用偶联液A(1mM HCl)清洗,再用偶联液B清洗3-4遍。
②偶联:用偶联液B溶解的蛋白与填料介质混合,室温旋转孵育1h或4℃过夜;再用偶联液B洗去多余的蛋白;③封闭:将上述填料转移到封闭液(0.1M Tris-HCl,pH8.0)中封闭,室温封闭2h或4℃过夜;④循环清洗:封闭液(pH8.0)和清洗液(pH4.0)交替清洗填料至少3个循环;⑤平衡:用1xPBS缓冲溶液(pH7.4)平衡填料⑥保存:4℃,20%乙醇;3.多抗亲和纯化(1)硫酸铵沉淀①样品预处理:取过滤后的血清,用1xPBS稀释4-5倍;②硫酸铵沉淀:称取终浓度为0.277mg/mL的硫酸铵(45%)。
层析填料的选择及其装柱技术介绍幻灯片

理想的基质应符合下面的要求: 1.极低的非特异性吸附。 2.高度的亲水性。 3.较好的理化稳定性。 4.大量的化学基团能被有效地活化,而且容易和配体结合。 5.适当的多孔性。
一般亲和吸附剂采用的基质有纤维素、聚丙烯酰胺凝胶、交联 葡聚糖、琼脂糖、交联琼脂糖、多孔玻璃珠等。
常用亲和吸附剂采用的基质
反相层析填料的选择原则
考虑样品组分的种类和性质、分离的规模及对分辨率 的要求、流动相条件:
待分离物分子量,对介质孔径的选择提供指导; 样品组分的疏水性质决定采用何种配基; 分离的规模及对分辨率的要求也是须考虑的因素,通
常分离规模和分辨率的关系是负相关的 ; 反相层析时的流动相条件很大程度上影响反相介质基
层析填料的选择 及装柱技术介绍
佳辰公司生物中心 陈阶
2012-04-18
如何选择填料?
一、依据所纯化的对象的各物理和化学特性。
(一般包括蛋白、酶、重组蛋白、单抗、抗体及抗原、肽类、病毒、 核酸等)
二、纯化所要达到的目的(粗提?中度纯化? 精细纯化?)。
三、所要选择的层析方法
1. 凝胶过滤 ( Gf)
易脱落。 4) 配体自身应具有较好的稳定性。
根据配体对待分离物质的亲和性的不同,可以将其分为 两类:特异性配体(specific ligand)和通用性配体 (general ligand)
亲和吸附介质的配基
(1)酶的抑制剂 (2)抗体 (3)A蛋白 A蛋白(protein A)为分子量约42KD的蛋白质,
Sephadex LH-20 同时具备亲水和亲脂双重性质, 且被分离物质的极性在分离过程中起着重要作 用。
Pharmadex LH-20同时适用于分子类别非常相似 的物质的分离和工业规模的制备,既可用于初 步纯化步骤,也可用于最终精制步骤,如非对 映同分异构体的分离。
蛋白纯化实验常见问题精华解析

蛋白纯化实验常见问题精华解析本文主要介绍了蛋白纯化过程中常见的问题.内容选择方面恐存众不足,望指正。
1)我现在手头有个融合蛋白,纯化的时候用助溶剂溶解后做了GST 亲和层析,之后用蛋白酶切割后却发现目的蛋白没有活性。
请问有哪些可能会出现这种原因?怎么解决?测过基因序列,没有问题。
细胞破碎后,融合蛋白在沉淀里,我用助溶剂提取后,可以用GST做亲和层析,但效率只有50~60%左右。
洗脱完融合蛋白不需要助溶剂,但这样的条件下切割完后目的蛋白会沉淀,我用0.5%CHAPS 助溶可勉强溶解部分,目的蛋白疏水性比较强。
既然是疏水性很强你可以加点甘油或者乙二醇降低极性,这样溶解就会好得多,此外你也直接加2%的PEG这样也降低极性,同时保护你的蛋白,你再做纯化就可以了,我以前遇到这样的蛋白是用10%的乙醇加3-5%的PEG5000,这样的情况下蛋白还是活性回收率很高,我觉得那些表面活性剂能不用还是不用的好,你失去活性你可以监测一下提取,纯化,酶切到底哪里失去了活性,这样更容易解决你的问题。
还有注意缓冲液PH值,看看改变它对溶解度有没有影响。
蛋白有沉淀那你可以稍微把蛋白的浓度降低点,这也是个办法。
2)请问楼主有没有合成过配体为FMN的亲和胶?我已经给你发了相关一些偶联的材料,请查收,现在一般要自己合成亲和填料,有很多公司都提供活化好的介质,自己偶联就可以,其中比较常用的有溴化氰琼脂糖凝胶,环氧琼脂糖凝胶,氨基琼脂糖凝胶,羧基琼脂糖凝胶,活化酯琼脂糖凝胶,以及巯基琼脂糖凝胶等,但是如果是小分子的配基最好选择有3-10碳作为手臂的活化介质为好,此外也曾经有文献报导固定辅酶时,偶联位置不同,选择性有差别,因此你可以选择自己适合的活化介质。
不同的活化的介质对偶联的配基有不同的要求,所以要对自己的配基了解比较清楚就可以选择合适的活化介质。
我一般在合成的时候大多选择环氧琼脂糖凝胶,它能应用的范围广氨基巯基羟基都可以,而且合成的介质刚性好,非特异吸附少,所以不需要特殊处理未反应基团。
CNBr活化琼脂糖凝胶 4FF

CNBr-Activated Crystarose 4FF活性基团:C≡N一、简介CNBr-Activated Crystarose 4FF 是CNBr预活化填料,它采用平均粒径为45-165μm的交联琼脂糖凝胶。
该活化填料的活化基团可以结合蛋白质、多肽以及核酸等其他含有氨基(-HN2)基团的配基。
其优点是具有反应条件温和,反应速度快、活性高、偶联效率高,与市场上同类产品不同的地方时偶联后的配基脱落极低。
二、产品技术指标:三、适用范围自制亲和层析填料,提供对不同用户具体的偶联方案。
四、应用实例实验名称:偶联牛血清白蛋白(BSA)实验步骤:1、取BSA 100mg 溶解于10ml 的0.1M 碳酸钠缓冲液中,pH8.5。
2、称量活化填料1g,(1g 干的活化的填料经完全溶胀为4-5ml)。
3、在抽滤瓶中用3倍体积1mM HCL清洗填料,或用5倍体积浸泡填料后丢弃上清,尽量吸干液体(可选方法:也可不用清洗直接加入偶联蛋白液进行偶联)。
4、将清洗后的填料加入BSA 溶液中混合,在摇床上振荡,室温偶联3 小时以上或者4℃过夜。
5、然后加用Tris-HCL缓冲液或者加入50mg 甘氨酸,室温反应2小时以上以封闭剩余的基团。
6、将偶联后的BSA 的填料装柱子,用5—10倍体积的0.5M NaCl溶液洗柱,收集洗脱液。
7、偶联量测定:计算方法:BSA 的偶联量=(B0—B1)/V。
注:B0:偶联前BSA 总蛋白量,单位:mg。
B1:偶联后(洗柱液)未偶联的BSA 总蛋白量,单位:mg。
V:填料的体积,单位:ml。
备注:该偶联方法可用于偶联单抗及其它蛋白,均能获得很好的偶联效果。
琼脂糖亲和层析填料

琼脂糖亲和层析填料琼脂糖亲和层析填料的应用与发展作者:AI写手【导语】琼脂糖亲和层析填料是一种常见的生物分离技术,在生物医药和生物工程领域得到广泛应用。
本文将从历史发展、原理、应用领域和优缺点等方面,全面介绍琼脂糖亲和层析填料的特点和意义。
【1. 历史发展】(1)亲和层析技术自1968年被发明以来,经历了多年的发展和改进。
琼脂糖亲和层析填料是亲和层析技术的一种重要形式,可以追溯到20世纪70年代初。
(提及主题文字:"琼脂糖亲和层析填料")(2)早期的琼脂糖亲和层析填料主要由琼脂糖与靛蓝等显色剂耦联形成,用于蛋白质的分离纯化。
然而,由于显色剂可能对蛋白质产生不可逆的结构变化和影响活性,这种方法并不理想。
(3)随着科技的进步和对生物分离技术的需求不断增长,琼脂糖亲和层析填料的制备和应用也得到了改进。
现在,常见的琼脂糖亲和层析填料采用葡萄糖基团与琼脂糖结合,具有更好的特异性和选择性。
【2. 原理】琼脂糖亲和层析填料的原理是基于分子之间的亲和性相互作用。
琼脂糖是一种多糖,具有高度可溶且无毒的特点,与许多生物大分子如蛋白质、肽等具有显著的亲和性。
当生物样品通过琼脂糖亲和层析填料时,目标分子会与填料表面的琼脂糖结合并附着在填料上,从而实现目标分子的分离纯化。
填料表面的琼脂糖能够选择性地结合目标分子,其它非目标分子会被洗脱掉,从而达到分离的目的。
【3. 应用领域】琼脂糖亲和层析填料在生物医药和生物工程领域有着广泛的应用。
主要应用领域包括:(1)蛋白质纯化:琼脂糖亲和层析填料能够选择性地捕获目标蛋白质,可用于纯化和富集特定蛋白质。
(2)药物研发:通过琼脂糖亲和层析填料,可以从复杂的药物样品中分离出目标化合物,用于药物研发与分析。
(3)基因工程:琼脂糖亲和层析填料可用于分离和纯化重组蛋白质,如表达的重组蛋白质、酶等。
(4)疾病诊断:利用琼脂糖亲和层析填料,可以从体液中快速、高效地分离出特定生物标志物,用于疾病诊断和监测。
CNBr 4FF 使用说明书

网址:CNBr 4FF使用说明书为确保产品的性能和无忧的操作,使用前请仔细阅读本手册,有任何疑问请咨询本公司售后技术支持或当地的销售人员。
1. 产品介绍CNBr 4FF是经过溴化氰活化后含有氰酸酯基团的快流速纯化介质,适用于偶联蛋白质、多肽、核酸等含有氨基的生物分子。
在生物医药纯化工艺中经过反复的验证,得到了广泛的应用。
特点如下:a. 应用广泛,可适用于偶联含氨基的生物类大分子。
b. 多点偶联,简单、灵活、快速、有效,能高效的维持生物分子的生物学活性及稳定性。
c. 流速快、产率高、易于放大。
表1:介质性能参数基质高度交联4%的琼脂糖粒径范围45-165µm平均粒径90µm结合载量30mg(Trypsinogen)/ml(介质)pH稳定性3-11(长期)2-11(短期)最大流速700cm/h操作压力≤0.3MPa贮存溶液100%丙酮贮存温度4-8℃2. 偶联条件a.(A液)清洗取适量的沉降胶(0.83g清洗完成后约为1.0ml),用5倍体积的A液将介质重悬,5min后将溶液抽干。
重复此步骤5次。
备注:此步骤用于激活介质,要保证A液的清洗体积要充足、清洗时间固定在0.5h左右(时间太久介质上面的基团会水解)。
b. 配基溶液的制备将要偶联的生物分子用B液溶解或者将生物分子置换到B液中(生物分子浓度为1-10mg/ml,建议为5mg/ml)。
备注:确保偶联时的pH和盐浓度与B液一致。
c. 偶联将清洗完的介质和准备好的样品按等比例(体积比)混合,室温条件下温和混匀3-4h。
确网址:定偶联成功(通过测定偶联前后生物分子含量确定偶联效率)后,将溶液抽干。
备注:偶联建议在室温条件下偶联3-4h。
对于不稳定的配基,建议4-8℃偶联过夜。
d.(C液)清洗用5倍体积的C液将偶联后的介质重悬,再将溶液抽干。
再重复此步骤3次。
备注:此步骤用于清洗介质中残留的生物分子,清洗要彻底。
e. 封闭用5倍体积的C液进行重悬,室温条件下温和混匀3-4小时后,将溶液抽干。
蓝胶说明书翻译

二乙基氨基乙基-亲和层析蓝胶使用说明书目录号码153-7307介绍二乙基氨基乙基亲和层析蓝胶是一种双功能基团离子交换/亲和层析介质,该介质是由同时偶联有Cibacron Blue F3GA和二乙基氨基乙基BIO-GEL A-5m的基团的琼脂糖。
Cibacron Blue F3GA是一种离子型的、憎水的基团,其空间活性结合位点对具有双核苷酸折叠的蛋白质具有吸附作用,如白蛋白。
二乙基氨基乙基通过阴离子交换,其结合等电点比流动相低的蛋白质。
通过这两种双功能基团制备成二乙基氨基乙基亲和层析蓝胶成为一种非常强有力纯化工具。
通过改变盐离子强度和流洗缓冲液的PH,可以从不同种类的血清中纯化出高纯度的IgG。
二乙基氨基乙基亲和层析蓝胶层析过程提供一种纯化血清蛋白的最简便方法。
免疫球蛋白被洗脱后,其它杂蛋白组分可以通过通过离子强度进行洗脱下来。
二乙基氨基乙基亲和层析蓝胶对血清白蛋白亲和层析能力比二乙基氨基乙基离子交换层析的能力更强,可以从混有白蛋白的血清中分离出其它组分的有效的方法。
产品描述填料Bio Gel A-5m 琼脂胶颗粒半径150-300纳米(50-100网孔)洗脱方法0.01MTris,pH8,0.15MNaCl,0.04%NaN3官能团Cibacron blue and diethylaminoethy1(亲和分离介质三嗪染料和DEAE)推荐流速范围15-25cm/hr压力限定15pis容量血清0.2-1ml血清/蓝胶IgG的收率>55%白蛋白的收率>90%蛋白酶的去除率100%稳定性pH 2-11有机溶剂酒精温度不耐高温高压存储在0.02% NaN3或其他防腐剂中,4℃保存,一年*使用1.5*20cm的柱子和流动相1:1的压力下,确定的流速。
**兔血清和人血清能力的结合能力有很大的批间差异性,应按照不同的批号,进行不同的选择。
没有提供的必须材料预洗缓冲液0.1M醋酸,pH为3,1.4M的NaCl,40%异丙醇流洗缓冲液参照表2再生缓冲液2M盐酸胍于+流洗缓冲液中或1.5MNaSCN布氏漏斗层析柱概述1、利用表格1所给出的信息来准备适当的缓冲液,准备准确缓冲液对最适IgG的回收是必须的。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
GE HealthcareInstructions 71-7086-00 AF Affinity mediaCNBr-activated Sepharose™ 4B CNBr-activated Sepharose 4B is a pre-activated medium for immobilization of ligands containing primary amines. It provides a very convenient way to immobilize ligands by the cyanogen bromide method. The coupling reaction is spontaneous, rapid and easy to carry out.The application area covers immobilization of proteins, peptides and nucleic acids.Table 1. Medium characteristics.Bead structure: 4% agaroseSpacer: NoneCoupling capacity: 25–60 mg α-chymotrypsinogen/ml drainedmediumBead size range: 45–165 μmAverage bead size: 90 μmMax linear flow rate*: 75 cm/h at 25°C, HR 16/10 column, 5 cm bed height pH stability**:Long term: 3–11Short term: 2–11Chemical stability***: Stable to all commonly used aqueous solutions.Can be used with non-ionic detergents, denaturingsolvents, e.g. 8 M urea and 6 M guanidinehydrochloride. Stable in organic solvents, such as50% dimethylformamide and 50% dioxane. Autoclavable: Not recommended* Linear flow rate = volumetric flow rate (cm3/h) column cross-sectional area (cm2)** The ranges given are estimates based on our knowledge and experience. Please note the following:pH stability, long term refers to the pH interval where the gel is stable over a longperiod of time without adverse effects on its subsequent chromatographic perfor-mance.pH stability, short term refers to the pH interval for regeneration, cleaning-in-place and sanitization procedures.*** Data refer to the coupled product, provided that the ligand can withstand the pH or chemical environment.p.Contents1. Preparing the medium 42. Coupling the ligand 43. Factors effecting the coupling efficiency 54. Packing Sepharose 4B 75. Binding 96. Elution 97. Regeneration 108. Storage 109. Further information 1010. Ordering information 11p.1. Preparing the mediumCNBr-activated Sepharose 4B is supplied lyophilized in the presence ofadditives. These additives must be washed away at low pH (pH 3) beforecoupling the desired ligand. The use of low pH (pH 3) preserves the activity ofthe reactive groups, which otherwise hydrolyze at high pH.Weigh out the required amount of powder (1 g lyophilized powder givesabout 3.5 ml final volume of medium) and suspend it in 1 mM HCl. Themedium swells immediately and should now be washed for 15 minutes with1 mM HCl on a sintered glass filter (porosity G3). Use approximately200 ml 1 mM HCl per gram freeze-dried powder, added in several aliquots. 2. Coupling the ligandGeneral ligand coupling procedure1. Dissolve the ligand to be coupled in coupling buffer, 0.1 M NaHCO3 pH 8.3 containing 0.5 M NaCl. Use about 5 ml coupling solution/glyophilized powder.About 5–10 mg protein per ml medium is recommended. For smallerligands add 1–10 μmoles per ml medium.2. Add the coupling solution containing the ligand with the preparedmedium suspension in a stoppered vessel.3. Rotate the mixture end-overend for 1 h at room temperature orovernight at 4 °C. Other gentle stirring methods may be employed.Do not use a magnetic stirrers as these can disrupt the Sepharosebeads.4. Wash away excess ligand with at least 5 medium (gel) volumes ofcoupling buffer.5. Block any remaining active groups. Transfer the medium to 0.1 M Tris-HCl buffer, pH 8.0 or 1 M ethanolamine, pH 8.0. Let it stand for 2 hours.p.6. Wash the medium with at least three cycles of alternating pH. Wash withat least 5 medium volumes of each buffer.Each cycle should consist of a wash with 0.1 M acetic acid/sodiumacetate, pH 4.0 containing 0.5 M NaCl followed by a wash with0.1 M Tris-HCl, pH 8 containing 0.5 M NaCl.3. Factors effecting the coupling efficiencypHThe coupling reaction proceeds most efficiently in the pH range 8–10 where the amino groups on the ligand are predominantly in the unprotonated form. A buffer at pH 8.3 is most frequently used for coupling proteins. Coupling at low pH is less efficient but may be advantegeous if the ligand looses biological activity when it is fixed firmly by multi-point attachmentor if steric hindrance between binding sites occurs when a large amount of high molecular weight ligand is immobilized. A buffer of approximately pH 6is used for coupling at low pH.Coupling solutionCoupling should be performed in bicarbonate or borate buffers. Tris andother buffer salts containing amino groups should not be used since thesewill couple to the medium.Organic solvents may be needed to dissolve the ligand. Dimethylformamide and dioxane may be used up to 50% of the final mixture. The same concentration of organic solvents should be included in the coupling buffer. Always adjust the pH after dissolving the ligand, since organic solvent usually lowers pH.p.SaltTo minimize protein-protein adsorption and the formation of proteinaggregates, it is recommended to have a high salt content, 0.5 M NaCl, in the coupling buffer.TemperatureCoupling is completed within 2 hours at room temperature, 20–25 °C. If cold room temperatures are necessary, coupling can be carried out overnight. Ligand concentrationA very high ligand concentration can have adverse effects on affinitychromatography. Firstly, the binding efficiency of the adsorbent maybe reduced due to steric hindrance between the active sites. Secondly,substances are more strongly bound to the immobilized ligand and this may result in difficult elution. Thirdly, the extent of non-specific binding increases at high ligand concentrations.For an efficient adsorbent, 1–10 μmoles ligand per ml medium isrecommended. For protein ligands, 5–10 mg protein per ml medium isrecommended.Controlling the coupling efficiencySometimes it may be necessary to reduce the number of coupling groups on the matrix to preserve the structure of the binding site in a labile molecule, or to facilitate elution when high binding constants make elution difficult or when steric effects reduce the binding efficiency of a large ligand.Reduced coupling activity may be achieved by controlled hydrolysis ofthe activated medium prior to coupling, or by coupling at a lower pH. Pre-hydrolysis reduces the number of active groups available for couplingand reduces the number of points of attachment between the protein and matrix as well as the amount of protein coupled. In this way a higher binding activity of the product is obtained. At pH 3, coupling activity is lost onlyslowly, whereas at pH 8.3 activity is lost fairly rapidly. A large molecule is coupled at only about half as many points after 4 h pre-hydrolysis at pH 8.3 as it is before the number of active groups was reduced.p. 6Blocking excess remaining groupsRemaining active groups on the medium should be deactivated or blocked after the coupling. These can be hydrolyzed in a mildly alkaline pH (2 hoursat room temperature or 16 h at 4 °C).Alternatively, these can also be blocked by adding an excess of a small primary amine (e.g. Tris-HCl, ethanolamine, glycine) at approximatelypH 8 (2 hours at room temperature or 16 h at 4 °C).These blocking agents introduce a small number of charged groups intothe medium. The effect of these charged groups is overcome by the useof a relatively high salt concentration (0.5 M NaCl) in the buffer for affinity chromatography.Washing the adsorbentTo remove excess of uncoupled ligand after coupling, the adsorbentis washed alternatively with high and low pH buffer solutions at leastthree times. Acetate buffer (0.1 M, pH 4) and coupling buffer (pH 8.3) each containing 0.5 M NaCl are suitable. This procedure ensures that no freeligand remains ionically bound to the immobilized ligand.4. Packing Sepharose 4BPrepare a slurry with binding buffer, see below, in a ratio of 75% settled medium to 25% buffer. The binding buffer should not contain agents which significantly increase the viscosity. The column may be equilibrated with viscous buffers at reduced flow rates after packing is completed.1. Equilibrate all material to the temperature at which the chromatographywill be performed.2. De-gas the medium slurry.3. Eliminate air from the column dead spaces by flushing the end pieceswith buffer. Make sure no air has been trapped under the column net.Close the column outlet with a few centimeters of buffer remaining in the column.p. 74. Pour the slurry into the column in one continuous motion. Pouring theslurry down a glass rod held against the wall of the column will minimize the introduction of air bubbles.5. Immediately fill the remainder of the column with buffer, mount thecolumn top piece onto the column and connect the column to a pump.6. Open the bottom outlet of the column and set the pump to run at thedesired flow rate. This should be at least 133% of the flow rate to beused during subsequent chromatographic procedures. However, themaximum flow rate, see Table 1, is typically employed during packing. Note: If you have packed at the maximum linear flow rate, do not exceed 75% of this in subsequent chromatographic procedures.7. Maintain the packing flow rate for 3 bed volumes after a constant bedheight is reached.Using an adapterAdapters should be fitted as follows:1. After the medium has been packed as described above, close thecolumn outlet and remove the top piece from the column. Carefully fillthe rest of the column with buffer to form an upward meniscus at thetop.2. Insert the adapter at an angle into the column, ensuring that no air istrapped under the net.3. Make all tubing connections at this stage. There must be a bubble-freeliquid connection between the column and the pump.4. Slide the plunger slowly down the column so that the air above thenet and in the capillary tubings is displaced by eluent. Valves on theinlet side of the column should be turned in all directions during thisprocedure to ensure that air is removed.5. Lock the adapter in position on the medium surface, open the columnoutlet and start the eluent flow. Pass eluent through the column at thepacking flow rate until the medium bed is stable. Re-position the adapter on the medium surface as necessary.The column is now packed and equilibrated and ready for use.p. 85. BindingConditions for binding depend on which ligand is used. Literature references and textbooks may give good guidelines.The adsorption will depend upon parameters such as sample concentration, flow rate, pH, buffer composition and temperature. General guidelines for adsorption are:• Sample pH should be the same as that of the binding buffer. Filter the sample through a 0.22 μm or 0.45 μm filter to prolong the working life of the medium.• After the sample has been loaded, wash the medium with binding buffer until the base line is stable.6. ElutionConditions for elution of bound substances depend on which ligand is used. Literature references and textbooks may give good guidelines.General guidelines are described below.• pH change: A change in pH alters the degree of ionization of charged groups at the binding sites. Elution is generally affected by a decrease in pH. The chemical stability of the matrix, ligand and adsorbed substances determines the limits of pH which may be used.•Ionic strength: A buffer with increased ionic strength is used. Elution with a continuous or step-wise gradient may be used. A gradient ofincreasing salt concentration can be used to separate substances bound to the adsorbent. NaCl is most frequently used and enzymes usuallyelute at a concentration of 1 M NaCl or less. If the interaction has a very high affinity, a chaotropic salt may be required.• Competitive elution: Competitive eluents are often used to selectively elute substances from a group specific adsorbent and also when theaffinities are relatively low. Selectively retained substances are usuallydisplaced at low concentrations of eluting agents, often less then 10 mM.Either continuous or step-wise gradients may be used.p.• Reduced polarity: Conditions which lower the polarity of the eluent to promote elution may be used if they do not inactivate elutedsubstances. Dioxane (up to 10%) or ethylene glycol (up to 50%) may beused.• Deforming eluents: If the elution methods described above fail to affect elution, deforming agents, such as chaotropic salts, guanidine-HCl orurea, which alter the structure of the proteins can be used.7. RegenerationConditions for regeneration depend on which ligand has been coupled.Literature references and textbooks may give good guideines.A general regeneration method is described below:An affinity medium may be regenerated for re-use by washing the medium with 2–3 column volumes of alternating high pH (0.1 M Tris-HCl, 0.5 M NaCl, pH 8.5) and low pH (0.1 M sodium acetate, 0.5 M NaCl, pH 4.5) buffers. This cycle should be repeated 3 times followed by re-equilibration in binding buffer.8. StorageLyophilized CNBr-activated Sepharose 4B should be stored below 8 °C.Swollen coupled medium should be stored at 4–8 °C in presence of abacteriostatic agent, e.g. 20% ethanol.9. Further informationCheck /protein-purification for more information.Useful information is also available in the Affinity ChromatographyHandbook, se ordering information.p. 1010. Ordering informationProduct Pack size Code No. CBNr-activated Sepharose 4B 15 g 17-0430-01250 g 17-0430-02 LiteratureAffinity Chromatography Handbook, 1 18-1022-29 Principles and MethodsAffinity Columns and Media, 1 18-1121-86 Product Profilep. 11GE, imagination at work and GE monogram are trademarks of General Electric Company.Drop Design and Sepharose are trademarks of GE Healthcare companies. © 1996-2009 General Electric Company – All rights reserved. Previously published Aug. 1996.All goods and services are sold subject to the terms and conditions of sale of the company within GE Healthcare which supplies them. A copy of these terms and conditions is available on request. Contact your local GE Healthcare representative for the most current information.GE Healthcare Europe GmbH Munzinger Strasse 5D-79111 Freiburg GermanyGE Healthcare UK Limited Amersham Place Little ChalfontBuckinghamshire, HP7 9NA UKGE Healthcare Bio-Sciences Corp.800 Centennial Avenue P.O. Box 1327Piscataway, NJ 08855-1327USAGE HealthcareJapan Corporation Sanken Bldg.3-25-1, HyakuninchoShinjuku-ku, Tokyo 169-0073JapanFor local offoce contact information, visit /contact GE Healthcare Bio-Sciences AB Björkgatan 30751 84 Uppsala Sweden/protein-purification71-7086-00 AF 12/2009imagination at work。