医用耗材、试剂申请准入审批流程
医用耗材管理制度

医用耗材管理(一)医用耗材管理委员会工作职责1、依据国务院第76号令《医疗器械管理监督条例》、国家药监局第4号令《一次性使用无菌医疗器械监督管理办法》,我院要建立并完善医用耗材管理制度、流程;2、审核科室新增医用耗材申请报告,并确定准入品种;3、分析、论证本院医用耗材使用情况,并提出淘汰品种;4、医用耗材管理委员会的日常工作由医用耗材管理管理办公室负责,办公室设在医学工程科。
(二)医用耗材试用制度1、科室须根据临床需求、新技术新业务开展的需要提出试用耗材;2、试用科室填写《医用耗材试用申请表》,并备齐以下资质;①医疗器械注册证及登记表②生产企业营业执照副本、生产许可证(进口产品不需要)③各级经营企业的营业执照副本、经营许可证④各级经销商及业务员的授权书⑤业务员身份证复印件、联系方式⑥报关单(进口且非中标产品需要)⑦小包装产品3、将《医用耗材试用申请表》和相关资质提交至护理部或医务处。
护理耗材由护理部,医用耗材由医务处论证试用的合理性、合法性,审批签字并确定试用数量。
4、医学工程科审核耗材相关资质的合法性并签字确认;5、一次性使用无菌器械还要由医院感染管理科喝茶确认是否符合感控规定;6、各部门审批完成后,将《医用耗材试用申请表》交至护理部或医务处,由其确定试用数量和费用。
供应商到财务部门交齐试用费用后,试用科室方可试用;7、试用完成,如科室评估结果为希望长期使用该耗材,则按新医院耗材申请流程接续进行。
(三)医用耗材申购制度为优化我院医院耗材的品种,在满足临床需求的前提下尽量压缩品种总量,进一步提高耗材管理水平,特制定医院耗材申购制度。
1、限量申请按照各科的业务特点及近几年医用耗材的用量品种、新技术开展情况,确定各科室提交申请的品种、数量限额。
科室提交上会讨论的医用耗材使用申请数必须小于或等于限额数。
2、替代原则除科室开展新技术(开展的新技术应由相关部门批准)所需耗材外,原则上不再增加新的耗材品种。
医疗器械审批流程法规

医疗器械审批流程法规医疗器械审批是确保医疗器械安全有效性的一项重要工作。
为了保护公众的健康和安全,各国都制定了一系列法规和规定来规范医疗器械的审批流程。
本文将介绍一些医疗器械审批流程的法规。
一、国内医疗器械审批法规在中国,医疗器械审批工作由国家药品监督管理局(NMPA)负责。
目前,中国实施的医疗器械审批流程可分为三个阶段:注册申请、技术评价和审批。
1. 注册申请阶段医疗器械注册申请是医疗器械企业向NMPA提交的一个重要阶段。
在这个阶段,企业需要提供详细的产品信息,包括但不限于产品名称、结构和工作原理、临床试验报告、使用说明书等。
2. 技术评价阶段技术评价是医疗器械审批流程中的一个重要环节。
NMPA将组织专家对医疗器械进行科学的技术评价,评估其安全性和有效性。
评价结果将作为批准或拒绝注册的依据。
3. 审批阶段在技术评价通过后,NMPA将对医疗器械进行审批。
审批结果将决定是否批准该产品上市销售。
如果通过审批,企业将获得医疗器械注册证书。
二、国际医疗器械审批法规除了国内法规外,国际上也有一些相关的医疗器械审批法规,例如美国FDA(食品和药物管理局)的审批程序。
下面是美国医疗器械审批法规的一般流程:1. 预市批准(PMA)或510(k) 申请在美国,医疗器械的审批分为两种主要类型:预市批准(PMA)和510(k) 申请。
针对不同的产品类别,FDA要求医疗器械企业申请相应的批准类型。
2. 临床试验医疗器械企业需要进行临床试验来评估其产品的安全性和有效性。
这些试验必须符合FDA的要求,并在合适的患者群体中进行。
试验结果将作为产品是否获得批准的依据。
3. 后市场监管医疗器械在上市销售后,也需要进行后市场监管。
FDA要求企业进行质量管理体系的建立,并及时报告任何产品缺陷或安全问题。
FDA 还会进行定期的检查和审查,以确保产品的安全性和质量。
三、国际合作和互认机制为了促进全球医疗器械审批的合作和互认,各国之间建立了一些合作机制。
医用耗材及其他耗材采购流程
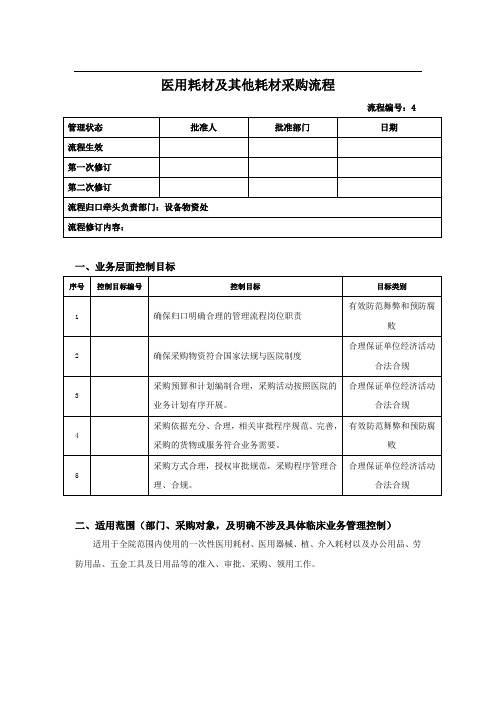
医用耗材及其他耗材采购流程流程编号:4管理状态批准人批准部门日期流程生效第一次修订第二次修订流程归口牵头负责部门:设备物资处流程修订内容:一、业务层面控制目标序号控制目标编号控制目标目标类别1 确保归口明确合理的管理流程岗位职责有效防范舞弊和预防腐败2 确保采购物资符合国家法规与医院制度合理保证单位经济活动合法合规3采购预算和计划编制合理,采购活动按照医院的业务计划有序开展。
合理保证单位经济活动合法合规4采购依据充分、合理,相关审批程序规范、完善,采购的货物或服务符合业务需要。
有效防范舞弊和预防腐败5采购方式合理,授权审批规范,采购程序管理合理、合规。
合理保证单位经济活动合法合规二、适用范围(部门、采购对象,及明确不涉及具体临床业务管理控制)适用于全院范围内使用的一次性医用耗材、医用器械、植、介入耗材以及办公用品、劳防用品、五金工具及日用品等的准入、审批、采购、领用工作。
三、内控制度制度及编号序号制度名称制度编号和文号发文部门发文日期1 医疗器械(耗材)管理制度设备物资处2014.1.12医学装备(医疗器械)管理委员会工作制度设备物资处2014.1.13 高值耗材采购制度和流程设备物资处2014.1.14 新增医疗器械管理制度设备物资处2014.1.15 物资招标采购实施办法设备物资处2014.1.16 一次性医疗用品采购制度设备物资处2014.1.17 一般物资评价采购制度设备物资处2014.1.1四、流程关键风险控制岗位及不相容职责分工(全过程)岗位姓名所属部门岗位职责说明科室负责人主任负责所有耗材采购审批。
业务部门负责人所有临床科室负责耗材付款申请审批。
关键岗位1 库房保管员负责耗材入库验收、出库、盘点。
关键岗位2 库房保管员负责耗材入库验收、出库、盘点。
关键岗位3 库房保管员负责耗材入库验收、出库、盘点。
关键岗位4 库房主管汇总领用申请,布置出库指令,转发采购需求,出入库信息管理。
产品资质定期审验关键岗位5 采购员汇总审核采购申请,制作电子订单并发送,审核出、入库单财务部门财务发票及入库单审核、付款计划制作、库房盘点、制作财务报表专业委员会审核新进耗材、试剂审批主管分院长审核新进耗材、试剂审批,每月耗材账务付款审批五、流程图说明1、总述(1)流程关键节点①建立健全耗材采购管理制度,包括采购目录建立、目录更改、目录作废、采购审批、采购执行、使用监控等制度。
医疗器械申报流程

医疗器械申报流程医疗器械申报是指将医疗器械产品提交给相关监管机构进行安全性和有效性评价的过程。
医疗器械的申报流程一般包括以下几个步骤:立项、研发、临床试验、技术文件编制、注册申报和审批。
下面将对这些步骤进行详细介绍。
首先是立项阶段。
在这个阶段,企业或个人需要相应的资金、技术和人力资源来定义医疗器械产品的研发目标和方向。
这一阶段通常包括前期调研、市场需求分析和技术可行性分析等工作,目的是为了明确申报的医疗器械的研发方向和目标。
其次是研发阶段。
在这个阶段,企业或个人需按照前期立项的目标和方案,进行医疗器械产品的原型开发和功能验证。
这一阶段的工作主要包括产品设计、零部件采购、工艺流程制定、生产线设计等,目的是制作出具备基本功能的医疗器械原型。
接下来是临床试验阶段。
在这个阶段,医疗器械原型需要进行临床试验,以评估其安全性和有效性。
临床试验分为人体试验和动物试验两个阶段。
人体试验需要申请伦理委员会的批准,并按照伦理委员会的规定进行试验;动物试验则需按照动物实验室的相关规定进行。
然后是技术文件编制阶段。
在这个阶段,企业或个人需要编制医疗器械的技术文件。
技术文件通常包括产品的构造、功能、性能、作用机制、用途范围、临床试验结果等信息。
技术文件的编制需要积累大量的实验数据,并按照相关标准和要求进行整理和归档。
接下来是注册申报阶段。
在这个阶段,企业或个人需要向相关监管机构申请医疗器械的注册。
申请注册的资料通常包括技术文件、临床试验报告、生产工艺和质量管理体系等信息。
相关监管机构将对申报资料进行审查,如发现问题或需要补充资料,申请人需要及时进行整改和重新提交。
最后是审批阶段。
在这个阶段,有关监管机构将对注册申请资料进行审批,以确定医疗器械是否符合相关安全和有效性要求,并决定是否批准注册。
审批时间通常需要几个月到数年不等,具体时间取决于医疗器械的分类、级别和监管机构的工作效能。
一旦通过审批,医疗器械就可以生产和销售。
总结起来,医疗器械申报流程一般包括立项、研发、临床试验、技术文件编制、注册申报和审批等步骤。
医用材料准入及供应商资质管理制度

医用材料准入及供应商资质管理制度
一、医用材料的购入应以保证质量为前提,从资质齐全的合格供应商进货,严格审核耗材、试剂及销售人员的资质证明。
二、新增的耗材和试剂,必须由使用科室填写〃新增医用材料审批表”,科主任签字,供应商提供真实有效的资质证明,经医学装备部和相关职能科室审核后,报呈院领导审批。
三、属于集中采购目录内的,医学装备部按照有关规定组织专家进行遴选。
不在集中采购目录内的,由医学装备部组织院所医学装备管理委员会的专家严格论证后,进行招标采购。
四、供应商应按规定提供合法、有效资质材料复印件,并加盖企业印章
1、《企业法人营业执照》
2、《医疗器械生产企业许可证》
3、《医疗器械经营企业许可证》
4、《医疗器械注册证》及其附件(《医疗器械注册证登记表》或《医疗器械产品制造认可表》)
5、原厂授权书
6、《消毒产品卫生许可证批件》(如果属于消毒产品)
7、产品合格证明
8、其他资质材料(如进口产品的检测鉴定报告、报关单等)
9、销售人员身份证明
10、产品介绍、使用说明等技术文件
五、供应商不得提供市场准入证件过期或超出规定范围的产品,若违反相关规定应承担相应责任。
六、医学装备部建立合格供应商目录并对上述相关资料存档。
2020医用耗材、试剂采购管理办法(精华版)

2020医用耗材、试剂采购管理办法为进一步规范医用耗材、试剂(含科研试剂)采购行为,加强对采购活动的管理工作,确保采购过程公开透明、公平公正、廉洁高效,根据《中华人民共和国政府采购法》、《省级医疗卫生计生单位政府采购实施细则》等有关法律法规和规章制度,结合医院工作实际,制定本管理办法。
1.招标采购办认真执行《医疗器械监督管理条例》,根据临床诊断和治疗的需要,进行计划采购。
2.医用耗材采购,供应商必须提供产品有效期内的《中华人民共和国医疗器械注册证》(提供注册证复印件),若注册证有附件的,还须提供附件(提供复印件);产品属三类医疗器械的,投标人须具备有效期内《医疗器械经营企业许可证》或《医疗器械经营许可证》;产品属二类医疗器械的,供应商具备有效期内《医疗器械经营企业许可证》或《第二类医疗器械经营备案凭证》等证件,复印件必须加盖经销单位公章,并核实证件的真实性与有效性,不得购买无证、缺证和过期产品,严格把好购买质量关。
3.医用耗材招标采购中,贯彻质量第一的原则,对医用耗材质量,临床医务人员有决定权,保证采购的医院耗材符合临床医疗活动质量要求,确保临床使用安全。
4.采购流程(1)招标采购办接到按程序审批后的医用耗材申请,由医院采购办实施。
使用科室未在3 个月内按要求提供相关技术材料,采购项目审批程序自动作废。
(2)年采购金额超过50 万元的医用耗材,医院采购办按规定以公开招标、竞争性比选、遴选、单一来源等采购方式确定入选产品及其供应商。
(3)年采购金额50 万以内医用耗材,医院采购办可直接在XX省药交所采购平台采购,产品信息比对(原则上三家以上),进行价格谈判,形成相关采购记录,签订网上采购协议;也可以参照本办法4.2 条款组织采购。
(4)医用耗材遴选,由使用科室推荐品牌(原则上三家及以上供招标采购办参考),经严格审核产品和供应商资质合格后,由采购小组进行价格谈判,遴选出价格性能优异的产品,形成相关采购记录交招标采购办采购。
医疗器械的申报和审核流程

医疗器械的申报和审核流程随着医疗技术的不断发展和医疗市场的不断扩大,医疗器械的需求也不断增长。
但是,任何一种医疗器械在进入市场之前,都需要经过一系列的申报和审核流程,以确保其安全性和有效性。
这篇文章将详细介绍医疗器械的申报和审核流程。
一、医疗器械的申报流程医疗器械的申报流程包括了多个环节,一般分为以下几个步骤:1. 器械研发和实验室检测在进行任何器械的申报之前,首先需要进行大量的研发和实验室检测。
这个过程需要具有高度专业技能和经验的团队,例如医学专业人士、化学工程师、生物医学工程师等。
2. 产品型号和规格确认在进行申报前,需要明确产品的型号和规格,以此建立技术文件并制定相关的标准。
3. 品质管理审核通过采取ISO9001认证或相似的质量管理体系,制定品质管理审核程序,确保产品的品质和性能符合相关标准和要求。
4. 适用性审查每种器械的适用性不同,因此需要对适用性进行审查。
这个过程包括识别适用范围,评估所需功能以及决定在不同条件下使用器械时会产生的风险和问题。
5. 使用说明书和标签符合性审查对器械的使用说明书和标签进行审查,确保这些文件和信息符合相关标准和规定。
6. 风险管理和质量控制计划确认对风险管理和质量控制计划进行确认,通过这些计划确定器械如何使用和维护,以及如何处理可能出现的问题。
7. 申请文件准备在完成以上几个步骤后,还需要准备完整的申请文件,以便进行下一步的审核流程。
二、医疗器械的审核流程医疗器械的审核流程涉及机构和人员的多个环节。
一般而言,医疗器械的审核流程包括了以下几个步骤:1. 申请文件递交首先,申请人需要将所有的申请文件提交给相应的机构或部门,这些机构或部门负责审核器械的安全性和有效性。
2. 评估核查在收到申请文件后,审核机构会进行评估核查。
这个过程包括对文件的完整性和可靠性进行确认,以及从技术和临床角度对器械的性能和安全性进行评估和审查。
3. 环境和设施审核审核机构还会对生产器械的环境和设施进行审核,确定它们是否符合相关标准和规定。
医用耗材验收入库管理制度、职责及流程

医用耗材验收入库管理制度、职责及流程1.目的医院SPD中心库房是储存、配送各类医用耗材的重要部门,各类高值耗材、低值耗材及其他医用物资的验收入库管理至关重要,必须建立科学、完善的制度和流程,以适应现代化医院管理的需要,达到服务临床需求、确保医疗安全的目的。
2.适用范围适用于医院入库的高值耗材、低值耗材及其他医用物资。
3.验收入库管理制度3.1 未经医院批准采购的商品,一律拒收;3.2 验收人员应严格按照国家相关法律法规标准以及医院采购合同约定的质量条款进行验收,如发现商品质量包装要求不符合规定,应立即拒收;3.3 供应商配送单、商品必须同时到达方可验收,否则拒收;配送单和采购订单不符,应立即拒收;若代为保管,所发生的一切后果和责任,由供应商自负;3.4 严格按规定的验收原则和抽样原则对待验收商品进行验收。
4.验收人员职责4.1 物资供应中心验收员负责验收,SPD验收人员协助完成验收,验收时,双方人员必须同时到场;4.2 严格按照国家相关法律法规标准、根据医院采购合同约定的质量条款、平台配送单、退货单内容对采购商品和科室退货商品进行验收;4.3 严格按规定的验收原则和抽样原则对待验收商品进行验收,并在规定的场所、时限内完成;4.4 对验收过程中发现质量可疑商品或不合格商品,及时与物资供应中心验收人员沟通上报,不合格商品退回供应商,并登记;4.5 科室退货商品,对外包装、标签等进行验收,合格后方可入库;4.6 加强业务学习,了解食药监械最新通知及规定、熟悉商品特性。
5.入库人员职责5.1 入库员根据已经验收的配送单,进行系统入库,确保入库商品信息与系统一致;5.2 加强业务学习,了解食药监械最新通知及规定,熟悉商品特性;5.3 保证数据安全,不泄露系统数据。
6.验收流程6.1 低值耗材验收流程6.1.1 供应商送配送单、商品至SPD中心库房,验收人员首先核对商品信息与字典是否一致,其次检查商品运输方式、包装、标识是否符合规定,再核对采购计划、品名、型号、规格、数量、批号、有效期、生产厂家、单价、金额、注册证等信息,准确无误后医院验收人员在配送单签字、盖验收合格章,供应商、SPD验收人员在配送单签字;6.1.2 SPD入库员根据平台订单办理入库,核对商品品名、型号、规格、数量、批号、有效期、生产厂家、单价、金额、注册证等信息,信息准确无误后,打印三联入库单,由供应商、入库员、仓管员签字,黄联由供应商保管,红联由入库员保管,白联由结算组保管;6.1.3 SPD入库员打印低值标签,交给仓管员赋码、上架。
医院新进设备耗材审批流程

医院新进设备耗材审批流程英文回答:Medical Equipment and Consumables Approval Process for New Hospital.1. Introduction.The acquisition of new medical equipment and consumables is a critical decision for hospitals, involving substantial investments and potential impact on patient care. Establishing a comprehensive approval process is essential to ensure the judicious use of resources, adherence to regulatory standards, and the selection of appropriate equipment and supplies for the hospital's needs.2. Process Outline.The proposed approval process encompasses the following key steps:1. Department Request:Clinical departments submit a written request to the procurement department, outlining the specific equipment or consumables required, along with justifications for the purchase.2. Technical Evaluation:A technical committee composed of clinicians and engineers evaluates the proposed purchase, assessing its clinical utility, technical specifications, and compatibility with existing systems.3. Financial Analysis:The finance department reviews the cost of the purchase and its impact on the hospital's budget, considering both capital and ongoing expenses.4. Regulatory Compliance Review:The legal team ensures that the proposed purchase complies with all applicable regulatory requirements, including those related to safety, infection control, and data privacy.5. Risk Assessment:A risk assessment is conducted to identify potential risks associated with the acquisition, such as patient safety concerns, operational challenges, or technological obsolescence.6. Purchase Approval:Based on the evaluations and assessments, a purchase decision is made by the hospital's executive leadership team.7. Vendor Selection:A competitive bidding process is conducted toselect the supplier that offers the best combination of quality, price, and delivery time.3. Key Considerations.In addition to the outlined steps, the approval process also incorporates the following key considerations:Clinical Needs: The primary focus of the process is to meet the clinical needs of the hospital, ensuring that patients have access to the appropriate equipment and supplies for their care.Cost-Effectiveness: Purchases are evaluated not only based on their initial cost but also on their long-term cost-effectiveness, considering factors such as ongoing maintenance and potential savings.Integration with Existing Systems: The acquisition of new equipment and consumables should be compatible with existing systems and infrastructure within the hospital, ensuring seamless integration and efficient workflow.Staff Training and Support: The process accounts for the training and support required for staff to effectively use and maintain the new equipment or consumables.Sustainability: Environmental and sustainability considerations are taken into account, promoting the selection of equipment and supplies that are environmentally friendly and minimize waste.中文回答:新医院设备耗材审批流程。
医疗耗材出入库管理制度

医疗耗材出入库管理制度购置规定:1、使用科室必须提出书面申请,包括所需产品型号、产地、技术参数和申请购置的设备经济效益预测。
2、使用科室申请后,中心召集医疗设备论证评估委员会进行可行性论证后,由药械科进行市场调查,报招标小组,采取公开、公正、公平的招标形式,选购性能良好价格适宜的仪器设备。
验收规定:1、设备到货后中心领导、药械科、档案室、使用科室等有关科室到场。
2、开箱验收设备时各种资料要齐全,否则不予签验收单。
3、设备安装调试必须由供方派熟练的工程技术人员进行现场调试、培训。
4、设备随机资料应采集整理归档。
管理规定:1、货到验收合格后办理出入库手续,由财务科负责固定资产帐、后勤保障部负责辅助帐及使用科室卡片帐同时帐、登记。
2、使用科室应选派责任心强、技术熟练的同志,严格按操作规程使用,凡实习生、进修人员不得单独操作,严禁非医技人员使用。
3、不能使用的设备由本科室提出书面请示,经后勤保障部核实转到财务报废库,每半年进行一次清查上报院领导批示后,按程序办理报废手续。
4、因工作需要医疗设备需要长期调整,应及时通知财务科开调拨单、药械科更改帐卡和科室之间相互验收后方可调整。
5、设备不经院领导允许,任何科室和个人不得私自外借、拆卸、维修。
6、各科设备原则上不能外借,如工作需要在正常工作期间由后勤保障部协调办理互借手续,下班后由总值班协调办理互借手续。
互借期间,借方收入除上交中心外,双方各得 50%。
7、设备效益分析每月一次单机分析,每季一次汇总分析,及时上报院领导。
8、固定资产每年盘点一次,做到帐物相符。
科室各种医疗设备管理保养规定:1、设备到位后,由后勤保障部会同相关人员安装、验收、调试、培训后办理手续,交2、科室应有专人负责保管、养护。
3、设备应建立操作规程、使用和养护记录。
各种设备操作人员应经过培训,熟悉设备使用科室,进行正常运行。
性能及操作规程后,才干上岗操作。
凡实习生、进修人员不得单独操作,严禁非医技人员使用。
医用耗材管理办法

医用耗材管理办法第一条为进一步规范医院医用耗材的管理,保障医用耗材使用合法、合理、安全,根据《医疗器械监督管理条例》(国务院令第650号)及相关法律法规,特制定本办法。
第二条医院医用耗材管理参照国家按照风险程度实行的医疗器械分类管理模式。
第一类是风险程度低,实行常规管理可以保证其安全、有效的医用耗材。
第二类是具有中度风险,需要严格控制管理以保证其安全、有效的医用耗材。
第三类是具有较高风险,需要采取特别措施严格控制管理以保证其安全、有效的医用耗材。
第三条管理原则(一)满足临床诊疗所需。
(二)确保质量与安全。
(三)实行比质比价的采购原则,即质量第一、价格合理、择优采购;省阳光采购平台挂网产品优先采购。
(四)实行《医用耗材常购目录》与《合格供应商目录》管理制度。
第四条职责分工(一)医学装备(医用耗材)管理委员会负责第三类医用耗材的准入论证,《医用耗材常购目录》的审定。
(二)设备科为医用耗材管理的主管部门,负责医用耗材的准入及临床的使用指导及管理。
(三)医务处、护理部作为医疗业务主管部门,负责临床科室医用耗材的使用合理性管理.(四)物流管理部为医用耗材的实物管理部门,负责医用耗材的质量管理与配送。
第五条准入与资质管理(一)临床、医技科室使用的医用耗材实行分类申报论证、审批准入管理。
第一、二类医用耗材的准入经相关部门、院领导审批。
第三类医用耗材需由医疗业务主管部门严格审批,医用耗材主管部门组织医学装备(医用耗材)管理委员会相关专家进行准入论证。
(二)医用耗材采购实施前,医用耗材主管部门必须查验申请准入的医用耗材供应商、生产厂商、产品的相关资质证件是否符合国家的有关要求,采购办复核相关资质文件的合法性。
(三)物流管理部建立医用耗材资质档案(供应商、生产厂商、产品资质档案),并实施动态效期管理。
(四)“三类”医用耗材技术档案应在院感科备案。
第六条采购管理(一)医用耗材首次进院,由采购办根据医院《采购管理办法》的要求实施采购,采购程序结束后,由物流管理部实行最低库存量和定期、定量补库模式管理.物流管理部日常采购执行医院《医用耗材常购目录》。
医用耗材及其他耗材库存管理流程
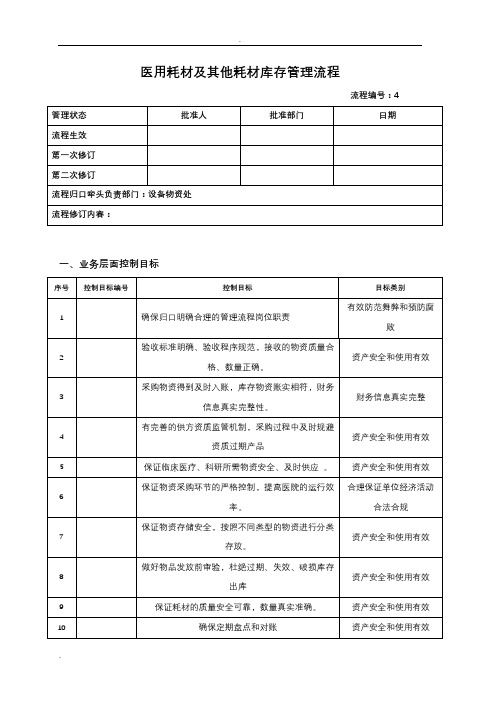
医用耗材及其他耗材库存管理流程流程编号:4一、业务层面控制目标二、适用范围(部门、采购对象,及明确不涉及具体临床业务管理控制)适用于全院范围内使用的一次性医用耗材、医用器械、植、介入耗材以及办公用品、劳防用品、五金工具及日用品等。
三、内控制度制度及编号四、流程关键风险控制岗位及不相容职责分工(全过程)五、流程图说明1、控制痕迹发票、入库单、库房台账(万达系统)、盘库清单、库存报表、领用单、库房台账(万达系统)2、流程详述每个库房设立待验区、不合格区和合格区三个独立区块。
所有货物到达库房后,先进入待验区。
库房管理人员验收后,合格品放入合格区储藏,等待配送与领用。
不合格品暂时堆放于不合格区,等待妥善处理。
日常盘库中清理出来的过期、损耗货品,也应及时存入不合格区,等待处理。
A1医疗器械送达库房待验区后,验收人员应按送货清单与发票核对货物的品名、规格数量、金额、质量,逐一核对,确保正确无误。
必须对每件产品进行查验,核对其产品注册证书、批准文号、条形码、有效期,并录入物资管理系统,完成入库数据记录。
对医疗器械产品外观包装及标识必须认真进行查验,进口产品应有中文标识,发现外包装破损,包装上无注册证书编号和产品批准文号的产品不予以验收。
植入性医疗器械要求必须附有使用病人的信息资料,植入材料的条形码要求在红会医疗器械追溯系统中扫描录入,形成追溯记录。
临床医生确认使用品种、规格,为病人植入后,临床医生在《植入性医疗器械使用登记表》上确认,由跟台护士将产品条形码粘贴到《植入性医疗器械使用登记表》上,手术室统一交至设备部库房入库。
《植入性医疗器械使用登记表》包括:科室名称、患者姓名、性别、年龄、住院号、床位号、手术日期、到货日期、产品名称、类型、编号/批号、规格、数量、有效日期、条形码、产品公司名称、代理公司名称、医疗器械生产企业许可证、医疗器械产品注册号、经营许可证等跟踪信息。
《植入性医疗器械使用登记表》整理归档,B1设备物资处库房应将送货清单、以备查询。
医院医用耗材准入管理规定

医院医用耗材准入管理规定模板1为了进一步加强医用耗材准入管理,严格规范医用耗材引进流程,满足临床工作需求,保障医疗安全和医疗质量,现对医用耗材的申请和准入制定以下管理规定。
1.组织机构(1)由分管医用耗材采购的院领导、医务处、护理部、医用耗材保障部门、计财/资产处、感染管理办公室等相关职能部门负责人组成医院医用耗材采购领导小组,具体负责医用耗材的引进审批。
分管院领导任组长,各职能部门负责人任组员,医务处医务科负责人任秘书。
(2)成立医院医用耗材准入评审专家库,成员包括各临床医技科室行政正、副主任、科护士长、各护理学组组长。
2.准入流程(1)医用耗材保障部门负责医用耗材新品的申请登记工作。
①各医用耗材生产(经营)企业委托人凭相关的授权委托和身份证明到医用耗材保障部门领取新品进院申请表格,按要求填写完整后在规定时限内提交。
②提交申请时需一并提供下列材料:营业执照;医疗器械生产或者经营的许可证或者备案凭证;医疗器械注册证或者备案凭证;销售人员身份证复印件(正反),加盖企业公章的授权书原件;授权书应当载明授权销售的品种、地域、期限,注明销售人员的身份证号码;进口产品英文授权需附中文翻译件;产品彩页;样品。
必要时,医疗机构可以派员对供货者进行现场核查,对供货者质量管理情况进行评价。
③每份表格仅限申报一种产品,填写不完整或附件材料不齐全的,不予受理。
(2)医用耗材保障部门、医务处、护理部、计财/资产处和感染管理办公室负责医用耗材新品申请的初筛工作。
①医用耗材保障部门对医用耗材、生产(经营)企业及其委托人的资质进行审核。
②医用耗材保障部门连同计财/资产处对同类产品价格、收费状况进行审核。
③医务处、护理部对医用耗材的临床应用价值、需求程度及使用医用耗材的相关技术进行审核。
④感染管理办公室负责审核医用耗材是否符合院感相关规定。
(3)通过初筛的品种,由医用耗材保障部门对根据应用范围的不同进行分类,一般分为两类:专科品种、多科和公用品种。
医用耗材准入制度

医用耗材准入制度1. 医用耗材准入制度啊,这可太重要了!就好比一道门,把不好的耗材挡在外面,让好的耗材进来为我们服务。
比如说,要是没有严格的准入,那质量差的口罩混进来了,我们怎么能放心地用呢?2. 你想想看,医用耗材准入制度不就是我们健康的一道保障吗?就像一个严格的守卫,仔细审查每一个要进入的耗材。
比如那些不合格的注射器,不就被它挡在外面了嘛!3. 医用耗材准入制度真的能决定很多啊!它就如同一个筛选器,把优质的留下,劣质的淘汰。
就像手术中要用的缝线,有了准入制度,我们才能用到可靠的呀!4. 哎呀呀,医用耗材准入制度可不能小瞧!这简直就是为我们的医疗安全保驾护航啊!好比建房子,没有好的材料怎么行?那些不合格的耗材不就像劣质砖头嘛!5. 医用耗材准入制度可是非常关键的呀!它就像一个把关人,只让合适的耗材进来。
比如说,要是没有它,那些容易引起过敏的绷带混进来了,那可咋办?6. 你们知道吗,医用耗材准入制度真的很厉害呢!它就好像一双敏锐的眼睛,能发现不好的耗材。
就像检测试剂,有了严格准入,我们才能相信它的准确性呀!7. 医用耗材准入制度可不是闹着玩的呀!这可是关系到我们每个人的健康呢!就像食品安全一样重要。
比如不合格的导管,没有准入制度能行吗?8. 哇塞,医用耗材准入制度真的太有必要了!它就如同一个精确的天平,衡量着每一种耗材。
就像心脏起搏器,得靠准入制度保证质量啊!9. 医用耗材准入制度啊,这可是我们的大救星呢!它把不好的东西都挡在了外面。
就像消毒用品,没有好的准入,怎么能放心消毒呢?10. 医用耗材准入制度真的是超级重要啊!这就像是一道坚固的防线,保护着我们的医疗安全。
比如不合格的骨科植入物,多亏了准入制度把它拦住了呀!我的观点结论:医用耗材准入制度是极其重要的,必须严格执行,才能确保我们用到安全、可靠的医用耗材,保障我们的健康和安全。
医用试剂准入管理制度范本

医用试剂准入管理制度第一章总则第一条为了加强医用试剂的管理,确保医用试剂的质量和安全,根据《医疗器械监督管理条例》、《药品管理法》等相关法律法规,制定本制度。
第二条本制度适用于医用试剂的采购、验收、储存、使用、销毁等环节的管理。
第三条医用试剂的管理应遵循合法、合规、科学、安全的原则。
第四条医疗机构应当设立医用试剂管理部门,负责医用试剂的管理工作。
第二章采购与验收第五条医用试剂的采购应选择具有合法资质的生产企业或经营企业。
第六条采购医用试剂时,应进行充分的市场调研,选择质量可靠、价格合理的试剂。
第七条采购合同应明确试剂的品种、规格、数量、质量标准、交货时间等内容。
第八条医用试剂到货后,应由医用试剂管理部门负责验收。
验收时,应核对试剂的品种、规格、数量、生产日期、有效期等信息,确保试剂质量符合要求。
第九条验收合格的医用试剂,应填写验收记录,并办理入库手续。
第三章储存与管理第十条医用试剂应存放在符合要求的储存设施中,确保试剂的安全和有效期内使用。
第十一条储存医用试剂的设施应具备以下条件:(一)温度、湿度等环境参数符合试剂的要求;(二)通风良好,避免阳光直射;(三)有防火、防盗、防潮、防爆、防泄漏等安全措施;(四)设置明显的警示标识和操作规程。
第十二条医用试剂的使用应严格按照试剂说明书和操作规程进行。
第十三条医疗机构应当定期对医用试剂进行质量检查,发现问题及时处理。
第四章使用与销毁第十四条医用试剂的使用应遵循“先进先出”的原则,确保试剂的新鲜度。
第十五条使用医用试剂时,应做好使用记录,包括试剂的名称、规格、数量、使用日期、使用人员等信息。
第十六条医用试剂在使用过程中,如发现质量问题,应立即停止使用,并及时报告医用试剂管理部门。
第十七条医用试剂的销毁应按照相关规定进行,确保不会对环境和人体造成危害。
第五章监督检查第十八条医疗机构应当定期对医用试剂的管理情况进行监督检查,发现问题及时纠正。
第十九条医用试剂管理部门应当定期对医用试剂的质量进行抽检,确保试剂质量安全。