医疗器械临床试验规定(局令第5号)
《体外诊断试剂注册管理办法》(国家食品药品监督管理总局局令第5号)

《体外诊断试剂注册管理办法》(国家食品药品监督管理总局局令第5号)国家食品药品监督管理总局令第 5 号《体外诊断试剂注册管理办法》已于2014年6月27日经国家食品药品监督管理总局局务会议审议通过,现予公布,自2014年10月1日起施行。
局长张勇2014年7月30日体外诊断试剂注册管理办法第一章总则第一条为规范体外诊断试剂的注册与备案管理,保证体外诊断试剂的安全、有效,根据《医疗器械监督管理条例》,制定本办法。
第二条在中华人民共和国境内销售、使用的体外诊断试剂,应当按照本办法的规定申请注册或者办理备案。
第三条本办法所称体外诊断试剂,是指按医疗器械管理的体外诊断试剂,包括在疾病的预测、预防、诊断、治疗监测、预后观察和健康状态评价的过程中,用于人体样本体外检测的试剂、试剂盒、校准品、质控品等产品。
可以单独使用,也可以与仪器、器具、设备或者系统组合使用。
按照药品管理的用于血源筛查的体外诊断试剂和采用放射性核素标记的体外诊断试剂,不属于本办法管理范围。
第四条体外诊断试剂注册是食品药品监督管理部门根据注册申请人的申请,依照法定程序,对其拟上市体外诊断试剂的安全性、有效性研究及其结果进行系统评价,以决定是否同意其申请的过程。
体外诊断试剂备案是备案人向食品药品监督管理部门提交备案资料,食品药品监督管理部门对提交的备案资料存档备查。
第五条体外诊断试剂注册与备案应当遵循公开、公平、公正的原则。
第六条第一类体外诊断试剂实行备案管理,第二类、第三类体外诊断试剂实行注册管理。
境内第一类体外诊断试剂备案,备案人向设区的市级食品药品监督管理部门提交备案资料。
境内第二类体外诊断试剂由省、自治区、直辖市食品药品监督管理部门审查,批准后发给医疗器械注册证。
境内第三类体外诊断试剂由国家食品药品监督管理总局审查,批准后发给医疗器械注册证。
进口第一类体外诊断试剂备案,备案人向国家食品药品监督管理总局提交备案资料。
进口第二类、第三类体外诊断试剂由国家食品药品监督管理总局审查,批准后发给医疗器械注册证。
国家药品监督的管理局令第5号医疗器械临床试验规定

《医疗器械临床试验规定》(局令第5号)国家食品药品监督管理局令第5号《医疗器械临床试验规定》于2003年12月22日经国家食品药品监督管理局局务会审议通过,现予发布。
本规定自2004年4月1日起施行。
局长:郑筱萸二00四年一月十七日医疗器械临床试验规定第一章总则第一条为加强对医疗器械临床试验的管理,维护受试者权益,保证临床试验结果真实、可靠,根据《医疗器械监督管理条例》,制定本规定。
第二条医疗器械临床试验的实施及监督检查,应当依照本规定。
第三条本规定所称医疗器械临床试验是指:获得医疗器械临床试验资格的医疗机构(以下称医疗机构)对申请注册的医疗器械在正常使用条件下的安全性和有效性按照规定进行试用或验证的过程。
医疗器械临床试验的目的是评价受试产品是否具有预期的安全性和有效性。
第四条医疗器械临床试验应当遵守《世界医学大会赫尔辛基宣言》(附件1)的道德原则,公正、尊重人格、力求使受试者最大程度受益和尽可能避免伤害。
第五条医疗器械临床试验分医疗器械临床试用和医疗器械临床验证。
医疗器械临床试用是指通过临床使用来验证该医疗器械理论原理、基本结构、性能等要素能否保证安全性有效性。
医疗器械临床验证是指通过临床使用来验证该医疗器械与已上市产品的主要结构、性能等要素是否实质性等同,是否具有同样的安全性、有效性。
医疗器械临床试用的范围:市场上尚未出现过,安全性、有效性有待确认的医疗器械。
医疗器械临床验证的范围:同类产品已上市,其安全性、有效性需要进一步确认的医疗器械。
第六条医疗器械临床试验的前提条件:(一)该产品具有复核通过的注册产品标准或相应的国家、行业标准;(二)该产品具有自测报告;(三)该产品具有国务院食品药品监督管理部门会同国务院质量技术监督部门认可的检测机构出具的产品型式试验报告,且结论为合格;(四)受试产品为首次用于植入人体的医疗器械,应当具有该产品的动物试验报告;其它需要由动物试验确认产品对人体临床试验安全性的产品,也应当提交动物试验报告。
医疗器械研发中的临床试验规范

医疗器械研发中的临床试验规范医疗器械的研发过程中,临床试验是至关重要的一环。
临床试验旨在评估医疗器械的安全性和有效性,为其上市提供科学依据和数据支持。
为了确保临床试验的科学性和规范性,各国都制定了相应的临床试验规范。
本文将介绍医疗器械研发中的临床试验规范。
一、临床试验的定义和分类临床试验是指通过人体实验对医疗器械进行安全性和有效性评价的科学活动。
根据试验目的和设计,临床试验可分为治疗性试验、诊断性试验、预防性试验和心理行为试验等。
不同类型的临床试验有不同的试验设计和要求。
二、临床试验的伦理和法律要求临床试验涉及人体,因此必须符合伦理和法律要求。
伦理要求包括保护试验对象的权益、获取知情同意、保护隐私和数据安全等。
法律要求包括符合国家规定的试验审批程序、药物管理法规等。
研发团队必须遵循相关政策和法规,确保试验合法合规。
三、试验设计和样本选择临床试验的设计和样本选择是确保试验结果准确可靠的重要因素。
试验设计包括试验目标、分组方式、随机化和盲法等。
样本选择要符合试验目的和研究假设,通常需要进行统计学分析来确定样本量。
四、试验方案和研究流程试验方案是对临床试验进行计划和指导的文件。
试验方案包括试验背景、目的、研究问题、试验设计、样本选择、试验终点等。
研发团队必须编写详细的试验方案,并进行严格的执行和监控,确保试验流程的顺利进行。
五、试验数据的收集和分析试验数据的收集和分析是临床试验的重要环节。
试验数据应严格按照试验方案和数据管理计划进行收集和记录,并进行质量控制。
数据分析应使用合适的统计学方法,进行准确的数据解读和结论推导。
六、不良事件和风险管理临床试验中可能发生不良事件和风险,研发团队应制定相应的风险管理计划和不良事件报告制度。
不良事件的定义、分级和报告要求应明确规定,且研发团队应及时采取措施对不良事件进行处理和管理。
七、试验报告和结果解读试验结束后,研发团队需要编写试验报告,对试验结果进行解读和分析。
《医疗器械临床试验规定》(局令第5号)英文版

(Order No. 15 of SDA)The Provisions for Medical Device Classification were passed by the State Drug Administration at the administration affairs meeting on February 17 of 2000, are hereby promulgated and shall go into effect as of April 10, 2000.Zheng XiaoyuDirector- General of State Drug AdministrationApril 5, 2000Provisions for Medical Device ClassificationArticle1 The Provisions are stipulated in accordance with the Regulation on Supervision and Administration of Medical Device to standardize the classification of medical devices.Article2 "Medical devices" refer to those instruments, equipment, tools, materials and other objects, including the software attached to them, that are designed to be used either independently or in combination on human body. These devices are used for:1.Prevention, diagnosis, treatment, monitoring or remission of diseases;2. Diagnosis, treatment, monitoring, remission or compensation of injury or physical disability;3.Research, replacement or adjustment of anatomical or physiological process;4. Control of pregnancy.Basically, the effect of these devices on human body is not achieved through means of pharmacology, immunology or metabolism; though they might be resorted to in order to bring about certain supplementary effect.Article3 The Provisions are meant to direct the formulation of The Category of Medical Device Classification as well as to determine the classes of newly registered products.Article 4 The classification of medical devices should be determined by a combined judgement on three respects: its structural characteristics, form of operation as well as conditions for use.Specifically, their classification can be based on Criteria for Medical Device Classification (see appendix).Article 5 Guidelines for Medical Device Classification1. The structural characteristics of medical devicesAccording to their respective structural characteristics, medical devices are divided into active and passive devices.2. The forms of operation of medical devicesMedical devices are designated into different forms of operation in accordance with their intended purposes.1) Passive devices in terms of their form of operation can be classified as device used for transportation and storage of pharmaceutical liquid, device for alteration of blood, body fluids, medical dressing, surgical instruments;reusable surgical instruments, disposable aseptic device, implantable device, device for contraception and birth control, device for sterilization and cleaning, patient care device, in vitro diagnostic reagent, as well as other passive contacting device or passive supplementary device.2) Active devices in terms of their form of operation can be classified as device for treatment through energy, diagnostic monitoring, body fluids transportation and ionized radiation, laboratory instruments and medical sterilizer; as well as other active contacting device or active supplementary device.3. The conditions for use of medical devices:Medical devices may be divided into contacting or inserted devices and non-contacting devices based on their conditions for use, which include the possible injuries they might entail as well as their impact on the medical treatment.1) Contacting or inserted devicesa. Term of use: temporary use, short - term use, long-term use;b. Particular parts of the human body being contacted:skin, cavity and tract; trauma or body tissue; blood circulation system or central nervous system;c. The degree of injuries caused by malfunction of active devices:minor injuries, injuries, serious injuries.2) Non-contacting DevicesThe impact these devices have on treatment ranges from: basically no impact, indirect impact, substantial impact.Article 6 Principles for Medical Device Classification1. The classification of medical devices should be conducted in accordance with The Criteria for Medical Device Classification.2. The criteria for medical device classification are based on the intended purpose and the function of a medical device. To the same product, if the intended purpose or form of operation be different, its class shall be determined respectively.3. For the medical device to be used in combination with another, the classification of each should then be dealt with separately. Accessories to medical devices should be classified independently from the master device respecting its own conditions.4. For the medical device to be used on several parts of the human body, the classification should be determined on the basis of the risks involved in its intendedpurposes and form of operation.5. Software that controls the functions of the medical device should be designated to the same class of its associated medical device.6. If one medical device pertains to two classes at the same time, the higher one is adopted.7. Those products that are designed to monitor or affect the major functions of a medical device should be designated to the same class of the device being monitored or affected.8. The State Drug Administration shall readjust as it sees fit the classification of certain medical devices that call for special administration.Article 7 The State Drug Administration is competent authority take charge of the classification of medical devices. In case a medical device fails to be designated according to The Category of Medical Device Classification, its classification then should be based on The Regulation for Medical Device Classification at the discretion of the provincial drug administration, the result of which should be submitted to the State Drug Administration for approval.Article 8 The terms used in the provisions are defined as follows:1. Intended purpose: the desired effect of a medical device that is illustrated in its product specification, label or materials for publicity.2. Risk: the possible injuries that may be caused by the medical device and the seriousness of the injury.3.Term of use:1) Temporary Use: the intended term for consecutive use of the device is within 24 hours;2) Short-term Use: the intended term for consecutive use of the device ranges from 24 hours to 30 days;3) Long-term Use: the intended term for consecutive use of the device is more than 30 days.4) Term for consecutive use: the actual working time of a device without any stop in accordance with its intended purpose.4. Parts being operated upon and the device:1) Non-contacting devices: devices that do not directly or indirectly contact the body of a patient;2) Surface contacting devices: including devices contacting the following parts of the human body:a. skin: devices that only contact the surface of the unwounded skin;b. mucous membrane: devices that contact the mucous membrane;c. wounded surface: devices that contact the wounded area or the surface of other injured areas.3) Devices for surgical insertion: devices that are entirely or partly inserted into the body through the surface of body by surgery contacting the following parts of the human body:a. blood vessel: inserted devices contacting a point on a blood vessel or as a channel to the blood vessel system.b. tissue/bone/dentinum: devices and materials that are inserted into the tissue, bones as well as endodontium/dentinum system.c. blood circulation: devices that contact the blood circulation system.5.Implantables: devices that are entirely or partly inserted into the cavity or tract of the human body by surgery. These devices either remain in the body over a long period of time, or partly remain in the body for at least 30 days.6. Active device: any medical device that operates on electric power or other forms of energy excluding those directly generated by human body or gravity.7. Reusable surgical instruments: devices that are used to conduct such procedures during a surgery as excision, boring, sawing, clutching, scraping, clipping, drawing and clamping without having to resort to any active device and that can be reused after certain treatment.8.Central Circulation System: referring to a number of vessels of the blood circulation system including pulmonary artery, aorta, coronary artery, carotid artery, cerebral artery, cardiac vena, upper cavity vena, and lower cavity vena.9. Central Nervous System: referring to cerebrum, meninx and medulla spinalis.Article 9 The State Drug Administration is responsible for interpretation of the provisions.Article 10 The Provisions shall go into effect as of April 10, 2000.Criteria for Medical Devices Classification download。
IVD及医疗器械注册_2

整理课件
9
IVD首次注册申报资料
8. 生产工艺及反应体系的研究资料: a.主要生产工艺描述及确定依据( 国标\行标) b.反应体系的组成 c.被测样本的要求 d.试剂用量 e.体系的反应条件 f.体系的有效性确定方法(校准、质 控方法) g.提供各种验证资料
整理课件
10
IVD首次注册申报资料
9. 分析性能评估资料:
■ 自2014年10月1日起施行。 ■ 对IVD产品分类, 产品标准、产品研制、临床试验和注册申请、注册
检测、注册变更、重新注册等相关问题作出明确规定。
整理课件
4
IVD按照风险分类
第一类产品:
微生物培养基(不用于微生物鉴别和药 敏试验);
样本处理用产品,如溶血剂、稀释液、 染色液等。
第三类产品:
与致病性病原体抗原、抗体以及核酸等 检测相关的试剂;
与血型、组织配型相关的试剂;
与人类基因检测相关的整理试课件剂;
5
IVD按照风险分类
■ 第二类产品: 蛋白质、糖类、激素等除已明确为第三类、第一类的 产品,其他为第二类产品
■ 由国家食品药品监督管理局负责体外诊断试剂产品的分类界定。 ■ 对于根据上述分类原则尚无法界定的体外诊断试剂产品,由注册申
请人向国家食品药品监督管理局提出分类界定的申请。
整理课件
8
IVD首次注册申报资料
4. 产品说明书: 两份说明书、一致性声明
5. 产品标准及编制说明: 符合《医疗器械 注册产品标准编写规范》和《医疗器 械标准管理办法》即可
6. 检测报告
7. 原材料的研究资料: a.主要原材料的选 择、制备及质量标准 b.质控品、校准 品的原料选择、制备、定值过程及试 验 c.标准品的溯源性文件
《医疗器械临床试验质量管理规范》解读(九)完结

《医疗器械临床试验质量管理规范》解读(九)完结亲爱的读者朋友们,大家好!从2016年3月25日分享的解读 (一) 到今天的解读 (九), 奥咨达对《医疗器械临床试验质量管理规范》的连载解读已到了尾声。
感谢大家这两个多月来对这篇连载解读的持续关注,后续我们将通过其他方式为大家完整呈现此篇原创解读,敬请关注,谢谢!第九章试验用医疗器械管理第八十七条申办者应当参照国家食品药品监督管理总局有关医疗器械说明书和标签管理的规定,对试验用医疗器械作适当的标识,并标注“试验用”。
解读参考《医疗器械说明书和标签管理规定》(2014年7月30日,食品药品监督管理总局令第6号),器械设备或包装上,贴上“XX临床试验专用”标签。
按照规范的临床试验监管规定,申办方同一产品同步进行的不同临床试验,不允许相互调用。
第八十八条试验用医疗器械的记录包括生产日期、产品批号、序列号等与生产有关的记录,与产品质量和稳定性有关的检验记录,运输、维护、交付各临床试验机构使用的记录,以及试验后回收与处置日期等方面的信息。
解读此条规定要求试验用医疗器械的生产、质检、运输、使用、回收、销毁等整个过程记录的完整性及可溯性。
所有到达研究机构的产品均需进行上述记录。
除伦理审核时申办方应提供研究用器械的注册检验合格报告和自测报告外,在试验进行过程中不同生产批次的研究产品到达研究机构时均需提交该批次的质检报告,以证明临床试验中所用试验用器械的质量合格。
如果研究产品的贮存有特定要求,如低温保存。
那么研究用产品从生产企业运输到研究机构、试验过程中产品的保存等,均应保留相关温度记录。
如果温度超出规定范围,则应由生产企业根据具体数据评估产品是否可继续使用。
除试验用器械外,已上市对照产品的相关记录均应按同样要求进行记录。
第八十九条试验用医疗器械的使用由临床试验机构和研究者负责,研究者应当保证所有试验用医疗器械仅用于该临床试验的受试者,在试验期间按照要求储存和保管试验用医疗器械,在临床试验后按照国家有关规定和与申办者的协议对试验用医疗器械进行处理。
《医疗器械临床试验规定》(局令第5号)

《医疗器械临床试验规定》(局令第5号)国家食品药品监督管理局令第5号《医疗器械临床试验规定》于2003年12月22日经国家食品药品监督管理局局务会审议通过,现予发布。
本规定自2004年4月1日起施行。
局长:郑筱萸二○○四年一月十七日医疗器械临床试验规定第一章总则第一条为加强对医疗器械临床试验的管理,维护受试者权益,保证临床试验结果真实、可靠,根据《医疗器械监督管理条例》,制定本规定。
第二条医疗器械临床试验的实施及监督检查,应当依照本规定。
第三条本规定所称医疗器械临床试验是指:获得医疗器械临床试验资格的医疗机构(以下称医疗机构)对申请注册的医疗器械在正常使用条件下的安全性和有效性按照规定进行试用或验证的过程。
医疗器械临床试验的目的是评价受试产品是否具有预期的安全性和有效性。
第四条医疗器械临床试验应当遵守《世界医学大会赫尔辛基宣言》(附件1)的道德原则,公正、尊重人格、力求使受试者最大程度受益和尽可能避免伤害。
第五条医疗器械临床试验分医疗器械临床试用和医疗器械临床验证。
医疗器械临床试用是指通过临床使用来验证该医疗器械理论原理、基本结构、性能等要素能否保证安全性有效性。
医疗器械临床验证是指通过临床使用来验证该医疗器械与已上市产品的主要结构、性能等要素是否实质性等同,是否具有同样的安全性、有效性。
医疗器械临床试用的范围:市场上尚未出现过,安全性、有效性有待确认的医疗器械。
医疗器械临床验证的范围:同类产品已上市,其安全性、有效性需要进一步确认的医疗器械。
第六条医疗器械临床试验的前提条件:(一)该产品具有复核通过的注册产品标准或相应的国家、行业标准;(二)该产品具有自测报告;(三)该产品具有国务院食品药品监督管理部门会同国务院质量技术监督部门认可的检测机构出具的产品型式试验报告,且结论为合格;(四)受试产品为首次用于植入人体的医疗器械,应当具有该产品的动物试验报告;其它需要由动物试验确认产品对人体临床试验安全性的产品,也应当提交动物试验报告。
医疗器械临床试验

定义
依据2004年《医疗器械临床试验规定》(局令第5号)第三条规定:医疗器械临床试验是指:获得医疗器械 临床试验资格的医疗机构(以下称医疗机构)对申请注册的医疗器械在正常使用条件下的安全性和有效性按照规 定进行试用或验证的过程。医疗器械临床试验的目的是评价受试产品是否具有预期的安全性和有效性 。
免于进行临床试验的第三类医疗器械目录
国家食品药品监督管理总局关于发布免于进行临床试验的第三类医疗器械目录的通告(2014年第13号)
为了做好医疗器械注册管理工作,根据《医疗器械监督管理条例》(国务院令第650号)和《医疗器械注册 管理办法》(国家食品药品监督管理总局令第4号 ),国家食品药品监督管理总局组织制定了《免于进行临床试 验的第三类医疗器械目录》,现予发布,自2014年10月1日起施行 。
医疗器械临床试验
试验名称
01 定义
03 临床试验
目录
02 版本 04 豁免目录
依据2004年《医疗器械临床试验规定》(局令第5号)第三条规定:医疗器械临床试验是指:获得医疗器械 临床试验资格的医疗机构(以下称医疗机构)对申请注册的医疗器械在正常使用条件下的安全性和有效性按照规 定进行试用或验证的过程。医疗器械临床试验的目的是评价受试产品是否具有预期的安全性和有效性。
定义
体外诊断试剂的临床试验(包括与已上市产品进行的比较研究试验)是指在相应的临床环境中,对体外诊断 试剂的临床性能进行的系统性研究。
临床试验的基本原则
(一)基本要求
医疗器械通用知识考试题库含答案

医疗器械通用知识考试题库含答案1、受理第三类医疗器械产品注册申请的部门是()。
A、县级食品药品监督管理部门B、市级食品药品监督管理部门C、省级食晶药品监督管理部门D、国务院食品药品监督管理部门答案:D2、参与构成喉结的是()。
A、杓状软骨B、甲状软骨C、会厌软骨D、环状软骨答案:B甲状软骨最大,位于喉的前上方,其前方最突出的部分为喉结。
环状软骨位于甲状软骨的下方,呈环形,前低后高,分别与杓状软骨、甲状软骨形成关节,对支持呼吸道有重要作用。
会厌软骨位于甲状软骨的后上方,形状如匙,上端游离,下端借韧带连于甲状软骨后面,为喉口的盖。
3、()指单独地连同辅助设备一起用以进行测量的器具。
A、学习仪器B、辅助仪器C、测量仪器D、计量仪器答案:C4、M32LH表示()。
A、公称直径为32,旋向为左旋的普通细牙螺纹B、公称直径为32,旋向为左旋的普通粗牙螺纹C、公称直径为32,旋向为右旋的普通粗牙螺纹D、公称直径为32,旋向为右旋的普通细牙螺纹答案:B5、在医疗器械安全性评估中安全的等级分为()。
A、绝对安全、相对安全及描述安全B、相对安全、有条件安全及记述安全C、绝对安全、有条件安全及记述安全D、无条件安全、有条件安全及彻底安全答案:C6、在公式pV=nRT中,n与T分别表示气体的()。
A、摩尔数、绝对温标B、密度、摄氏温度C、单位体积的摩尔数、摄氏温度D、单位体积的摩尔数、绝对温标答案:A7、机件的内外结构均需表达,而又不对称时应采用()。
A、全剖视图B、半剖视图C、局部剖视图D、基本视图答案:C8、无菌器械生产企业的企业名称发生变更时,需经下列哪一部门批准()。
A、县级(食品)药品监督管理部门B、市级(食品)药品监督管理部门C、省级工商行政管理部门D、国务院(食品)药品监督管理部门答案:D9、颅顶骨形态上属()。
A、长骨B、短骨C、扁骨D、不规则骨答案:C骨骼分长骨、短骨、扁骨和不规则骨。
长骨主要部分在四肢;短骨主要包括腕骨、跗骨;扁骨主要包括颅顶骨、额骨、颞骨、蝶骨、枕骨、肩胛骨、胸骨、肋骨、盆骨;不规则骨主要包括椎骨、含气骨。
《医疗器械临床试验质量管理规范》

《医疗器械临床试验质量管理规范》(国家食品药品监督管理总局中华人民共和国国家卫生和计划生育委员会令第25号)2016年03月23日发布第25号《医疗器械临床试验质量管理规范》已经国家食品药品监督管理总局局务会议、国家卫生和计划生育委员会委主任会议审议通过,现予公布,自2016年6月1日起施行。
局长毕井泉主任李斌2016年3月1日医疗器械临床试验质量管理规范第一章总则第一条为加强对医疗器械临床试验的管理,维护医疗器械临床试验过程中受试者权益,保证医疗器械临床试验过程规范,结果真实、科学、可靠和可追溯,根据《医疗器械监督管理条例》,制定本规范。
第二条在中华人民共和国境内开展医疗器械临床试验,应当遵循本规范。
本规范涵盖医疗器械临床试验全过程,包括临床试验的方案设计、实施、监查、核查、检查,以及数据的采集、记录,分析总结和报告等。
第三条本规范所称医疗器械临床试验,是指在经资质认定的医疗器械临床试验机构中,对拟申请注册的医疗器械在正常使用条件下的安全性和有效性进行确认或者验证的过程。
第四条医疗器械临床试验应当遵循依法原则、伦理原则和科学原则。
第五条省级以上食品药品监督管理部门负责对医疗器械临床试验的监督管理。
卫生计生主管部门在职责范围内加强对医疗器械临床试验的管理。
食品药品监督管理部门、卫生计生主管部门应当建立医疗器械临床试验质量管理信息通报机制,加强第三类医疗器械、列入国家大型医用设备配置管理品目的医疗器械开展临床试验审批情况以及相应的临床试验监督管理数据的信息通报。
第二章临床试验前准备第六条进行医疗器械临床试验应当有充分的科学依据和明确的试验目的,并权衡对受试者和公众健康预期的受益以及风险,预期的受益应当超过可能出现的损害。
第七条临床试验前,申办者应当完成试验用医疗器械的临床前研究,包括产品设计(结构组成、工作原理和作用机理、预期用途以及适用范围、适用的技术要求)和质量检验、动物试验以及风险分析等,且结果应当能够支持该项临床试验。
医疗器械不良事件监测法规要求

•
由药品管理当局批准上市的医疗器械是否就是“绝 对”安全的?
“安全”意味着:对于用械和治疗的人群而言,只是“ 效益大于风险”,且仅是风险评价的“阶段性结论” 。一些发生率较低的长期效应或者已知风险的实 际发生频次或程度,只有在产品投入市场、大量 人群长期使用后才可能被逐渐发现或认识。
国家对医疗器械实施再评价及淘汰制度。
•
医疗器械注册管理办法
第十三条 申请第二类、第三类医疗器械注册,同时满足 以下条件的,可以免予注册检测: (四)已经获准注册的本企业同类产品按照规定进行医疗 器械不良事件监测,并且未发现严重不良事件; 第十四条 申请第二类、第三类医疗器械产品重新注册, 同时满足以下条件的,可以免予注册检测: (四)申请重新注册的医疗器械在原医疗器械注册证书有 效期内按照规定进行医疗器械不良事件监测,并且未发现 不良事件;
① 对疾病的预防、诊断、治疗、监护、缓解; ② 对损伤或者残疾的诊断、治疗、监护、缓解、补偿; ③ 对解剖或者生理过程的研究、替代、调节; ④ 妊娠控制。
----《医疗器械监督管理条例》,国务院276号令, 2000年颁布实施
•
医疗器械的管理类别
我国参照美国FDA的分类方式将医疗器械分 为三类:
• 一类:通过常规管理足以保证其安全性、 有效性的医疗器械;(低风险)
•
医疗器械注册管理办法
第三十七条 有下列情形之一的医疗器械,不予 重新注册: (二)经国家食品药品监督管理局再评价属于淘汰 品种的;
•
医疗器械生产企业监督管理办法 第三十九条 县级以上地方(食品)药品监督管 理部门应当在法律、法规、规章赋予的权限内, 建立本行政区域内医疗器械生产企业的监管档案 。监管档案应当包括医疗器械注册审批、生产许 可、生产监督检查、产品质量监督抽查、不良事 件监测、不良行为记录和投诉举报等内容。 第四十条 县级以上地方(食品)药品监督管理 部门应当将监督检查中发现的生产企业的以下行 为记入生产企业监管档案: (五)未按规定建立并有效实施质量跟踪和不良 事件监测制度的;
国家药监局、国家卫生健康委关于发布《医疗器械临床试验质量管理规范》的公告

医疗器械临床试验质量管理规范文章属性•【制定机关】国家药品监督管理局,国家卫生健康委员会•【公布日期】2022.03.24•【文号】国家药监局、国家卫生健康委令2022年第28号•【施行日期】2022.05.01•【效力等级】部门规章•【时效性】现行有效•【主题分类】医疗质量正文医疗器械临床试验质量管理规范(2022年3月24日国家药品监督管理局、国家卫生健康委员会令第28号公布自2022年5月1日起施行)第一章总则第一条为加强对医疗器械临床试验的管理,维护受试者权益和安全,保证医疗器械临床试验过程规范,结果真实、准确、完整和可追溯,根据《医疗器械监督管理条例》,制定本规范。
第二条在中华人民共和国境内,为申请医疗器械(含体外诊断试剂,下同)注册而实施的医疗器械临床试验相关活动,应当遵守本规范。
本规范涵盖医疗器械临床试验全过程,包括医疗器械临床试验的方案设计、实施、监查、稽查、检查以及数据的采集、记录、保存、分析,总结和报告等。
第三条医疗器械临床试验应当遵守《世界医学大会赫尔辛基宣言》的伦理准则和国家涉及人的生物医学研究伦理的相关规范。
参与医疗器械临床试验的各方应当按照试验中各自的职责承担相应的伦理责任。
第四条实施医疗器械临床试验应当有充分的科学依据和明确的试验目的,权衡受试者和社会预期的风险和获益。
只有当预期的获益大于风险时,方可实施或者继续实施临床试验。
第五条医疗器械临床试验应当在具备相应条件并且按照规定备案的医疗器械临床试验机构实施。
第六条医疗器械临床试验应当获得伦理委员会的同意。
列入需进行临床试验审批的第三类医疗器械目录的,还应当获得国家药品监督管理局的批准,并且在符合要求的三级甲等医疗机构实施临床试验。
第七条医疗器械临床试验的申办者应当建立覆盖医疗器械临床试验全过程的质量管理体系,确保医疗器械临床试验符合相关法律法规,保护受试者权益和安全。
第二章伦理委员会第八条伦理委员会的职责是保护受试者合法权益和安全,维护受试者尊严。
医疗器械临床试验常见问题解析

医疗器械临床试验常见问题解析近两年来,山东省食品药品监督管理局两次发布了《山东省食品药品监督管理局关于开展医疗器械临床试验监督抽查工作的通告》,通过对山东省内申办方二类医疗器械临床试验的监督抽查,发现存在临床试验过程不规范、存在个别真实性和一系列规范性问题。
其主要原因有临床试验机构办公室、伦理办公室相关人员和研究者对医疗器械临床试验质量的重要性意识薄弱,对临床试验的很多重要环节上不够重视等。
要解决这些问题,需要临床试验机构和研究者形成统一共识,重视临床试验的发展,进而探讨提出建议。
1.医疗机构及研究者存在的主要问题1.1 临床试验机构检查发现医疗机构存在问题:①所选机构不符合要求,非药物临床机构,《医疗器械临床试验质量管理规范》(局令第5 号)对医疗机构有相关要求[1];②临床试验机构无法提供临床试验用仪器设备;③机构工作人员对临床试验相关法规理解不够,对医疗器械相关的规范及检查要点理解不一致或者不到位;④医疗机构对器械临床试验缺乏不够重视,部分机构的机构办公室及伦理办公室无明确分工职责,大部分的医疗机构未建立完善的质量管理制度和标准操作规程。
1.2 临床试验人员检查发现临床试验人员存在如下不足:①临床试验人员对于自己参与的临床试验不够理解,培训不到位,对方案、职责等不清楚,本身投入临床试验的时间有限;②部分临床试验参与人员执行力不强,不能严格按照方案检验样本;③部分研究者不能正确执行知情同意,导致有知情同意书填写不完整或者不规范的现象,或者有些受试者虽然签署了知情同意书,但也违背了伦理的要求;④部分参与临床试验的人员责任感不够,无法使医疗器械临床试验达到科学化、统一化运行,也不能保证试验结果的科学性、可靠性;⑤部分研究者对不良事件、严重不良事件概念的认识以及对上报程序的不够了解,导致不能正确地反应不良事件。
2.临床试验机构和研究者存在的问题针对以上问题对医疗机构展开调研,实地调研选取省内药物临床试验机构,通过调研发现实施医疗器械临床试验的机构存在如下问题:第一,接受《医疗器械临床试验质量管理规范》及相关法律法规的培训相对较少。
医疗器械医疗器械文件目录

医疗器械文件目录一类、体系文件组织机构与职责文件编号文件名称质量手册HJ-SC-001-01 质量手册管理职责HJ-ZZ-001-01 质量方针和目标HJ-ZZ-002-01 组织机构设置及规定HJ-ZZ-003-01 质量管理体系HJ-ZZ-004-01 部门与人员职责管理制度HJ-ZD-001-01 文件控制程序HJ-ZD-002-01 记录控制程序HJ-ZD-003-01 培训管理制度HJ-ZD-004-01 管理评审控制程序HJ-ZD-005-01 设计开发和验证控制程序HJ-ZD-006-01 产品风险控制程序HJ-ZD-007-01 采购控制程序HJ-ZD-008-01 仓库管理制度HJ-ZD-009-01 原材料库管理程序HJ-ZD-010-01 成品库管理程序HJ-ZD-011-01 包装、标签、合格证管理规定HJ-ZD-012-01 检验和试验状态控制程序HJ-ZD-013-01 生产过程控制程序HJ-ZD-014-01 生产作业环境与人员卫生管理制度HJ-ZD-015-01 产品批号的管理规程HJ-ZD-016-01 产品标识和可追溯性管理制度HJ-ZD-017-01 产品防护控制程序HJ-ZD-018-01 过程和产品的监视和测量控制程序HJ-ZD-019-01 设备管理制度HJ-ZD-020-01 检验仪器设备管理制度HJ-ZD-021-01 化学试剂管理制度HJ-ZD-022-01 计量器具管理制度HJ-ZD-023-01 顾客反馈监视和测量控制程序HJ-ZD-024-01 内部审核控制程序HJ-ZD-025-01 销售管理制度HJ-ZD-026-01 产品退/换管理制度质量HJ-ZD-027-01 合同评审和顾客沟通控制程序HJ-ZD-028-01 不合格品控制程序HJ-ZD-029-01 质量事故与不良事件报告控制程序HJ-ZD-030-01 数据统计与分析控制程序HJ-ZD-031-01 产品召回制度HJ-ZD-032-01 纠正和预防措施控制程序二类、技术文件技术标准检验规程作业指导书仪器与设备操作规程三类、外来文件1、医疗器械监督管理条例(国务院令第276号)2、医疗器械生产监督管理办法(局令第12号)3、医器械生产企业质量体系考核办法(局令第22号)4、医疗器械说明书、标签和包装标识管理规定(局令第10号)5、医疗器械注册管理办法(局令第16号)6、医疗器械临床试验规定(局令第5号)7、医疗器械生产质量管理规范(试行)国食药监械[2009]833号8、医疗器械生产质量管理规范检查管理办法(试行)国食药监械[2009]834号。
二类医疗器械企业标准、注册检验、临床试验、产品注册等流程
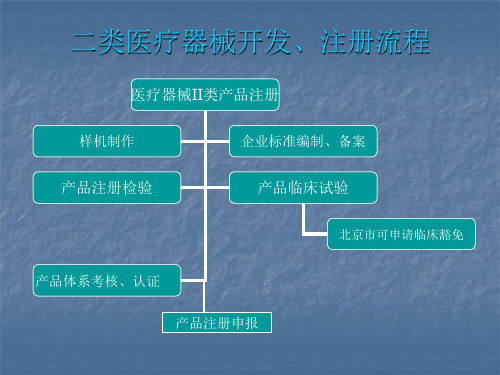
医疗器械II类产品注册
样机制作
企业标准编制、备案
产品注册检验
产品临床试验
北京市可申请临床豁免
产品体系考核、认证 产品注册申报
样机制作
根据市场需求、设计要求、各级标准要求制作样机并对样机进行评审;
1、市场需求:前期市场调查并编制《市场调研报告》、《技术方案调研报告》; 2、设计要求:按照市场需求编制《设计任务书》或《产品立项报告》,并依据
验方案、临床试验报告;
体系考核
体系考核(认证)流程 ■认证资料准备; ■联系公告机构(华光或药监局),确认审核时间、费用、合同; ■现场体系审核; ■不符合项整改(若有); ■取得证书(报告);
体系考核(认证)资料 1、设计任务书(包括项目开发方案及需求分析); 2、风险管理(分析)文档 3、产品技术报告; 4、产品自测报告; 5、使用说明书; 6、临床资料; 7、企业标准; 8、生产工艺文件:包括焊接、安装、连线、调试、软件烧写等; 9、检验规程:进货检验、(性能、安全、外观)检验、过程检验、周期检验、成品检验、出厂检验; 10、特殊过程、关键过程文档; 11、产品结构、总装、零部件图纸; 12、产品原理图、PCB图、接线图等电气图纸; 13、产品原材料、部件的采购清单或配置清单; 14、设计评审记录;(最少包括三次评审记录:设计输入、样机、最终) 15、其他文件:如车间、库房等管理文件;安全设备运行检查文件、设备维护保养文件、环境记录表
企业标准编制及备案
标准编制 依据医疗器械国家标准、行业标准、通用标准结合产品及企业自身情
况编制适用的企业标准;
企业标准备案
备案流程
●网上申报 ●资料准备 ●药监局受理申报 ●技术审评 ●获得审批后的标准
免于进行临床试验的体外诊断试剂临床评价资料基本要求(试行)

精心整理附件免于进行临床试验的体外诊断试剂临床评价资料基本要求(试行)根据《体外诊断试剂注册管理办法》(国家食品药品监督管理总局令第5号)第二十九条的规定,无需进行临床试验的体外诊断试剂,申请人(三)消费者自测用的体外诊断试剂。
二、基本要求(一)体外诊断试剂临床评价由申请人自行或委托其他机构或实验室在中国境内完成,试验过程由申请人进行管理,试验数据的真实性由申请人负责。
境外申请人可通过其在中国境内的代理人,开展相关临床评价工作。
(二)申请人可根据产品特点自行选择试验地点完成样本检测,检测地点的设施、试验设备、环境等应能够满足产品检测要求。
(三)申请人应在试验前建立合理的临床评估方案并遵照执行。
(四)实验操作人员应为专业技术人员。
(五)评价用样本应为来源于人体的样本,样本来源应可追溯。
评价记录)校准品的溯源情况、推荐的阳性判断值或参考区间等,应提供已上市产品的境内注册信息及说明书。
(二)与参考方法进行比较研究试验,考察待评价试剂与参考方法的符合率/一致性。
应选择参考实验室进行研究,参考实验室应具有中国合格评定国家认可委员会(CNAS)认可的相关检测资质。
四、试验方法试验方法的建立可参考相关方法学比对的指导原则,并重点关注以下内容:(一)样本要求1.选择涵盖预期用途和干扰因素的样本进行评价研究,充分考虑试验人群选择、疾病选择等内容,样本应能够充分评价产品临床使用的安全性、有效性。
2./,亦3.4.行符合统计学意义数量的评估。
(二)试验要点1.在试验操作的过程中应采用盲法。
待评价试剂和对比试剂/参考方法应平行操作,整个试验应有内部质量控制。
2.建议定量产品试验检测周期至少5天,定性产品试验检测周期10—20天,以客观反映实际情况。
3.扩大样本量和延长实验时间将提高试验的可靠性,申请人应选择适当的样本量进行充分的临床评价。
(三)数据收集和处理1.对于定量检测产品,应首先进行离群值观察,离群值的个数不得超过限值。
西格玛医学洞见:新法规下浅谈医疗器械临床试验

西格玛医学洞见:新法规下浅谈医疗器械临床试验讲师:南京西格玛医学一、什么叫做医疗器械?根据《医疗器械监督管理条例》739号令,医疗器械:是指直接或者间接用于人体的仪器、设备、器具、体外诊断试剂及校准物、材料以及其他类似或者相关的物品,包括所需要的计算机软件;其效用主要通过物理等方式获得,不是通过药理学、免疫学或者代谢的方式获得,或者虽然有这些方式参与但是只起辅助作用;其目的是:(一)疾病的诊断、预防、监护、治疗或者缓解;(二)损伤的诊断、监护、治疗、缓解或者功能补偿;(三)生理结构或者生理过程的检验、替代、调节或者支持;(四)生命的支持或者维持;(五)妊娠控制;(六)通过对来自人体的样本进行检查,为医疗或者诊断目的提供信息。
医疗器械种类很多,但我认为主要可以分为治疗类和监测诊断类。
二、医疗器械按照风险程度是如何分类的?按照风险程度实行分类管理。
第一类是风险程度低,实行常规管理可以保证其安全、有效的医疗器械。
第二类是具有中度风险,需要严格控制管理以保证其安全、有效的医疗器械。
第三类是具有较高风险,需要采取特别措施严格控制管理以保证其安全、有效的医疗器械。
三、我们国家对于医疗器械的备案与注册管理第一类医疗器械实行产品备案管理。
第二类、第三类医疗器械实行产品注册管理。
境内第一类医疗器械备案,备案人向设区的市级负责药品监督管理的部门提交备案资料。
境内第二类医疗器械由省、自治区、直辖市药品监督管理部门审查,批准后发给医疗器械注册证。
境内第三类医疗器械由国家药品监督管理局审查,批准后发给医疗器械注册证。
进口第一类医疗器械备案,备案人向国家药品监督管理局提交备案资料。
进口第二类、第三类医疗器械由国家药品监督管理局审查,批准后发给医疗器械注册证。
四、医疗器械临床评价医疗器械临床评价是指采用科学合理的方法对临床数据进行分析、评价,以确认医疗器械在其适用范围内的安全性、有效性的活动。
有下列情形之一的,可以免于进行临床评价:(一)工作机理明确、设计定型,生产工艺成熟,已上市的同品种医疗器械临床应用多年且无严重不良事件记录,不改变常规用途的;(二)其他通过非临床评价能够证明该医疗器械安全、有效的。
医疗器械临床试验流程

*****医疗器械临床试验管理规范根据国家食品药品监督管理局颁布的《医疗器械临床试验规定》(局令第5号)的要求,为了进一步加强对医疗器械临床试验的管理,强化对临床试验过程的监督,保证临床试验结果真实、可靠,特对本院医疗器械临床试验的流程及管理作如下操作规范:一。
医疗器械临床研究流程二、医疗器械临床研究的管理1. 试验器械的管理临床试验所需器械实行专项管理,必要时需经设备科审核批准方可进入临床科室。
医疗器械到达时,应通知临床药理基地办公室(电话-87783891)会同试验负责人、厂方负责人一起验证器械型号、拍摄实物照片,填写“*****医疗器械临床试验产品型号核对单”,并签字,备查。
试验器械应专物专用,不得挪作他用。
2. 临床试验阶段的管理进入实施阶段的临床试验,全面实行项目负责人负责制。
试验负责人应加强管理,向患者及家属充分告知相关事宜并填写临床试验知情同意书;试验过程中应及时、准确地填写观察记录表,如实记录试验结果,在病历上做相关记录.承担临床试验的科室建立试验器械使用登记本,门诊患者内容应包括姓名、住址、单位、联系电话、门诊病历号、诊断、器械名称、规格型号、序列号、使用日期、时间等;住院患者应包括姓名、住院号、联系电话、诊断、器械名称、规格型号、序列号、使用日期、时间等,承担临床试验人员应仔细核对并在登记本上双签名,登记本应由专人保管备查,试验结束后一同存档.住院患者进行的临床试验还需详细填写“*****医疗器械临床试验登记表”(附件4),并将登记表保存至住院病历最后一页,备查。
3. 不良事件的报告临床试验过程中出现不良事件的,应如实填写“*****医疗器械临床试验不良反应报告表”(附件5),及时按医院规定程序向临床药理基地办公室、药品监督管理部门、医院伦理委员会上报,并积极采取补救措施,分析不良事件原因。
发生严重不良反应的应在24小时内报告.4。
试验资料的存档管理临床试验结束后,承担试验人员应及时总结材料,按期完成临床试验报告。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
【发布单位】国家食品药品监督管理局【发布文号】国家食品药品监督管理局令第5号【发布日期】2004-01-17【生效日期】2004-04-01【失效日期】-----------【所属类别】国家法律法规【文件来源】-----------医疗器械临床试验规定(国家食品药品监督管理局令第5号)《医疗器械临床试验规定》于2003年12月22日经国家食品药品监督管理局局务会审议通过,现予发布。
本规定自2004年4月1日起施行。
局长:郑筱萸二○○四年一月十七日医疗器械临床试验规定第一章总则第一条为加强对医疗器械临床试验的管理,维护受试者权益,保证临床试验结果真实、可靠,根据《医疗器械监督管理条例》,制定本规定。
第二条医疗器械临床试验的实施及监督检查,应当依照本规定。
第三条本规定所称医疗器械临床试验是指:获得医疗器械临床试验资格的医疗机构(以下称医疗机构)对申请注册的医疗器械在正常使用条件下的安全性和有效性按照规定进行试用或验证的过程。
医疗器械临床试验的目的是评价受试产品是否具有预期的安全性和有效性。
第四条医疗器械临床试验应当遵守《世界医学大会赫尔辛基宣言》(附件1)的道德原则,公正、尊重人格、力求使受试者最大程度受益和尽可能避免伤害。
第五条医疗器械临床试验分医疗器械临床试用和医疗器械临床验证。
医疗器械临床试用是指通过临床使用来验证该医疗器械理论原理、基本结构、性能等要素能否保证安全性有效性。
医疗器械临床验证是指通过临床使用来验证该医疗器械与已上市产品的主要结构、性能等要素是否实质性等同,是否具有同样的安全性、有效性。
医疗器械临床试用的范围:市场上尚未出现过,安全性、有效性有待确认的医疗器械。
医疗器械临床验证的范围:同类产品已上市,其安全性、有效性需要进一步确认的医疗器械。
第六条医疗器械临床试验的前提条件:(一)该产品具有复核通过的注册产品标准或相应的国家、行业标准;(二)该产品具有自测报告;(三)该产品具有国务院食品药品监督管理部门会同国务院质量技术监督部门认可的检测机构出具的产品型式试验报告,且结论为合格;(四)受试产品为首次用于植入人体的医疗器械,应当具有该产品的动物试验报告;其它需要由动物试验确认产品对人体临床试验安全性的产品,也应当提交动物试验报告。
第二章受试者的权益保障第七条医疗器械临床试验不得向受试者收取费用。
第八条医疗器械临床试验负责人或其委托人应当向受试者或其法定代理人详细说明如下事项:(一)受试者自愿参加临床试验,有权在临床试验的任何阶段退出;(二)受试者的个人资料保密。
伦理委员会、(食品)药品监督管理部门、实施者可以查阅受试者的资料,但不得对外披露其内容;(三)医疗器械临床试验方案,特别是医疗器械临床试验目的、过程和期限、预期受试者可能的受益和可能产生的风险;(四)医疗器械临床试验期间,医疗机构有义务向受试者提供与该临床试验有关的信息资料;(五)因受试产品原因造成受试者损害,实施者应当给予受试者相应的补偿;有关补偿事宜应当在医疗器械临床试验合同中载明。
第九条受试者在充分了解医疗器械临床试验内容的基础上,获得《知情同意书》。
《知情同意书》除应当包括本规定第八条所列各项外,还应当包括以下内容:(一)医疗器械临床试验负责人签名及签名日期;(二)受试者或其法定代理人的签名及签名日期;(三)医疗机构在医疗器械临床试验中发现受试产品预期以外的临床影响,必须对《知情同意书》相关内容进行修改,并经受试者或其法定代理人重新签名确认。
第三章医疗器械临床试验方案第十条医疗器械临床试验方案是阐明试验目的、风险分析、总体设计、试验方法和步骤等内容的文件。
医疗器械临床试验开始前应当制定试验方案,医疗器械临床试验必须按照该试验方案进行。
第十一条医疗器械临床试验方案应当以最大限度地保障受试者权益、安全和健康为首要原则,应当由负责临床试验的医疗机构和实施者按规定的格式(附件2)共同设计制定,报伦理委员会认可后实施;若有修改,必须经伦理委员会同意。
第十二条市场上尚未出现的第三类植入体内或借用中医理论制成的医疗器械,临床试验方案应当向医疗器械技术审评机构备案。
第十三条已上市的同类医疗器械出现不良事件,或者疗效不明确的医疗器械,国家食品药品监督管理局可制订统一的临床试验方案的规定。
开展此类医疗器械的临床试验,实施者、医疗机构及临床试验人员应当执行统一的临床试验方案的规定。
第十四条医疗器械临床试验方案应当针对具体受试产品的特性,确定临床试验例数、持续时间和临床评价标准,使试验结果具有统计学意义。
医疗器械临床试用方案应当证明受试产品理论原理、基本结构、性能等要素的基本情况以及受试产品的安全性有效性。
医疗器械临床验证方案应当证明受试产品与已上市产品的主要结构、性能等要素是否实质性等同,是否具有同样的安全性、有效性。
第十五条医疗器械临床试验方案应当包括以下内容:(一)临床试验的题目;(二)临床试验的目的、背景和内容;(三)临床评价标准;(四)临床试验的风险与受益分析;(五)临床试验人员姓名、职务、职称和任职部门;(六)总体设计,包括成功或失败的可能性分析;(七)临床试验持续时间及其确定理由;(八)每病种临床试验例数及其确定理由;(九)选择对象范围、对象数量及选择的理由,必要时对照组的设置;(十)治疗性产品应当有明确的适应症或适用范围;(十一)临床性能的评价方法和统计处理方法;(十二)副作用预测及应当采取的措施;(十三)受试者《知情同意书》;(十四)各方职责。
第十六条医疗机构与实施者签署双方同意的临床试验方案,并签订临床试验合同。
第十七条医疗器械临床试验应当在两家以上(含两家)医疗机构进行。
第四章医疗器械临床试验实施者第十八条实施者负责发起、实施、组织、资助和监查临床试验。
实施者为申请注册该医疗器械产品的单位。
第十九条实施者职责:(一)依法选择医疗机构;(二)向医疗机构提供《医疗器械临床试验须知》;(三)与医疗机构共同设计、制定医疗器械临床试验方案,签署双方同意的医疗器械临床试验方案及合同;(四)向医疗机构免费提供受试产品;(五)对医疗器械临床试验人员进行培训;(六)向医疗机构提供担保;(七)发生严重副作用应当如实、及时分别向受理该医疗器械注册申请的省、自治区、直辖市(食品)药品监督管理部门和国家食品药品监督管理局报告,同时向进行该医疗器械临床试验的其他医疗机构通报;(八)实施者中止医疗器械临床试验前,应当通知医疗机构、伦理委员会和受理该医疗器械注册申请的省、自治区、直辖市(食品)药品监督管理部门和国家食品药品监督管理局,并说明理由;(九)受试产品对受试者造成损害的,实施者应当按医疗器械临床试验合同给予受试者补偿。
第二十条《医疗器械临床试验须知》应当包括以下内容:(一)受试产品原理说明、适应症、功能、预期达到的使用目的、使用要求说明、安装要求说明;(二)受试产品的技术指标;(三)国务院食品药品监督管理部门会同国务院质量技术监督部门认可的检测机构出具的受试产品型式试验报告;(四)可能产生的风险,推荐的防范及紧急处理方法;(五)可能涉及的保密问题。
第五章医疗机构及医疗器械临床试验人员第二十一条承担医疗器械临床试验的医疗机构,是指经过国务院食品药品监督管理部门会同国务院卫生行政部门认定的药品临床试验基地。
第二十二条医疗器械临床试验人员应当具备以下条件:(一)具备承担该项临床试验的专业特长、资格和能力;(二)熟悉实施者所提供的与临床试验有关的资料与文献。
第二十三条负责医疗器械临床试验的医疗机构及临床试验人员职责:(一)应当熟悉实施者提供的有关资料,并熟悉受试产品的使用;(二)与实施者共同设计、制定临床试验方案,双方签署临床试验方案及合同;(三)如实向受试者说明受试产品的详细情况,临床试验实施前,必须给受试者充分的时间考虑是否参加临床试验;(四)如实记录受试产品的副作用及不良事件,并分析原因;发生不良事件及严重副作用的,应当如实、及时分别向受理该医疗器械注册申请的省、自治区、直辖市(食品)药品监督管理部门和国家食品药品监督管理局报告;发生严重副作用,应当在二十四小时内报告;(五)在发生副作用时,临床试验人员应当及时做出临床判断,采取措施,保护受试者利益;必要时,伦理委员会有权立即中止临床试验;(六)临床试验中止的,应当通知受试者、实施者、伦理委员会和受理该医疗器械注册申请的省、自治区、直辖市(食品)药品监督管理部门和国家食品药品监督管理局,并说明理由;(七)提出临床试验报告,并对报告的正确性及可靠性负责;(八)对实施者提供的资料负有保密义务。
第二十四条负责医疗器械临床试验的医疗机构应当确定主持临床试验的专业技术人员作为临床试验负责人。
临床试验负责人应当具备主治医师以上的职称。
第六章医疗器械临床试验报告第二十五条医疗器械临床试验完成后,承担临床试验的医疗机构应当按医疗器械临床试验方案的要求和规定的格式(附件3)出具临床试验报告。
医疗器械临床试验报告应当由临床试验人员签名、注明日期,并由承担临床试验的医疗机构中的临床试验管理部门签署意见、注明日期、签章。
第二十六条医疗器械临床试验报告应当包括以下内容:(一)试验的病种、病例总数和病例的性别、年龄、分组分析,对照组的设置(必要时);(二)临床试验方法;(三)所采用的统计方法及评价方法;(四)临床评价标准;(五)临床试验结果;(六)临床试验结论;(七)临床试验中发现的不良事件和副作用及其处理情况;(八)临床试验效果分析;(九)适应症、适用范围、禁忌症和注意事项;(十)存在问题及改进建议。
第二十七条医疗器械临床试验资料应当妥善保存和管理。
医疗机构应当保存临床试验资料至试验终止后五年。
实施者应当保存临床试验资料至最后生产的产品投入使用后十年。
第七章附则第二十八条本规定由国家食品药品监督管理局负责解释。
第二十九条本规定自2004年4月1日起施行。
附件:1.世界医学大会赫尔辛基宣言2.医疗器械临床试验方案(略)3.医疗器械临床试验报告(略)。