(工艺技术)输液品种和水针品种的灭菌工艺要求问题
化学药品注射剂工艺研究的基本技术要求

放大生产阶段验证
工艺研究阶段的验证
通过对多批样品制备过程的分析,以及对制
剂中间产品及终产品质量的分析,对工艺过 程本身是否稳定,是否易于控制进行验证和 评价。
PPT文档演模板
化学药品注射剂工艺研究的基本技术 要求
工艺验证
放大生产阶段的验证
目的:考察所采用的制备工艺在规模化生
产时的可行性,对工艺是否适合工业化生 产进行验证和评价
无菌保证水平 杂质的控制水平 工业生产的可行性 临床使用的方便性
PPT文档演模板
选择最优剂型
化学药品注射剂工艺研究的基本技术 要求
剂型选择合理性和必要性
(三)无菌保证水平的具体考虑
首先要考虑被选剂型可采用的灭菌工艺的无菌 保证水平的高低,原则上首选剂型应能采用终 端灭菌工艺(F0≥8),并保证SAL≤10-6
处方研究
处方组成的考察
• 原料药的理化性质、生物学性质,为处方设
计提供依据。
理化性质:外观色泽、pH、pKa、熔点、
水分、溶解度、油/水分配系数等,以及在 固态和/或溶液状态下对光、热、湿、氧等 的稳定性情况、所含杂质情况
生物学性质(如在生理环境下的稳定性、
药代动力学性质、毒副作用及治疗窗等)
PPT文档演模板
以验证在所选用量下的安全性。
改变给药途径——充分证明在注射途径下的安全
性
PPT文档演模板
化学药品注射剂工艺研究的基本技术 要求
处方研究
处方设计
处方设计应在上述对药物和辅料有关研究
的基础上,根据具体剂型的特点及临床应用 的需要,结合相关文献及具体工作实践,先 设计几种基本合理的处方,然后结合制备工 艺研究,以制剂的外观、色泽、澄明度、 pH、含量、有关物质、细菌内毒素或热原、 不溶性微粒等为评价指标,对不同处方进行 考察。通过考察确定初步处方,并明确影响 制剂质量的关键因素。
注射剂灭菌工艺及输液剂灭菌需关注的问题

注射剂灭菌工艺及输液剂灭菌需关注的问题审评四部张玉琥高青摘要灭菌是注射剂制备工艺的重要步骤。
本文对注射剂灭菌工艺涉及的无菌保证水平、常用灭菌方法以及灭菌工艺验证等进行了简要介绍,并对输液剂灭菌需特殊关注的问题进行了讨论。
关键词注射剂;输液剂;灭菌注射剂的灭菌是关乎药品质量、保证用药安全的重要工艺步骤之一。
灭菌工艺研究中应根据药物和制剂的具体情况,选择适宜的灭菌方法,做到既杀死或除去微生物,达到灭菌目的,又保证药物的治疗作用和稳定性。
审评工作中注意到申报的部分注射剂品种其灭菌工艺存在问题,如灭菌方法选择不当、采用的灭菌温度偏低或灭菌时间偏短等,不能达到灭菌要求。
本文对注射剂灭菌工艺涉及的有关问题做简要介绍,供注册申请人选择研究制剂灭菌工艺时参考。
1 无菌保证水平和无菌检查灭菌制剂的无菌保证水平(SAL, sterility assurance level)是指灭菌后制剂中微生物的残存概率。
为保证用药安全,注射剂等要求无菌的制剂中微生物的存活概率(SAL)不得高于10-6(即每一百万注射剂终产品中存活微生物的不得多于1个)。
灭菌制剂的无菌保证水平可通过验证确定。
无菌检查是灭菌制剂质量控制的一个重要指标。
但无菌检查是抽样检查,而微生物污染属非均匀污染,由于抽样的概率问题,无菌保证水平不符合要求往往难以用无菌检查结果反映出来,除非是较严重污染的情况。
因此,灭菌制剂的无菌保证不能依靠终产品的无菌检查结果,而是取决于生产过程中采用合格的灭菌工艺、严格的GMP管理和良好的无菌保证体系。
2 注射剂常用灭菌工艺注射剂常用灭菌方法包括湿热灭菌和过滤除菌法。
2.1 湿热灭菌法系指将物品放在灭菌器内,利用高压饱和蒸汽等手段进行灭菌的方法。
蒸汽潜热大,穿透力强,容易使微生物蛋白质变性凝固,灭菌能力强,因此湿热灭菌是最有效及用途最广的灭菌方法。
注射剂制备中常用的湿热灭菌法包括热压灭菌和流通蒸汽灭菌。
2.1.1 热压灭菌法系在密闭的灭菌容器内,用压力大于常压的饱和水蒸气加热进行灭菌的方法。
氯化钠注射液灭菌工艺验证项目要求

氯化钠注射液灭菌工艺验证项目要求一、验证背景我公司申报的氯化钠注射液(10ml:90mg)灭菌工艺变更补充申请被国家局不予批准,理由是:灭菌工艺验证不全面,如未提供各取样点不同时间的温度统计数据、灭菌曲线、灭菌平台期数据等,对灭菌验证结果无法评价。
经讨论,现准备重新做氯化钠灭菌工艺变更的灭菌工艺验证,主要针对变更后的灭菌工艺即121℃15min进行验证。
二、验证对象普药车间灭菌柜性能确认和氯化钠注射液(10ml:90mg)工艺验证同步进行。
放样产品:氯化钠注射液(10ml:90mg)批量:1.9万支(灭菌柜10m满载能力为:244支/盘×36盘/车×2车/柜,共约17568支)批次:三批灭菌柜:普药车间B236-O003灭菌条件:121℃15min进行灯检、不印包三、验证项目要求1、普药车间灭菌柜性能确认同以前做的灭菌柜性能确认相同,空载、满载热分布均运行三次。
唯一不同的是此次满载样品采用氯化钠注射液(10ml:90mg)。
验证所需项目如下:1.1热分布(1)每次运行均需列出最高温度,最低温度,平均温度,F0最大值,F最小值,对应探头位置;计算出最高温度和最低温度的温差,最高温度和平均0温度的温差,最低温度和平均温度的温差。
可列表如下:(2)最后将三次运行的数据按照下表进行总结:验证内容项目结果数值位置空载热分布最高温度和位置最低温度和位置最高温度和最低温度的最大波动值/ 最高温度和平均温度的最大波动值/ 最低温度和平均温度的最大波动值/满载热分布最高温度和位置最低温度和位置最高温度和最低温度的最大波动值/最高温度和平均温度的最大波动值/ 最低温度和平均温度的最大波动值/F最小值(冷点)及位置F最大值(热点)及位置注:a、需提供取样点(冷、热点)的灭菌曲线、灭菌平台期(平衡时间+保温时间)数据;验证试验所用热电偶除需提供试验前校验结果外,还需提供试验后的比对结果。
b、平均温度是指保温阶段各点的平均温度。
水针的生产技术——灭菌与检漏

学习目标
知识目标
热原检查方法种类 水针的生产流程图
能力目标
能正确进行灭菌与检漏 操作 能对水针进行质量判断 能找出出现不合格产品 的原因, 的原因,并进行解决
适用的岗位
灭菌检漏岗位
质量检查岗位
水针料
注射剂的检漏
温度稍降 有色溶液 淹没安瓿
水淋洗 瓶洁净 灯检
注射剂的检漏
注射剂的质量判断
澄明度检查
无菌检查
注射剂的质量判断
热原检查 热原检查
装量的检查
其他
常见问题的原因及解决办法
澄明度
热原
无菌
污 严 当 处
污 热
产过
控制
污染严重 灭菌不当
污 灭菌
灭菌与检漏小结
重要概念
灭菌
重要技能
灭菌检漏操作 质量问题解决
检漏操作 灭菌 检漏操作
清场 填写记录
注射剂的灭菌
1 2 3 4
1~5ml安瓿:流通蒸汽 ~ 安瓿: 安瓿 流通蒸汽100℃、30min ℃ 10~20ml安瓿:流通蒸汽 ~ 安瓿: 安瓿 流通蒸汽100℃、45min ℃ 以油为溶剂的注射剂, 以油为溶剂的注射剂,选用干热灭菌法
灌封后一般12小时内灭菌 灌封后一般 小时内灭菌
关于中试及工艺验证要求的法规信息汇总
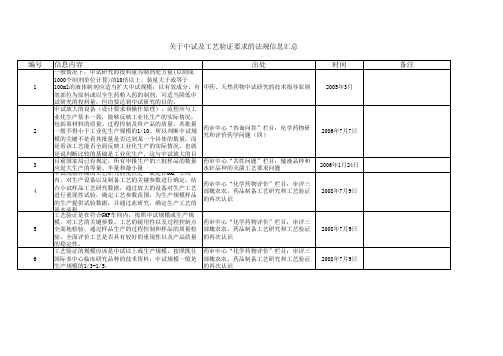
国家药监局:化学药品注射剂基本技术要 求(试行)
2008年1月10日
国食药监注[2006]265号:关于进一步加 强药品注册核查工作的通知
2006年6月26日
11
国家药监局:药品注册核查和生产现场检 查要点及判定原则 山东省药监局:药品注册申请自查自纠工 申报批准文号所需样品的试制应在本企业生产车间内进 作要求及标准附件1,药品注册现场核查 行;量按生产规模比例的大、中、小。 要点及判定标准 【关键词】药品注册管理办法,修订,工艺研究,中试放 大,工艺验证;初步阐述了工艺研究与验证各阶段的研 药审中心:审评四部,黄晓龙,《论坛》 究目的、内容与重要意义,从技术层面提出了如何通过 2008年2期总第20期 深入细致的研究工作来保证药品注册获得批准后能够按 照申报工艺组织生产。
输液品种和水针品种的灭菌工艺要求问题2006年1月24日中试规模样品的工艺研究的重点之一就是在gmp车间内对生产设备以及制备工艺的关键参数进行确定结合小试样品工艺研究数据通过放大的设备对生产工艺进行重现性试验确定工艺参数范围为生产规模样品的生产提供试验数据并通过此研究确定生产工艺的基本流程药审中心化学药物评价栏目
ห้องสมุดไป่ตู้
2008年5月23日 2006年11月21日 此项为核查的否决项。
2008年6月5日
该刊物应该涉及有中试放大、工 艺验证内容,但未看到全文。
12
13
14
15
中试研究是对实验室工艺合理性研究的验证与完善,是 保证制剂【制法】达到生产可操作性的必经环节。供质 量标准、稳定性、药理与毒理、临床研究用样品应是经 中试研究的成熟工艺制备的产品。中试规模应为制剂处 方量的10倍以上。中试过程中应考察工艺、设备及其性 能的适应性,加强制备工艺关键技术参数考核,修订、 完善适合生产的制备工艺。应提供至少三批中试生产数 据,包括投料量、半成品量、质量指标、辅料用量、成 品量及成品率等。提供制剂通则要求的一般质量检查、 微生物限度检查和含量测定结果。 仿制药进行质量研究和临床试验应采用中试以上规模的 样品。为了使进行质量研究和临床试验的样品具有代表 性,可以代表将来上市产品的质量,仿制药在临床前就 应对工艺放大进行充分的研究和初步的验证,以确保上 市产品与临床批样品质量一致。 中试研究是指在实验室完成系列工艺研究后,采用与大 生产基本相符的条件进行工艺放大研究的过程。中试研 究是小型生产模拟试验。 稳定性试验药物制剂的供试品应是放大试验的产品(如 片剂在10000片左右),其处方与生产工艺应与大生产 一致。
注射剂无菌保证工艺研究与验证常见技术问题

注射剂无菌保证工艺研究与验证常见技术问题1. 引言在医疗领域中,注射剂作为常见的药物给药方式之一,在临床应用中扮演着重要的角色。
为保证注射剂的使用安全和有效性,无菌保证工艺的研究与验证成为关键的环节。
本文将介绍注射剂无菌保证工艺研究与验证中常见的技术问题。
2. 注射剂无菌保证工艺研究2.1 微生物检测方法为了保证注射剂的无菌性,常采用微生物检测方法。
常见的微生物检测方法包括滴定法、膜过滤法和厌氧检测法等。
滴定法是一种快速而经济高效的微生物检测方法,通过将样品与营养培养基混合,观察并计数生长的微生物菌落来确定样品的微生物含量。
膜过滤法则是通过将样品通过滤膜,将微生物截留在滤膜上,然后将滤膜放置在富含营养物的培养基上进行培养并计数。
厌氧检测法主要用于检测厌氧菌和厌氧情况下的微生物污染。
2.2 热灭菌和灭菌验证热灭菌是注射剂无菌保证工艺中常用的一种控制微生物污染的方法。
热灭菌通过高温高压的条件来杀灭微生物,常用的方法包括蒸汽灭菌和干热灭菌。
灭菌验证是验证灭菌过程是否能够有效地杀灭微生物的过程。
常用的验证方法有生物指示器法和物理化学指标法。
生物指示器法是将含有特定微生物的指示器放置在被灭菌物品中进行灭菌,然后观察是否生长。
物理化学指标法则是通过监测灭菌过程中的温度、压力和湿度等指标来验证灭菌的效果。
3. 注射剂无菌保证工艺验证常见问题3.1 多次灭菌对药物质量的影响在注射剂的生产过程中,可能需要进行多次的灭菌操作,比如药物的配制、容器的灌装等。
然而,多次灭菌操作可能对药物的质量产生不良影响,比如药物的降解、颗粒形成、药效丧失等。
因此,在注射剂无菌保证工艺验证中,需要对多次灭菌操作进行评估和优化。
3.2 瓶封焊接质量的控制在注射剂的生产中,瓶封焊接是防止外界微生物污染的重要环节。
然而,瓶封焊接的质量可能会影响到注射剂的无菌性。
常见的焊接问题包括焊接不牢固、焊接不均匀等。
因此,瓶封焊接质量的控制是注射剂无菌保证工艺验证中需要注意的问题之一。
注射剂灭菌和灭菌工艺研究及验证指导原则

注射剂灭菌和灭菌工艺研究及验证指导原则引言:注射剂的灭菌过程是确保药品质量和安全的重要环节。
灭菌工艺的研究和验证对于保证注射剂的无菌性至关重要。
本文旨在探讨注射剂灭菌和灭菌工艺的研究以及验证指导原则,以提供指导和参考。
一、注射剂灭菌工艺研究1. 灭菌方法的选择:在注射剂灭菌工艺研究中,首先需要选择合适的灭菌方法。
常见的灭菌方法包括热灭菌、辐射灭菌和化学灭菌等。
根据不同的药品特性和使用要求,选择适当的灭菌方法,确保药品的无菌性。
2. 灭菌工艺参数的确定:灭菌工艺参数的确定是灭菌工艺研究的关键环节。
包括灭菌温度、灭菌时间、灭菌压力等参数的确定。
通过实验研究和数据分析,得出最佳的灭菌工艺参数,以确保药品在灭菌过程中的无菌性。
3. 灭菌效果的评价:灭菌效果的评价是灭菌工艺研究的重要内容。
通过微生物学方法对灭菌后的样品进行检测,评估灭菌效果。
常用的评价指标包括菌落总数、大肠杆菌和金黄色葡萄球菌等。
评价结果的合格性将直接影响灭菌工艺的验证结果。
二、灭菌工艺验证指导原则1. 灭菌工艺验证的目的:灭菌工艺验证的目的是验证所设计的灭菌工艺是否能够确保注射剂的无菌性。
通过严格的验证过程,评估灭菌工艺的有效性和可靠性,为生产实践提供科学依据。
2. 验证样品的选择:灭菌工艺验证需要选择具有代表性的样品进行验证。
样品的选择应考虑到药品的不同特性和使用要求,确保验证结果具有可靠性和可行性。
3. 验证方法的确定:灭菌工艺验证的方法应结合实际情况进行选择。
常用的验证方法包括生物指示物法、物化指标法和微生物学检测法等。
根据不同的药品特性和验证要求,选择适合的验证方法。
4. 验证参数的确定:灭菌工艺验证需要确定合适的验证参数。
包括灭菌温度、灭菌时间和灭菌压力等参数的确定。
通过合理的参数选择,验证灭菌工艺的可行性和有效性。
5. 验证结果的评估:灭菌工艺验证的结果需要进行评估。
根据验证结果,评估灭菌工艺的合格性和可行性。
如果验证结果符合要求,说明灭菌工艺设计合理;如果验证结果不符合要求,需要进行工艺调整和改进。
注射剂灭菌和灭菌工艺研究及验证指导原则

注射剂灭菌和灭菌工艺研究及验证指导原则引言:注射剂灭菌是制药行业中至关重要的步骤之一,其目的是确保注射剂产品在使用过程中不会引入任何微生物污染。
灭菌工艺的研究和验证是保证注射剂质量和安全性的关键环节。
本文旨在探讨注射剂灭菌和灭菌工艺的研究方法和验证原则,以指导相关工作的进行。
一、灭菌工艺研究灭菌工艺研究是确定注射剂灭菌工艺的基础,其主要包括以下几个方面:1.1 灭菌方法的选择:常见的注射剂灭菌方法包括高温灭菌、化学灭菌和辐射灭菌等。
在选择灭菌方法时,需要考虑到注射剂的特性、产品稳定性和灭菌效果等因素。
1.2 灭菌参数的确定:灭菌参数包括温度、时间、湿度等。
通过研究不同参数对灭菌效果的影响,确定最佳的灭菌参数组合,以保证注射剂的灭菌效果和产品质量。
1.3 灭菌容器的选择:灭菌容器的选择对于灭菌工艺的成功实施至关重要。
需要考虑容器的材质、密封性和透气性等因素,以确保容器不会对灭菌工艺产生干扰。
二、灭菌工艺验证灭菌工艺验证是验证灭菌工艺的有效性和可靠性,确保注射剂产品在实际应用中达到预期的灭菌效果。
其主要包括以下几个方面:2.1 合适的验证方法选择:根据注射剂产品的特性和灭菌工艺的要求,选择适合的验证方法。
常用的方法有生物指示物法、物理法和化学法等。
2.2 灭菌效力的验证:通过验证实验,确定灭菌工艺对于不同种类的微生物的灭菌效力,包括细菌、真菌和孢子等。
2.3 灭菌工艺监控:建立灭菌工艺的监控系统,定期对灭菌工艺进行监测和评估,以确保其持续有效。
2.4 灭菌工艺变更的验证:如果需要对灭菌工艺进行变更,需要进行相应的验证实验,以确保变更后的灭菌工艺仍然能够有效灭菌。
三、验证指导原则在注射剂灭菌和灭菌工艺的研究和验证过程中,需要遵循以下指导原则:3.1 合规性:灭菌和灭菌工艺的研究和验证需要符合国家和行业相关法规和标准的要求,确保产品的合规性和安全性。
3.2 综合性:灭菌和灭菌工艺的研究和验证需要综合考虑注射剂产品的特性、灭菌方法和灭菌参数等多个因素,以确保验证结果的可靠性和适用性。
(工艺技术)注射剂无菌保证工艺研究与验证常见技术问题

注射剂无菌保证工艺研究与验证常见技术问题[注:以下均为2008年度第一期讲习班(注射剂无菌保证工艺研究与验证技术要求)参会代表所提问题,本期讲习班讲习组根据目前的有关技术要求,经过认真梳理、分析、总结后,现予以发布,并就相关问题进行讨论与交流。
]1、按照欧盟决策树的要求,不能达到121℃,15分钟灭菌,可选择F0≥8的残存概率法。
请问,若产品能达到121℃,12分钟灭菌,是否就不能选择121℃,10分钟,同样,能达到10分钟,就不能选择8分钟,都是F0≥8的情况。
答:从微生物杀灭的数学模型可知,在初始污染相同的情况下,灭菌F0值越大,无菌保证水平越高。
因此,显然为降低产品残留微生物的风险,尽量选择高的F0值是顺理成章的。
2、在产品质量稳定的条件下,均能满足121℃,8分钟和115℃,30分钟,哪个条件应该优先选择呢?答:不考虑产品理化质量稳定性,理论上这两种条件达到的F0值几乎相等,无所谓优选哪个。
但实际生产中,还要考虑灭菌器内产品中热穿透的情况,灭菌器内不同部位的产品实际获得的F0值的差异,不同灭菌批次间产品的F0的差异等。
应该选择热分布差异小,产品F0值差异较小的灭菌工艺。
2℃,灭菌30分钟”,这种表示法是否规范?±3、申报资料中的灭菌条件为“101℃40min。
⨯15min或116℃⨯2℃,灭菌30分钟”几乎不能计算F0值。
灭菌条件的表示可以参照中国药典2005年版二部附录168灭菌法,121℃±2℃,灭菌30分钟”本身不能称为终端灭菌,因“101℃±2℃,灭菌30分钟”是否规范,“101℃±答:暂不说灭菌条件为“101℃4、同品种10ml、20ml注射剂,采取相同的灭菌方式是否合适?答:同品种10ml、20ml注射剂,可以采取相同的灭菌方式,但应进行热穿透试验,考察不同体积样品的热穿透是否有一致,同时考虑采用的灭菌方式应能保证大体积产品的无菌保证水平。
注射剂灭菌工艺研究存在的常见问题及几点思考

[作者简介] 陈晓莉,执业药师,副教授,主要从事药品认证管理工作。
联系电话:(010)67102284-1002,E 2mail:yji1955@ 。
・新药申报与审评技术・注射剂灭菌工艺研究存在的常见问题及几点思考陈晓莉,赵慧玲(1国家食品药品监督管理局药品认证中心,北京100061;2国家食品药品监督管理局药品审评中心,北京100038) [摘要] 总结了注射剂灭菌工艺研究中存在的常见问题,如处方研究前未了解药物的稳定特性,忽视了附加剂和灭菌工艺的筛选、无菌保证值和F 0值的考察以及工艺验证研究等,并针对上述问题提出了保证注射剂安全性和稳定性的若干建议。
[关键词] 注射剂;无菌保证值;F 0值;灭菌工艺;灭菌工艺验证[中图分类号]R944.11 [文献标识码]C [文章编号]1003-3734(2009)09-0780-04Co mm on problem s and rel a ted con si dera ti on s ofster ili za ti on process of i n jecti onCHEN Xiao 2li,Z HAO Hui 2ling(1Cen ter for Certification,S tate Food and D rug A dm inistration,B eijing 100061,China;2Center for D rug Eva lua tion,S tate Food and D rug A dm inistration,B eijing 100038,China )[Abstract] This article summarized the common p r oble m s occurring in sterilizati on p r ocess of injecti on p r oducts,such as lacking sufficient knowledge of drug stability,ignoring screening of exci p ients and sterilizati on p r ocess,neglecting sterilizing ensuring level and F 0study and p r ocess validati on .It als o p r ovided s ome considera 2ti ons for ensuring the safety and stability of injecti on p r oducts .[Key words] injecti on;sterilizing ensuring level;F 0value;sterilizati on p r ocess;sterilizati on p r ocess vali 2dati on 在药品研究申报资料中,部分注射剂研究由于忽视了药物结构和稳定性、无菌保证值以及灭菌工艺的筛选和优化,从而可能会影响其最终产品的质量。
输液针灭菌工艺守则

1、现场不得有明火,配电设备及易产生火源的设备,应远离操作现场。
2、加药时,药量应准确。
3、加药时,打开钢瓶阀门时,要慢慢操作,确保进入灭菌柜的环氧乙烷全部气化。
4、勿使环氧乙烷出气口朝向人的面部,如不慎皮肤粘膜或眼睛沾上环氧乙烷液体,应立即用水冲洗15分钟,防止灼伤,并作好个人防护。
5、扑灭环氧乙烷小面积着火,可以用CO2、干粉、卤素灭火机,也可以用水扑灭。
编制人(日期)
审核人(日期)
批准人(日期)
灭菌通用工艺守则
文件代号
共2页
第2页
六、操作人员的责任
1、对灭菌产品的批号、规格、数量要准确,对灭菌质量负责。
2、做好灭菌记录,签全名字,对所填写的原始记录真实性、正确性负责。
3、对违章作业造成的设备事故负责。
4、有权拒绝执行更改灭菌条件的口头指令。
编制人(日期)
审核人(日期)
批准人(日期)
三、操作环境的清理
1、应严格区分待灭菌产品和已灭菌产品,不得混放在一起。
2、操作环境不能堆放杂物,余钢瓶应存放在通风阴暗的专用房间内。
3、操作场所应有醒目的严禁烟火警告牌,并备有灭火器材,做好防火防爆。
四、原材料质量要求
1、灭菌药为纯环氧乙烷。
2、当更换其它厂家灭菌药时,应对灭菌效果用生物指示片予以验证。
灭菌通用工艺守则
文件代号
共2页
第1页
一、操作前的准备
1、确认环氧乙烷是否符合要求,并
1、灭菌工艺参数应事先设定,严格按灭菌操作规程进行。
2、炉门导轨及门封条每炉涂20#机械油润滑。
3、加药及出炉时,应注意通风,防止环氧乙烷中毒。
4、若发现人员中毒,其轻度症状为头晕或呕吐,应将中毒人员移至空气新鲜处即可,重度中毒者送医院治疗。
05灭菌注射用水工艺规程分则

主题内容与适用范围1. 本标准在小容量注射剂工艺规程通则的基础上,规定了灭菌注射用水的处方与依据、原辅材料、中间产品、成品质量标准、半成品的检验方法和控制、原辅材料的消耗定额等内容。
2. 本标准适用于灭菌注射用水的生产质量控制及检查。
产品名称、剂型、规格1. 产品名称:灭菌注射用水1.1. 通用名称:灭菌注射用水1.2. 汉语拼音:Miejun Zhusheyong Shui1.3. 英文名:Sterile Water for Injection2. 剂型:注射剂3. 规格:1ml 2ml4. 性状:本品为无色的澄明液体,无臭,无味。
产品处方和依据1. 处方:注射用水适量全量1000ml2. 依据:中国药典2005年版二部3. 批准文号:(1)1ml国药准字H37023022 (2)2ml国药准字H370230234. 滤器要求:除炭过滤(5μm钛棒),粗滤(5μm钛棒),保证过滤(0.45μm聚砜),精滤(0.22μm聚砜).5. 装量限度:每个容器装量不得低于标示装量。
6. 灭菌要求:灭菌程序100±1℃30分钟。
7. 贮藏:密闭保存。
8. 有效期:3年。
成品和半成品质量标准1. 半成品质量标准:标准依据:中国药典2005年版二部标准来源:中国药典2005年版二部4. 成品质量标准原辅材料的消耗定额物料平衡计算方法1. 配制工序:实际配制量滤器储存转交灌封药液数量+×100%2. 灌封工序:()()%100/⨯÷+支配制转交量支不合格品数支灌封合格品数ml3. 灯检工序:%100⨯+灭菌转交数不合格品数灯检合格品数4. 印字包装工序:%100⨯+灯检转交数破损数包装完成数附录1. 常用法定计量单位:1.1. 容量:ml 表示:1L=1000ml 1升=1000毫升 1.2. 重量:g 表示:1kg=1000g 1公斤=1000克1g=1000mg 1公斤=1000克1.3. 压力:Pa(帕) Kpa (千帕) MPa (兆帕)注:1atm (标准大气压)=760mmHg 柱=101.3kPa 1mmHg=133.322Pa 0.1MPa=1kg/cm 2 1Pa=7.5×10-3mmHg 1atm=1.031g/cm 21.4. 标准溶液浓度:mol/L (摩尔/升) 1.5. 温度:“℃”(摄氏度)1.6. 百分比“%”纯度百分比系指重量的比例(g/g )溶液的百分比系指100ml 溶液含溶质的克数(g/ml )。
注射剂灭菌和灭菌工艺研究及验证指导原则

注射剂灭菌和灭菌工艺研究及验证指导原则
注射剂是一种常见的药物剂型,其灭菌是保证药品质量和安全的重要
环节。
注射剂灭菌工艺研究及验证指导原则是指在注射剂生产过程中,对灭菌工艺进行研究和验证,以确保注射剂的灭菌效果符合要求,从
而保证药品的质量和安全。
注射剂灭菌工艺研究及验证指导原则包括以下几个方面:
一、灭菌工艺研究
灭菌工艺研究是指对注射剂灭菌工艺进行研究,以确定最佳的灭菌工
艺参数。
灭菌工艺参数包括灭菌温度、灭菌时间、灭菌压力、灭菌介
质等。
在灭菌工艺研究中,需要进行灭菌效果试验,以确定最佳的灭
菌工艺参数。
二、灭菌工艺验证
灭菌工艺验证是指对注射剂灭菌工艺进行验证,以确保灭菌效果符合
要求。
灭菌工艺验证需要进行灭菌效果试验和生物指示剂试验。
灭菌
效果试验是指对注射剂进行灭菌处理后,检测灭菌效果的试验。
生物
指示剂试验是指使用生物指示剂对注射剂进行灭菌处理,以检测灭菌
效果的试验。
三、灭菌工艺指导原则
灭菌工艺指导原则是指对注射剂灭菌工艺进行指导的原则。
灭菌工艺指导原则包括以下几个方面:
1. 灭菌工艺应符合国家相关标准和规定。
2. 灭菌工艺应根据注射剂的特点进行研究和验证。
3. 灭菌工艺应确保灭菌效果符合要求。
4. 灭菌工艺应定期进行验证,以确保灭菌效果符合要求。
总之,注射剂灭菌工艺研究及验证指导原则是保证注射剂质量和安全的重要环节。
在注射剂生产过程中,需要严格按照灭菌工艺研究及验证指导原则进行操作,以确保注射剂的灭菌效果符合要求,从而保证药品的质量和安全。
药学专业知识:输液的一般生产工艺及质量检查

药学专业知识: 输液的一般生产工艺及质量检查今天我们总结输液的一般生产工艺及质量检查, 具体内容包括:(一)输液车间的要求(净化要求)1.一般洗涤、配液罐封、室内洁净度为10000级, 温度为18℃~28℃, 相对湿度为50%~65%, 室内正压﹥4.9Pa。
2.洗瓶机、传送机、罐封机、盖膜、盖胶塞等关键部分, 采用局部层流净化。
洁净度要求10000级或100级。
(二)输液瓶的质量要求和清洁处理1.输液瓶的质量要求(1)光滑圆整、大小合适、瓶口内径必须符合要求;(2)输液瓶应采用硬质中性玻璃, 理化性质稳定符合国家标准;(3)塑料输液瓶应耐水、耐腐蚀, 无毒、质轻、耐热性好、机械强度高、化学稳定性好、可热压灭菌。
2.输液瓶的清洁处理通常有直接水洗、酸洗、碱洗等方法, 一般认为用重铬酸钾清洁液洗涤效果较好。
3.胶塞与隔膜的质量要求与清洁处理(1)胶塞的质量要求:①富于弹性与柔软性;②针头刺入与拔出后应立即闭合;③具有耐溶性, 不增加药液的杂质;④可耐受高温灭菌;⑤有高度化学稳定性;⑥对药液中药物或附加剂的吸附作用达最低程度;⑦无毒性无溶血作用。
(2)涤纶膜的特点:①对电解质无通透性, 理化性质稳定, 用稀酸或水洗无脱落物;②耐热性好, 并有一定的机械强度, 灭菌后不易破碎。
(3)涤纶膜的处理:逐张分散, 用药用乙醇浸泡或放入蒸馏水中于112℃~115℃或煮沸30分钟。
再用滤清的注射用水动态飘洗备用。
(三)输液的配制1.注射用水必须新鲜、无热原, pH、铝盐符合要求;2.原料应选用优质的注射用原料;3.输液可采用0.01%~0.5%针用活性炭吸附热原、色素及其他杂质, 并有助滤作用。
(四)输液的滤过滤过装置与注剂基本相同。
精滤目前多采用微孔滤膜。
(五)输液的灌封由药液灌注、加膜、盖橡胶塞和轧铝盖四步组成, 严格控制室内洁净度等。
(六)输液的灭菌1.及时灭菌:一般从配制药液至灭菌在4h内完成;2.F0大于8分, 常用12分钟。
输液品种和水针品种的灭菌工艺要求问题

输液品种和水针品种的灭菌工艺要求问题编者按:为进一步促进药品技术审评工作的开放性,进一步有效整合社会资源,搭建与业内同道交流、讨论问题的互动平台,我中心于2006年4月在网站上公布了第一批与申办人共同研究的“共性问题”。
第一批发布的“共性问题”有:1、输液品种和水针品种的灭菌工艺的问题;2、含绿原酸中药注射剂的安全性问题;3、透皮吸收促进剂在皮肤外用制剂中的问题。
这些问题分别来自于中药、化药审评中在某一领域具有一定普遍性或某一专业现阶段较为突出存在的问题。
第一批共性问题发布后,陆续收到了来自研发一线的研究者、生产者提出的诸多回馈意见和建议。
我中心有关部门认真研究了大家的回馈意见,并再次进行了文献调研,在此基础上结合审评实际,形成第一批“共性问题”阶段性研究总结。
有些阶段总结是基于问题中的重点关注环节进行总结的,有些是基于现阶段对此问题的认识进行总结的,无论何种总结方式其目的旨在与广大业内同道进行更加深入的讨论交流,分享我们对此问题的认识。
需要说明是,日前所形成的阶段性研究结果,仅代表中心现阶段对该问题的认识,并非定论性研究成果,亦非技术要求。
以网站这一平台同步公布与申办人共同研究的“共性问题”是一种新探索。
第二批共性问题现已在中心网站发布。
今后中心还将就审评工作中发现的一些普遍性问题与申办人以多种有效的形式加以研究,竭诚希望广大从事药品研发和注册的业内同道,积极参与上述讨论,提出您对这些问题的意见和建议。
让我们大家共同努力,为推动我国药品研究与评价,为保证公众用药安全有效,促进医药事业健康协调发展,做出我们应有的贡献!一、对反馈信息的整理和分析(一)共收到反馈信息23条,具体内容总结如下:问题1:大输液和小水针品种确定灭菌工艺的基本原则是什么?反馈信息中提到的原则包括:保证产品质量的稳定,保证产品无菌和细菌内毒素符合要求,选择效果显著的灭菌方式,选择具有现实性、可验证性的灭菌工艺。
问题2:对于不同的灭菌工艺,是否都进行灭菌工艺的验证?是否针对每个不同品种都需进行验证?验证的侧重点(工艺条件、生产设备等)有何不同?是在申报前还是获得生产批文后再进行验证?是在生产规模还是中试规模上进行验证?反馈信息中基本所有的信息均认为不同的工艺均需要进行验证,还有的提到发生设备变更、系统改造、灭菌条件(包括设备、包装容器、容器位置、灭菌参数等)改变等情况都需要进行验证。
阐述注射水针剂生产车间工艺设计问题

阐述注射水针剂生产车间工艺设计问题伴随着科学技术的飞速发展,医学科学技术日新月异,新的医疗器械和药品不断涌现。
注射水针剂作为常用的急救药品,是目前临床治疗中使用最多、应用最广的剂型。
药品是人类健康得以保障基础,药品的生产、经营和使用单位对药品管理以及药品质量优劣都直接关系着人民用药的有效性和安全性,因此就需要我们生产和经营中完善各项管理制度,并不断完善生产工艺。
一、注射水针剂概念注射水针剂是指水溶性注射剂,针剂是由溶液型针剂、注射用灭菌粉末、混悬剂和乳剂四种形式构成的,注射水针剂也是针剂中采用最为广泛,最具有代表性的一种注射剂。
注射水针剂生产是通过采用注射用水为媒介和溶解物,将药物进行溶解,注入安瓿瓶内生产出来的,因此这一过程也被称之为注射剂生产流程。
注射水针剂的生产工艺主要可以分为灭菌工艺和无菌工艺两种。
在目前我国的注射水针剂生产之中采用最为广泛的为灭菌生产工艺,其主要生方法是通过将已经灌封的半成品采用高壓蒸汽或者水浴式方法灭菌,进而达到成品无菌要求和确保药品的使用质量要求。
二、注射水针剂生产在目前的注射水针剂生产中,对车间管理要严格遵守《药品生产质量管理规范》,该规范是目前制药企业和单位的工作依据,也是确保药品质量的基础。
在药品生产中,采用《药品生产质量管理规范》对于指导医药工业厂房设计有着重要作用和意义,在生产车间设计中除了考虑到车间大小、各生产要素流动的合理安排之外,还要对房间布局、内部结构和生产流程进行全面考虑与处理,分析相关的生产流程和设计要求,进而提出合理有效的管理控制策略。
1、工艺流程简述注射水针剂的原辅料经称量配料后,加入浓配罐,与注射用水充分搅拌,辅料活性炭将吸附脱去杂质与色素,药液由卫生泵输送,经钛棒过滤器过滤进入稀配罐,再往稀配罐加入注射用水,将药液稀释至工艺所需浓度,由卫生泵输送经钛棒过滤器二次过滤送灌装岗位。
安瓿瓶经超声波清洗、多针水汽交替清洗、热层流灭菌、层流净化,经多针灌装稀配药液后,拉丝封口,再经水浴式灭菌柜灭菌、灯检合格后外包入库。
输液工艺规程通则

目录1.输液工艺流程图及环境区域划分2.输液质量控制要点3.常用原辅料质量标准4.内包材质量标准5.操作过程及工艺条件6.管道器具清洗7.设备一览表及主要设备生产能力8.包装要求,标签、说明书管理与存放9.技术经济指标计算及物料衡算10.生产注意事项1.输液工艺流程图及环境区域划分1.1输液一线工艺流程图及环境区域划分1.2输液二线工艺流程图及环境区域划分2. 输液质量控制要点3.常用辅料质量标准3.1 注射用水质量标准依据Ch•P2000版二部。
3.2 药用炭质量标准依据Ch•P2000版二部。
3.3 氢氧化钠质量标准依据Ch•P2000版二部。
3.4 盐酸质量标准依据Ch•P2000版二部。
4. 内包材质量标准及检验操作程序4.1 输液瓶质量标准依据GB2639-90、GB12425、GB12416-1。
4.2 用涤纶薄膜质量标准4.3 输液用胶塞质量标准依据GB9890-88、GB3512。
4.4 输液用铝盖质量标准依据GB5197-85。
5. 操作过程及工艺条件5.1 配料本工序包括备料、称量、复核、配制、检验、粗滤、精滤等过程。
5.1.1 备料5.1.1.1 按生产指令限额备料。
5.1.1.2 核对品名、批号、生产厂家、规格及数量,应与物料交接单相符。
5.1.1.3核对无误后存放于备料室。
5.1.2称量5.1.2.1天平、磅称应在校验周期内使用,使用前进行校正,称量时专人唱读复核做好记录。
5.1.2.2 原料及附加剂启封前、称量前后应严格核对名称、含量、数量。
原装药品不得以标签量为准。
5.1.2.3 投料计算5.1.2.3.1原料不纯时折合投料的计算公式:原料理论用量(g)×中间产品标示量%原料实际用量(g)=实测原料含量%×(1-干燥失重)%注:原料理论用量(g)=实际配液数(ml)×单位容积处方规定量(g/ml)5.1.2.3.2 含有结晶水或纯盐基类药物投料的计算公式:实际原料的分子量中间产品标示量% 原料实际用量(g)=×原料理论用量(g)×处方规定的分子量实测原料含量%5.1.2.3.3 按效价单位抗生素的投料计算公式:抗生素的理论用量总单位数(μ)×中间产品标示量% 原料实际用量(g)=原料效价(μ/mg)×10005.1.2.4 剩余的原辅料应封口贮存,在容器外注明品名、日期、剩余量及使用人签名。
注射剂无菌保证工艺研究及评判的原那么要求

注射剂无菌保证工艺研究及评判的原那么要求注射剂无菌保证工艺是指为实现规定的无菌保证水平所采取的通过充分验证后的灭菌(无菌)生产工艺。
目前,注射剂的无菌保证工艺要紧有两种:1.终端灭菌工艺:在操纵微生物污染量的基础上,在药品灌封后,通过湿热灭菌方式除菌。
一样来讲,本方式本钱低,无菌保证水平高,适宜于大容量注射剂和小容量注射剂的灭菌。
2.无菌生产工艺:在无菌系统环境下,通过除菌过滤法或无菌操作法,以避免污染为目的,排除致使污染的各类可能性来保证无菌水平。
一样来讲,由于本方式对环境系统的要求高,且阻碍无菌操作的因素多而使得无菌保证水平比终端灭菌工艺低。
无菌生产工艺一样适宜于粉针剂,亦可适宜于临床需要但不能进行终端灭菌的小容量注射剂。
由此,终端灭菌工艺和无菌生产工艺具有不同的系统要求、不同的除菌方式和不同的无菌保证结果。
评判无菌保证工艺是不是有效曾一度要紧通过对终产品抽样进行无菌查验来判定;由于微生物在产品中的散布是不均匀的,且抽检样品的数量有限,故抽检的结果不能真实代表整批产品的无菌状态。
国际上更为注重无菌保证工艺的设计是不是合理、所用的设备与工艺是不是通过充分的验证,在此基础上,切实依照验证后的工艺进行生产,如此才能保证灭菌(无菌)工艺的靠得住性。
在业界,经常使用“无菌保证水平”(Sterility AssuranceLevel,SAL)概念来评判灭菌(无菌)工艺的成效,SAL的概念为产品经灭菌/除菌后微生物残余的概率。
该值越小,说明产品中微生物存在的概率越小。
为了保证注射剂的无菌平安性,国际上一致规定,采纳湿热灭菌法的SAL不得大于10-6,即灭菌后微生物存活的概率不得大于百万分之一;而采纳无菌生产工艺的产品,其SAL一样只能达到10-3,故仅限于临床必需注射给药而确实无法耐受终端灭菌的产品。
无菌生产工艺只适用于粉针剂或部份小容量注射剂。
一、注射剂剂型选择的原那么注射剂包括大容量注射剂(50ml以上)、小容量注射剂(20ml以下)和粉针剂三种剂型。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
输液品种和水针品种的灭菌工艺要求问题编者按:为进一步促进药品技术审评工作的开放性,进一步有效整合社会资源,搭建与业内同道交流、讨论问题的互动平台,我中心于2006年4月在网站上公布了第一批与申办人共同研究的“共性问题”。
第一批发布的“共性问题”有:1、输液品种和水针品种的灭菌工艺的问题;2、含绿原酸中药注射剂的安全性问题;3、透皮吸收促进剂在皮肤外用制剂中的问题。
这些问题分别来自于中药、化药审评中在某一领域具有一定普遍性或某一专业现阶段较为突出存在的问题。
第一批共性问题发布后,陆续收到了来自研发一线的研究者、生产者提出的诸多回馈意见和建议。
我中心有关部门认真研究了大家的回馈意见,并再次进行了文献调研,在此基础上结合审评实际,形成第一批“共性问题”阶段性研究总结。
有些阶段总结是基于问题中的重点关注环节进行总结的,有些是基于现阶段对此问题的认识进行总结的,无论何种总结方式其目的旨在与广大业内同道进行更加深入的讨论交流,分享我们对此问题的认识。
需要说明是,日前所形成的阶段性研究结果,仅代表中心现阶段对该问题的认识,并非定论性研究成果,亦非技术要求。
以网站这一平台同步公布与申办人共同研究的“共性问题”是一种新探索。
第二批共性问题现已在中心网站发布。
今后中心还将就审评工作中发现的一些普遍性问题与申办人以多种有效的形式加以研究,竭诚希望广大从事药品研发和注册的业内同道,积极参与上述讨论,提出您对这些问题的意见和建议。
让我们大家共同努力,为推动我国药品研究与评价,为保证公众用药安全有效,促进医药事业健康协调发展,做出我们应有的贡献!一、对反馈信息的整理和分析(一)共收到反馈信息23条,具体内容总结如下:问题1:大输液和小水针品种确定灭菌工艺的基本原则是什么?反馈信息中提到的原则包括:保证产品质量的稳定,保证产品无菌和细菌内毒素符合要求,选择效果显著的灭菌方式,选择具有现实性、可验证性的灭菌工艺。
问题2:对于不同的灭菌工艺,是否都进行灭菌工艺的验证?是否针对每个不同品种都需进行验证?验证的侧重点(工艺条件、生产设备等)有何不同?是在申报前还是获得生产批文后再进行验证?是在生产规模还是中试规模上进行验证?反馈信息中基本所有的信息均认为不同的工艺均需要进行验证,还有的提到发生设备变更、系统改造、灭菌条件(包括设备、包装容器、容器位置、灭菌参数等)改变等情况都需要进行验证。
大部分信息认为不同品种也需要进行验证,仅有两条信息认为应针对工艺而非针对品种进行验证。
反馈信息中基本所用的信息均认为应在生产规模上进行验证,原因:考虑到验证的准确性(需要对制水、空调、灭菌、罐装等均需要验证,而小量生产和静态验证都有局限性)和成本问题(即便在中试规模进行了验证,由于中试生产与生产规模的生产的设备等不同,故转为大生产时,仍还需要验证)。
较多的反馈信息认为工艺的验证主要与GMP有关,与审评关系不大,审评时需要重点关注采用的灭菌工艺对产品质量的稳定性的影响,所以在注册申请时不应对工艺验证提出过多的要求。
同时提出应依靠加强市场监督和抽检来保障产品的质量(包括无菌检查)。
也有的提出要加强现场考核工作。
也有反馈信息提出目前国家局已有规定,所有申报生产的三批样品的数量应是大生产的等量、半量和最小量,所以验证工艺是在申报时就可以进行的。
反馈信息中有两条提出应在注册申报时进行验证,审评时应结合GMP知识,对验证工作提出要求。
反馈信息中还提到应停止部分小水针与大输液的改剂型研究,很多小水针制成大输液后质量不稳定,不能采用常规灭菌方法,如硫酸奈替米星注射液、克林霉素磷酸酯注射液等。
反馈信息中仅有一条信息认为灭菌工艺不需验证,终产品按照中国药典2005年版要求无菌检验合格即可。
问题3:灭菌工艺确定后,是否进行回顾性验证?多久进行一次验证?基本上所有回馈信息均认为回顾性验证时需要的,但对于多久需要验证一次有两种观点,一种提出了基本的时间控制点,如获得批件后的大生产的前三批是要验证的,此后每三个月进行重点项目的检查,一年进行一次回顾性验证,还有的认为一年1-2次,另一种认为是回顾性验证的时间确定非常复杂,不同品种的情况也不相同,所以回顾性验证的时间间隔是需要在实际大生产中摸索的,是GMP控制的内容。
也正因为如此,在注册申报时无法提出具体的回顾性验证的时间。
对以上信息的分析:问题1:与中心的认识基本一致,中心目前主要关心的是灭菌的有效性和对产品质量的影响,至于现实性等成本方面的问题考虑得较少。
问题2:对于灭菌工艺需要验证这一点,与中心的认识也比较一致。
但关于注册申请时对工艺验证要求到何种程度的问题,目前中心也在调研讨论中。
确实如反馈信息中提到,中试规模和将来的生产规模存在着生产条件上的不同,中试规模的验证工作不能完全代表生产规模的验证工作,但灭菌工艺是生产工艺的重要部分,尤其对于采用了非常规灭菌工艺的品种,评价其灭菌工艺是否合理,是评价其生产工艺是否合理的重要部分,而灭菌工艺的评价是需要工艺验证资料的支持的,所以,对于审评工作,需要在对灭菌工艺验证有一定认识的基础上,提出合理的技术要求。
关于各种灭菌工艺验证的侧重点,可能由于问题表述得不够清晰,仅有两条反馈信息提到工艺条件是指产品要求,生产设备是指设备能达到什么能力。
反馈信息中均没有具体提到不同灭菌工艺验证的侧重点是什么。
问题三:对于需要进行回顾性验证这一点,与中心的认识也是一致的。
但对于回顾性验证的具体内容是什么,在灭菌工艺验证中是何种地位,在注册申请时是否要求提供相关的资料,目前中心也在调研讨论中。
二、我们调研的情况小组成员与无锡华瑞、北京双鹤、上海信谊、常州四药等国内一些大型企业的相关技术人员进行了电话交流和书面交流,获得的信息主要内容如下:1、生产单位对产品的灭菌工艺是非常关注的。
在选择灭菌工艺时,都是非常慎重的。
2、如果样品中各成分稳定,生产企业会优先采用常规条件的湿热灭菌法,而不采用非常规的湿热灭菌法(如温度较低或者时间较短等),或者过滤除菌的方法,原因是从方法的可靠性上,湿热灭菌至少可以保证将染菌率控制在10-6,而过滤除菌一般只能控制在10-3;从生产成本和可控性方面,湿热灭菌都优于其他灭菌方式。
3、对于常规的灭菌工艺的验证主要是对设备的验证,即生产设备能否达到生产的需要,主要包括热分布试验和热穿透试验等。
此外,还包括生物指示剂试验(也可称为挑战性试验),即将生物指示剂放入灭菌柜,按照生产条件进行灭菌,取出后检查应无微生物生长。
对于非常规的灭菌工艺的验证还需要进行另一项挑战性试验,主要内容为培养基试验,即在生产条件下进行培养基的生产,然后对培养基进行无菌性试验,即取一定数量培养基,封口于30-35℃培养7天后于20-25℃培养7天,应确认无任何微生物生长。
4、对于采用过滤除菌的产品,需要严格控制过滤器的质量。
应选择绝对0.22um(即每一孔径均小于0.22um)的微孔滤膜,通过起泡点试验进一步验证滤器的孔径,通过挑战性试验(挑战性试验所用溶液为含有细菌的溶液,细菌浓度为100个孢子/cm2,收集过滤液培养应为阴性结果)考察过滤性能,在过滤后再次通过起泡点试验验证滤膜的完整性。
5、对于采用常规灭菌条件的产品和采用非常规灭菌条件的产品的生产时各方面的生产条件的控制是不同的,如灌装区空气洁净度的控制不同,生产管道的控制不同(如常规灭菌工艺下一个批号产品生产结束后消毒灭菌一次,过滤除菌的每日消毒除菌一次)、控制灭菌前的染菌情况等等。
两种情况的GMP 要求是不同的。
对于采用非常规灭菌条件的产品,需要通过对整个生产过程的控制实现终产品的无菌,而灭菌工艺仅能作为提高产品无菌保证水平的一部分手段。
6、对于采用常规灭菌条件的产品和采用非常规灭菌条件的产品,在进行无菌的抽样检查时,抽样量时不同的,后者抽样量较大。
7、关于回顾性验证,一般会随着生产时间的不同而不断调整,起初一般是3个月一次,然后逐步变为6个月一次,12个月一次。
需要视品种的具体情况和生产的具体情况确定。
三、对此问题的初步认识结合上述调研情况,对小水针与大输液的灭菌工艺问题提出以下意见:1、首先需要对灭菌方法是否合理进行判断。
如果样品中各成分稳定,优先采用常规条件的湿热灭菌法,而不采用非常规的湿热灭菌法(如温度较低或者时间较短等),或者过滤除菌的方法。
如果已上市同产品可以采用常规的灭菌方法,而在研产品由于处方等原因导致无法采用常规的灭菌方法的,则建议完善处方后采用常规灭菌的方法。
如果是小水针、大输液、冻干粉的改剂型后,原剂型可采用常规灭菌方法,而改剂型后不能采用常规灭菌方法的,应属于剂型改变不合理。
对于注册分类3.1的品种或其他注册分类的新药,也建议在确定剂型时考虑灭菌工艺的问题,建议优选能够采用常规灭菌工艺的剂型。
2、如果确因产品对热不稳定而无法采用常规的湿热灭菌法的产品,在申请生产时需要提供工艺验证的关键性的资料,并在生产批件中注明在大生产时进行全面的验证(包括回顾性验证),在质量标准转正或者再注册时提供全面的工艺验证资料。
3、可以结合GMP的要求一并进行对产品的无菌情况进行控制,生产企业需具备相应生产工艺的GMP证书。
对于采用了非常规灭菌工艺的产品,需要在批件中注明灭菌工艺,并要求在相应的GMP条件下生产。
对于目前无法提供相应生产工艺的GMP证书的,可在质量标准转正或者再注册时提供。
四、下一步工作内容1、关注药学评价组将在9月份组织的关于“无菌药品无菌的安全性评估”的讲座,并根据讲座情况完善此课题。
2、继续进行调研工作,并注意寻找具有非常规灭菌工艺实际生产经验的企业或相关技术人员进行交流。
3、调研的重点问题包括:非常规灭菌工艺验证的主要方法、步骤、结果评价问题;常规灭菌工艺、非常规灭菌工艺验证的方法是否有所不同,各自的侧重点是什么;灭菌前的染菌水平如何控制、测定和评价,与确定灭菌工艺的关系如何。