他克莫司软膏英文说明书
皮肤科常用药物说明

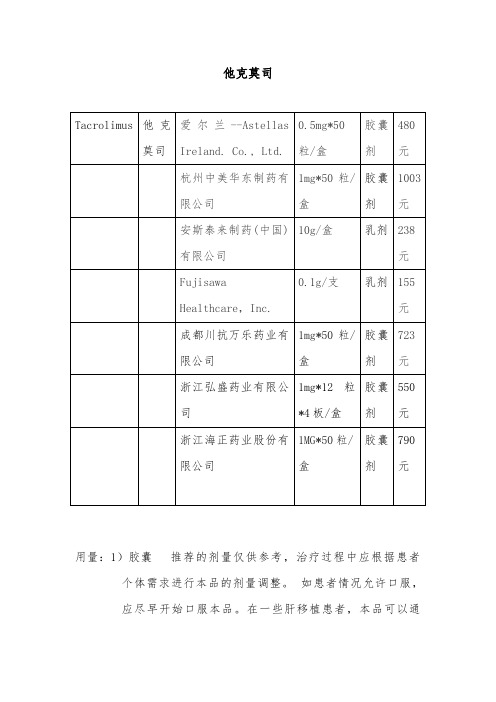
1药品信息【药品名称】他克莫司软药品包装正面图片膏【常用名称】【英文名称】 Tacrolimus Ointment【汉语拼音】 Ta Ke Mo Si Ruan Gao【拼音码】ptptkmsrg【规格】10g:3mg%); 10g:10mg%)【剂型】软膏【【主要成分】他克莫司【性状】本品为白色至淡黄色软膏。
【处方组成】每克本品含他克莫司%或%(w/w),软膏基质为矿物油、石蜡、碳酸丙烯酯、白凡士林和白蜡。
【有效期】 30个月【贮藏】室温25℃保存;允许的温度范围是15-30℃。
【批准文号】: H; H适应症适用于因潜在危险而不宜使用传统疗法、或对传统疗法反应不充分、或无法耐受传统疗法的中到重度特应性皮炎患者,作为短期或间歇性长期治疗%和%浓度的本品均可用于成人,但只有%浓度的本品可用于2岁及以上的儿童。
目前皮肤科用于治疗:特应性皮炎(AD),银屑病,白癜风,单纯糠疹,硬化萎缩性苔藓,激素依赖皮炎,龟头炎,接触性皮炎,系统性红斑狼疮,蕈样肉芽肿!可能对下列疾病有效:扁平苔藓、血管炎、硬皮病、皮肌炎、毛囊角化病、鱼鳞病、斑秃、结节病等。
用法用量成人 %和%他克莫司软膏在患处皮肤涂上一薄层本品,轻轻擦匀,并完全覆盖,一天两次,持续至特应性皮炎症状和体征消失后一周。
封包疗法可能会促进全身性吸收,其安全性未进行过评价。
本品不应采用封包敷料外用。
儿童 %他克莫司软膏在患处皮肤涂上一薄层本品,轻轻擦匀,并完全覆盖,一天两次,持续至特应性皮炎症状和体征消失后一周。
封包疗法可能会促进全身性吸收,其安全性未进行过评价。
(他克莫司软膏) 不应采用封包敷料外用。
注意事项【禁忌】对他克莫司或制剂中任何其他成分有过敏史的患者禁用本品。
【注意事项】 1、外用本品可能会引起局部症状,如皮肤烧灼感(灼热感、刺痛、疼痛)或瘙痒。
局部症状最常见于使用本品的最初几天,通常会随着特应性皮炎受累皮肤好转而消失。
应用%浓度的本品治疗时,90%的皮肤烧灼感持续时间介于2分钟至3小时(中位时间为15分钟)之间,90%的瘙痒症状持续时间介于3分钟至10小时(中位时间为20分钟)之间。
普特彼(他克莫司软膏)简要说明书

普特彼(他克莫司软膏)说明书【普特彼药品名称】通用名:他克莫司软膏商品名:普特彼英文名:TacrolimusOintment汉语拼音:TaKeMoSiRuanGao【普特彼成份】普特彼主要成份为:他克莫司。
【普特彼性状】普特彼为白色至淡黄色软膏。
【普特彼处方组成】每克普特彼含他克莫司0.03或0.1(w/w),软膏基质为矿物油、石蜡、碳酸丙烯酯、白凡士林和白蜡。
【普特彼药代动力学】综合对49例成年特应性皮炎患者进行的两项药代动力学研究的结果表明,局部应用0.1浓度的普特彼后,他克莫司会被吸收。
单次或多次应用0.1浓度的普特彼后,血中他克莫司峰浓度介于检测不出至20ng/ml之间,49例患者中有45例血药峰浓度值低于5ng/ml。
对20例儿童特应性皮炎患者(年龄6-13岁)进行的药代动力学研究结果表明,应用0.1浓度的普特彼后,所有患者血中他克莫司峰浓度均低于1.6ng/ml。
从血药浓度来看,间歇性局部应用普特彼长达一年也不会导致他克莫司在全身蓄积。
局部应用他克莫司的生物利用度尚不清楚。
以静脉注射他克莫司的历史数据作对比,特应性皮炎患者局部应用普特彼的相对生物利用度低于0.5。
在平均治疗体表面积(BSA)达53的成人中,局部应用普特彼后的吸收量(即AUC)约比肾或肝移植患者将他克莫司作为免疫抑制剂口服的吸收量低30倍。
能引起全身性作用的他克莫司血药浓度目前尚不清楚。
【普特彼适应症】普特彼适用于因潜在危险而不宜使用传统疗法、或对传统疗法反应不充分、或无法耐受传统疗法的中到重度特应性皮炎患者,作为短期或间歇性长期治疗。
0.03和0.1浓度的普特彼均可用于成人,但只有0.03浓度的普特彼可用于2岁及以上的儿童。
【普特彼用法用量】成人0.03和0.1他克莫司软膏在患处皮肤涂上一薄层普特彼,轻轻擦匀,并完全覆盖,一天两次,持续至特应性皮炎症状和体征消失后一周。
封包疗法可能会促进全身性吸收,其安全性未进行过评价。
他克莫司 用法(一)

他克莫司用法(一)他克莫司1. 介绍他克莫司(英文名称:Tacrolimus),是一种免疫抑制剂,广泛应用于器官移植手术后预防和治疗排斥反应。
2. 适应症•器官移植术后的免疫抑制:他克莫司可以用于肾脏、心脏、肺、肝脏、胰腺等器官移植手术后免疫抑制的预防和治疗。
3. 用法用量•肾移植患者:术后第1天开始,根据患者情况,每日口服他克莫司/kg分2次服用,调整剂量以维持血药浓度在正确的范围内。
4. 注意事项•高血压:长期使用他克莫司可能导致高血压,需要定期监测血压情况。
•肾功能不全:由于他克莫司主要通过肾脏代谢,肾功能不全的患者需要调整剂量,避免药物积累导致毒副作用。
•多种药物相互作用:他克莫司与某些药物(如氯霉素、酮康唑等)相互作用,可能影响药物的疗效或增加毒副作用,务必告知医生正在使用的其他药物。
5. 不良反应•肾功能损害:部分患者在使用他克莫司期间可能出现肾功能不全,需要定期监测肾功•皮肤反应:少数患者可能出现皮肤病变、瘙痒等不良反应,应立即停药并就医。
•高血糖:他克莫司使用时间较长的患者可能出现高血糖,需要监测血糖情况。
6. 禁忌症•对他克莫司过敏者禁用。
•孕妇和哺乳期妇女慎用。
7. 药物储存•存放在阴凉干燥处,避免阳光直射和高温。
以上就是关于他克莫司的一些用法和注意事项的简要介绍,希望可以对大家有所帮助。
请在使用药物前务必咨询医生,并仔细阅读药品说明书。
1. 药物副作用•恶心和呕吐:他克莫司治疗期间,部分患者可能出现恶心和呕吐的副作用,可适量进食并与医生沟通寻求缓解措施。
•腹泻:有些患者可能出现腹泻症状,应保持充足的水分摄入并避免高脂高纤维食物。
•高血压:他克莫司可能导致一些患者的血压升高,需要定期监测,并根据医生建议进行控制。
•感染:免疫抑制作用可能增加患者感染的风险,特别是真菌感染,应避免接触病原体,并及时发现和治疗感染。
2. 药物相互作用•他克莫司可能与一些药物相互作用,包括其他免疫抑制剂、抗生素、抗真菌药物等,应在使用其他药物前告知医生,避免药物相互作用导致不良反应。
他克莫司

他克莫司别名:他克莫司,大环哌南,普乐可复【外文名】Tacrolimus, Prograf, FK506【药理作用】在分子水平,他克莫司的作用显然是利用与细胞性蛋白质(FKBP12)相结合,而在细胞内蓄积产生效用。
FKBP12-他克莫司复合物会专一性地结合以及抑制calcinurin,其会抑制T细胞中所产生钙离子依赖型讯息传导路径作用,因此防止不连续性淋巴因子基因的转录。
本药是具有高度免疫抑制的药物,其活性在体外及体内实验中都已被证实。
本药抑制形成主要移值排斥作用之细胞毒性淋巴球的生成。
本药是具有高度免疫抑制的药物,其活性在体外及体内实验中都已被证实。
本药抑制形成主要移植排斥作用之细胞毒性淋巴球的生成。
本药抑制T细胞的活化作用以及T辅助细胞依赖B细胞的增生作用。
也会抑制如白介素-2、白介素-3及γ-干扰素等淋巴因子的生成与白介素-2受体的表达。
在分子水平,本药的效应似乎是由结合到细胞性蛋白质(FKBP)所产生,此蛋白质也会造成该化合物累积在细胞间。
在体内试验中发现,本药显示出对肝脏及肾脏移植有效。
【适应症】肝脏及肾脏移植的首选免疫抑制药物,肝脏及肾脏移植后排斥反应对传统免疫抑制方案耐药者,也可选用该药物。
【用法用量】下列口服及静脉注射给药之建议剂量只是概略指标,本药的实际剂量应依据别病人的需要而加以调整,建议剂量只有起始剂量,因此治疗过程中应藉由临床判断并辅以他克莫司血中浓度的监测以调整剂量。
口服给药每日剂量分两次投予。
最好是在空腹或至少进食前1hr或进食后2-3hr服用胶囊,以达到最大吸收量。
口服胶囊时,通常须连续服用以抑制移植排斥作用。
并没有治疗期间的限制。
静脉注射给药输注用浓缩液必须在聚乙烯或玻璃瓶中用5%葡萄糖注射或者生理盐水稀释。
所形成的最终输注用溶液的浓度必须在0.004-0.1mg/ml范围间。
24hr内输注20-250ml。
此溶液不可以一次全量快速注释给药。
当患者的状况允许时,应尽快将静脉注射疗法改为口服疗法。
他克莫司胶囊说明书

他克莫司胶囊以下内容仅供参考,请以药品包装盒中的说明书为准。
妊娠:慎用哺乳:服用本药后应停止哺乳核准日期:2009年09月30日修改日期:2009年10月23日2016年05月31日2016年08月29日2017年04月21日他克莫司胶囊说明书请仔细阅读说明书并在医师指导下使用警示语:由于免疫抑制,发生淋巴瘤和其他恶性肿瘤,尤其是皮肤癌的风险增加;对细菌、病毒、真菌和原虫感染包括机会感染在内的易感性增加。
本品应由有免疫抑制治疗和器官移植病人管理经验的医师处方。
服用本品的患者应由配备足够实验室设备和医护人员的医疗机构进行随访。
负责维持治疗的医师应掌握进行随访所需的全部信息。
【药品名称】通用名称:他克莫司胶囊英文名称:Tacrolimus Capsules汉语拼音:Takemosi Jiaonang【成份】本品主要成份为他克莫司。
【性状】本品为硬胶囊,内容物为白色或类白色粉末。
【适应症】预防肝脏或肾脏移植术后的移植物排斥反应。
治疗肝脏或肾脏移植术后应用其他免疫抑制药物无法控制的移植物排斥反应。
【规格】0.5mg(以他克莫司计)【用法用量】他克莫司的治疗需要在配备有充足实验设备和人员的条件下密切监测。
只有在免疫抑制治疗和移植患者管理方面有经验的医师才可处方他克莫司和改变免疫抑制治疗方案。
不慎、无意或在无监督下的他克莫司胶囊和他克莫司缓释胶囊之间的转换是不安全的。
这可能导致移植物排斥或增加不良反应发生,包括由于他克莫司全身暴露的临床相关差异而导致的免疫抑制不足或过度。
患者应维持他克莫司单一剂型及相应的日给药方案进行治疗。
改变剂型或调整剂量只能在移植专家严密的监督下进行。
任何剂型转换后,都需要监测治疗药物,并调整剂量以保证他克莫司的全身暴露前后一致。
以下推荐起始剂量仅作一般指导。
给药剂量主要基于对个体患者排斥反应和耐受性的临床评价辅以血药浓度监测(参见以下推荐目标全血谷浓度)。
如果排斥反应临床症状明显,则应考虑改变免疫抑制治疗方案。
他克莫司说明书

孚诺(复方多粘菌素B软膏),他克莫司(普特彼)说明书如下:【普特彼药品名称】通用名:他克莫司软膏商品名:普特彼英文名:Tacrolimus Ointment汉语拼音:TaKeMoSiRuanGao【普特彼成份】普特彼主要成份为:他克莫司。
【普特彼性状】普特彼为白色至淡黄色软膏。
【普特彼处方组成】每克普特彼含他克莫司0.03或0.1(w/w),软膏基质为矿物油、石蜡、碳酸丙烯酯、白凡士林和白蜡。
【普特彼药理毒理】药理作用他克莫司治疗特应性皮炎的作用机制还不清楚。
虽然对他克莫司的作用机制已有一定了解,但是这些发现与特应性皮炎的临床关系还不明确。
他克莫司已被证实可以抑制T淋巴细胞活化,首先与细胞内蛋白FKBP-12结合,形成由他克莫司-FKBP-12、钙、钙调蛋白和钙调磷酸酶构成的复合物,从而抑制钙调磷酸酶的磷酸酶活性,阻止活化T细胞核转录因子(NF-AT)的去磷酸化和易位,NF-AT这种核成分会启动基因转录形成淋巴因子(例如IL-2,γ干扰素)。
他克莫司还可以抑制编码IL-3、IL-4、IL-5、GM-CSF和TNF-?的基因的转录,所有这些因子都参与早期阶段的T细胞活化。
此外,他克莫司可以抑制皮肤肥大细胞和嗜碱性粒细胞内已合成介质的释放,下调朗格罕细胞表面FCεRI的表达。
毒理作用在为期26周的大鼠实验和为期28天的家兔实验中,每天外用他克莫司软膏(0.03-1)后,在显微镜下观察到皮肤变化(增生、表皮空泡形成、棘层肥厚、浅表炎症)。
由于这些皮肤变化与他克莫司浓度不相关,也见于赋形剂组,而空白对照组极少见,因而被认为与赋形剂有关而与他克莫司本身无关。
在大鼠中外用高浓度软膏(基本上≥0.3)观察到全身毒性反应,与经口服或静脉摄入后相似。
在为期52周的尤卡坦微型猪局部实验中,肉眼或显微镜下所见的改变均被认为与外用他克莫司(0.03?0.3)无关,因为在赋形剂对照组也观察到同样的改变。
在对豚鼠进行的实验中,他克莫司软膏(0.03?3)不诱发接触过敏或光敏化反应,对白化无毛小鼠也不诱发皮肤光毒性。
他克莫司软膏治湿疹的原理

他克莫司软膏治湿疹的原理他克莫司软膏(Tacrolimus ointment)是一种非激素的外用药物,被广泛用于治疗湿疹。
其主要成分是他克莫司(也被称为FK506),是一种新型的免疫调节剂。
他克莫司软膏的治疗原理与传统的激素药物不同,它能够通过抑制免疫反应和炎症反应,减轻湿疹患者的症状,并且不会引起激素依赖性。
湿疹是一种常见的皮肤病,通常由免疫系统的异常活化和过度炎症反应引起。
湿疹患者的皮肤经常出现红肿、瘙痒和皮疹等症状,且反复发作。
传统的治疗方法主要是使用激素类药物,如皮质类固醇,来减轻炎症和症状。
然而,长期使用皮质类固醇可能引起一系列的副作用,如皮肤变薄、血管扩张和皮肤萎缩等。
他克莫司软膏的作用机制主要通过抑制免疫反应来减轻湿疹炎症并改善患者的症状。
具体来说,他克莫司可以通过抑制T细胞的活化和分泌干扰素γ等炎症介质的产生来调节免疫反应。
T细胞是免疫系统中的重要细胞类型,它们在湿疹患者的皮肤中过度活跃,导致炎症反应的增强。
他克莫司具有选择性地结合并抑制T细胞激活所需的信号通路,从而有效地减少T细胞的活性和炎症介质的释放。
此外,他克莫司还可以抑制巨噬细胞和其他免疫细胞的活化和炎症反应。
巨噬细胞是一类重要的免疫细胞,它们在湿疹的发病过程中起着重要作用。
巨噬细胞的活化会产生一系列的炎症介质,如肿瘤坏死因子α和白细胞介素1等,进一步增加炎症反应的程度。
他克莫司能够抑制巨噬细胞的激活和炎症介质的产生,从而减轻湿疹患者的症状。
此外,他克莫司还具有一定的抗氧化作用。
氧化应激是湿疹患者皮肤损伤和炎症加重的重要因素之一。
通过减少氧化应激反应的发生,他克莫司能够有效地保护湿疹患者的皮肤,减轻炎症和症状。
总结起来,他克莫司软膏治疗湿疹的原理是通过抑制免疫反应和炎症反应来缓解湿疹患者的症状。
它通过抑制T细胞和巨噬细胞的活化和产生炎症介质,减少炎症反应的程度。
此外,他克莫司还具有抗氧化作用,能够减少氧化应激的发生,保护湿疹患者的皮肤。
Besponsa (inotuzumab ozogamicin) 产品说明书

Besponsa™ (inotuzumab ozogamicin)(Intravenous)Document Number: IC-0317 Last Review Date: 10/22/2021Date of Origin: 09/19/2017Dates Reviewed: 09/2017, 11/2018, 11/2019, 11/2020, 11/2021I.Length of AuthorizationCoverage will be provided for 6 months (for up to a maximum of 6 cycles) and may not berenewed.II.Dosing LimitsA.Quantity Limit (max daily dose) [NDC Unit]:•Besponsa 0.9 mg powder for injection: 7 vials per 21 daysB.Max Units (per dose and over time) [HCPCS Unit]:Cycle 1•27 billable units (2.7 mg) on Day 1, 18 billable units (1.8 mg) on Days 8 and 15 of a 21 to 28-day cycleSubsequent Cycles (maximum of 5 cycles)•27 billable units (2.7 mg) on Day 1, 18 billable units (1.8 mg) on Days 8 and 15 of a 28-day cycle for up to 2 cycles•18 billable units (1.8 mg) on Day 1, Day 8, and Day 15 of a 28-day cycle for up to 3 cycles III.Initial Approval Criteria 1Coverage is provided in the following conditions:•Baseline electrocardiogram (ECG) is within normal limits; AND•Patient has not previously received inotuzumab ozogamicin; ANDUniversal Criteria 1-3•Patient has CD22-positive disease; ANDAdult B-Cell Precursor Acute Lymphoblastic Leukemia (ALL) †Ф 1-3•Patient is at least 18 years of age; ANDo Patient has relapsed or refractory disease; AND▪Used as single agent therapy or in combination with mini-hyper CVD(cyclophosphamide, dexamethasone, vincristine, methotrexate, cytarabine); AND➢Patient is Philadelphia chromosome (Ph)-negative; OR➢Patient is Philadelphia chromosome (Ph)-positive and is intolerant orrefractory to prior tyrosine kinase inhibitor therapy (e.g., imatinib,dasatinib, ponatinib, nilotinib, bosutinib, etc.); OR▪Used in combination with bosutinib; AND➢Patient is Philadelphia chromosome (Ph)-positive; ORo Used as induction therapy in patients ≥65 years of age or with substantialcomorbidities; AND▪Used in combination with mini-hyper CVD; AND▪Patient is Philadelphia chromosome (Ph)-negativePediatric B-Cell Precursor Acute Lymphoblastic Leukemia (ALL) ‡ 3,4•Patient is at least 2 years of age; AND•Patient has relapsed or refractory disease; AND•Used as single agent therapy; ANDo Patient is Philadelphia chromosome (Ph)-negative; ORo Patient is Philadelphia chromosome (Ph)-positive and is intolerant or refractory to prior tyrosine kinase inhibitor therapy (e.g., imatinib, dasatinib, etc.) †FDA Approved Indication(s); ‡ Compendium Recommended Indication(s);Ф Orphan Drug IV.Renewal CriteriaCoverage cannot be renewed.V.Dosage/AdministrationVI.Billing Code/Availability InformationHCPCS Code:•J9229 − Injection, inotuzumab ozogamicin, 0.1 mg: 1 billable units = 0.1 mgNDC:•Besponsa 0.9 mg lyophilized powder in single-dose vial: 00008-0100-xxVII.References1.Besponsa [package insert]. Philadelphia, PA; Pfizer Inc., March 2018. Accessed October2021.2.Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab Ozogamicin versus StandardTherapy for Acute Lymphoblastic Leukemia. N Engl J Med. 2016 Aug 25;375(8):740-53.3.Referenced with permission from the NCCN Drugs & Biologics Compendium (NCCNCompendium®) inotuzumab ozogamicin. National Comprehensive Cancer Network,2021. The NCCN Compendium® is a derivative work of the NCCN Guidelines®.NATIONAL COMPREHENSIVE CANCER NETWORK®, NCCN®, and NCCNGUIDELINES® are trademarks owned by the National Comprehensive Cancer Network,Inc. To view the most recent and complete version of the Compendium, go online to. Accessed October 2021.4.Bhojwani D, Sposto R, Shah NN, et al. Inotuzumab ozogamicin in pediatric patients withrelapsed/refractory acute lymphoblastic leukemia [published correction appears inLeukemia. 2019 Mar 7;:]. Leukemia. 2019;33(4):884–892. doi:10.1038/s41375-018-0265-z.Appendix 1 – Covered Diagnosis CodesAppendix 2 – Centers for Medicare and Medicaid Services (CMS)Medicare coverage for outpatient (Part B) drugs is outlined in the Medicare Benefit Policy Manual (Pub. 100-2), Chapter 15, §50 Drugs and Biologicals. In addition, National Coverage Determination (NCD), Local Coverage Articles (LCAs), and Local Coverage Determinations (LCDs) may exist and compliance with these policies is required where applicable. They can be found at: https:///medicare-coverage-database/search.aspx. Additional indications may be covered at the discretion of the health plan.Medicare Part B Covered Diagnosis Codes (applicable to existing NCD/LCA/LCD): N/Aw。
他克莫司
他克莫司用量:1)胶囊推荐的剂量仅供参考,治疗过程中应根据患者个体需求进行本品的剂量调整。
如患者情况允许口服,应尽早开始口服本品。
在一些肝移植患者,本品可以通过鼻饲来口服给药。
本品通常与其他免疫抑制药物一起使用,亦出现有单独使用本品的个例报道。
本品不能与环孢素并用。
如出现排斥反应或不良事件发生,需考虑更改免疫抑制治疗方案。
在维持治疗阶段,建议持续使用本品来维持移植物的存活。
如患者病情恶化(如出现急性排斥反应的征兆),应考虑改变免疫抑制剂用药方案。
多种方案均可用于控制排斥反应,如增加类固醇激素用量、加用短期的单克隆或多克隆抗体、增加本品的用量。
如出现中毒征兆(如明显的不良事件),应减少本品的用量。
并应告诉患者,在未经主管医师同意的情况下,不应擅自减量。
在移植术后患者的情况改善期内,本品的药代动力学可能会发生改变,需要调整本品的剂量。
每日服药两次(早晨和晚上),最好用水送服。
建议空腹,或者至少在餐前1小时或餐后2-3小时服用。
如必要可将胶囊内容物悬浮于水,经鼻饲管给药。
若患者临床状况不能口服,首剂须静脉给药。
2)乳膏成人0.03%和0.1%他克莫司软膏在患处皮肤涂上一薄层本品,轻轻擦匀,并完全覆盖,一天两次,持续至特应性皮炎症状和体征消失后一周。
封包疗法可能会促进全身性吸收,其安全性未进行过评价。
本品不应采用封包敷料外用。
儿童0.03%他克莫司软膏在患处皮肤涂上一薄层本品,轻轻擦匀,并完全覆盖,一天两次,持续至特应性皮炎症状和体征消失后一周。
封包疗法可能会促进全身性吸收,其安全性未进行过评价。
普特彼(他克莫司软膏) 不应采用封包敷料外用。
市场他克莫司在中国上市的短时间内,市场份额迅速上升,已成为肝脏及肾脏移植后排斥反应的临床一线药物。
目前我国免疫抑制剂市场规模为50~60亿,其中国外公司将的他克莫司免疫抑制剂市场份额占80%,开发我国内他克莫司具有自主知识产权的生产技术及开发技术迫在眉急。
他克莫司软膏说明书(英文PDR)
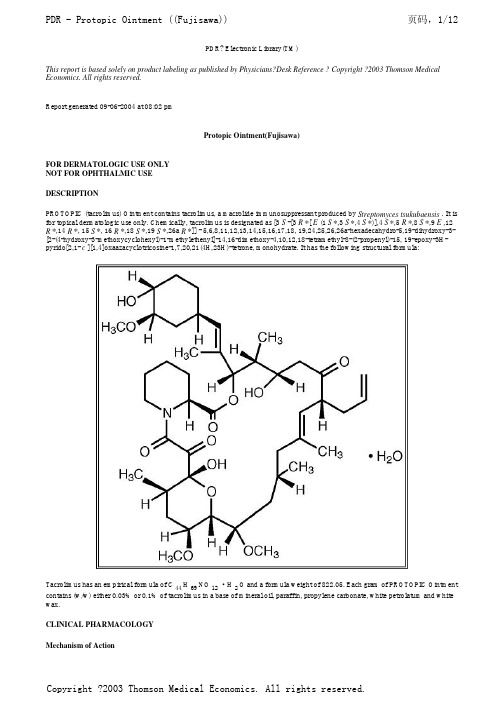
PDR? Electronic Library(TM)This report is based solely on product labeling as published by Physicians?Desk Reference ? Copyright ?2003 Thomson Medical Economics. All rights reserved.Report generated 09-06-2004 at 08:02 pmProtopic Ointment(Fujisawa)FOR DERMATOLOGIC USE ONLYNOT FOR OPHTHALMIC USEDESCRIPTIONPROTOPIC (tacrolimus) Ointment contains tacrolimus, a macrolide immunosuppressant produced by Streptomyces tsukubaensis . It is for topical dermatologic use only. Chemically, tacrolimus is designated as [3 S -[3 R *[ E (1 S *,3 S *,4 S *)],4 S *,5 R *,8 S *,9 E ,12 R *,14 R *, 15 S *, 16 R *,18 S *,19 S *,26a R *]] - 5,6,8,11,12,13,14,15,16,17,18, 19,24,25,26,26a-hexadecahydro-5,19-dihydroxy-3-[2-(4-hydroxy-3-methoxycyclohexyl)-1-methylethenyl]-14,16-dimethoxy-4,10,12,18-tetramethyl-8-(2-propenyl)-15, 19-epoxy-3H-pyrido[2,1- c ][1,4]oxaazacyclotricosine-1,7,20,21(4H,23H)-tetrone, monohydrate. It has the following structural formula:Tacrolimus has an empirical formula of C44 H69NO12·H2O and a formula weight of 822.05. Each gram of PROTOPIC Ointmentcontains (w/w) either 0.03% or 0.1% of tacrolimus in a base of mineral oil, paraffin, propylene carbonate, white petrolatum and white wax.CLINICAL PHARMACOLOGYMechanism of ActionThe mechanism of action of tacrolimus in atopic dermatitis is not known. While the following have been observed, the clinicalsignificance of these observations in atopic dermatitis is not known. It has been demonstrated that tacrolimus inhibits T-lymphocyte activation by first binding to an intracellular protein, FKBP-12. A complex of tacrolimus-FKBP-12, calcium, calmodulin, and calcineurin is then formed and the phosphatase activity of calcineurin is inhibited. This effect has been shown to prevent the dephosphorylation and translocation of nuclear factor of activated T-cells (NF-AT), a nuclear component thought to initiate genetranscription for the formation of lymphokines (such as interleukin-2, gamma interferon). Tacrolimus also inhibits the transcription for genes which encode IL-3, IL-4, IL-5, GM-CSF, and TNF-(alpha), all of which are involved in the early stages of T-cell activation. Additionally, tacrolimus has been shown to inhibit the release of pre-formed mediators from skin mast cells and basophils, and to downregulate the expression of Fc[egr ]Rl on Langerhans cells. PharmacokineticsThe pooled results from two pharmacokinetic studies in 49 adult atopic dermatitis patients indicate that tacrolimus is absorbed after the topical application of 0.1% PROTOPIC Ointment. Peak tacrolimus blood concentrations ranged from undetectable to 20 ng/mL after single or multiple doses of 0.1% PROTOPIC Ointment, with 45 of the 49 patients having peak blood concentrations less than 5ng/mL. The results from a pharmacokinetic study of 0.1% PROTOPIC Ointment in 20 pediatric atopic dermatitis patients (ages 6-13 years), show peak tacrolimus blood concentrations below 1.6 ng/mL in all patients.There was no evidence based on blood concentrations that tacrolimus accumulates systemically upon intermittent topical application for periods of up to 1 year. The absolute bioavailability of topical tacrolimus is unknown. Using IV historical data for comparison, the bioavailability of tacrolimus from PROTOPIC in atopic dermatitis patients is less than 0.5%. In adults with an average of 53% BSA treated, exposure (i.e., AUC) of tacrolimus from PROTOPIC is approximately 30-fold less than that seen with oralimmunosuppressive doses in kidney and liver transplant patients. The lowest tacrolimus blood level at which systemic effects can be observed is not known. CLINICAL STUDIESThree randomized, double-blind, vehicle-controlled, multi-center, phase 3 studies were conducted to evaluate PROTOPIC Ointment for the treatment of patients with moderate to severe atopic dermatitis. One (Pediatric) study included 351 patients 2-15 years of age, and the other two (Adult) studies included a total of 632 patients 15-79 years of age. Fifty-five percent (55%) of the patients were women and 27% were black. At baseline, 58% of the patients had severe disease and the mean body surface area (BSA) affected was 46%. Over 80% of patients had atopic dermatitis affecting the face and/or neck region. In these studies, patients applied eitherPROTOPIC Ointment 0.03%, PROTOPIC Ointment 0.1%, or vehicle ointment twice daily to 10%-100% of their BSA for up to 12 weeks.In the pediatric study, a significantly greater (p < 0.001) percentage of patients achieved at least 90% improvement based on the physician's global evaluation of clinical response (the pre-defined primary efficacy end point) in the PROTOPIC Ointment 0.03% treatment group compared to the vehicle treatment group, but there was insufficient evidence that PROTOPIC Ointment 0.1% provided more efficacy than PROTOPIC Ointment 0.03%.In both adult studies, a significantly greater (p < 0.001) percentage of patients achieved at least 90% improvement based on the physician's global evaluation of clinical response in the PROTOPIC Ointment 0.03% and PROTOPIC Ointment 0.1% treatmentgroups compared to the vehicle treatment group. There was evidence that PROTOPIC Ointment 0.1% may provide more efficacy than PROTOPIC Ointment 0.03%. The difference in efficacy between PROTOPIC Ointment 0.1% and 0.03% was particularly evident in adult patients with severe disease at baseline, adults with extensive BSA involvement, and black adults. Response rates for each treatment group are shown below by age groups. Because the two adult studies were identically designed, the results from these studies were pooled in this table.A statistically significant difference in the percentage of adult patients with >/= 90% improvement was achieved by week 1 for those treated with PROTOPIC Ointment 0.1%, and by week 3 for those treated with PROTOPIC Ointment 0.03%. A statistically significant difference in the percentage of pediatric patients with >/= 90% improvement was achieved by week 2 for those treated with PROTOPIC Ointment 0.03%.In adult patients who had achieved >/= 90% improvement at the end of treatment, 35% of those treated with PROTOPIC OintmentGlobal Improvement over Baseline at the End-of-Treatment in Three Phase 3 Studies Physician's Global Evaluation of Clinical Response (% Improvement)Pediatric Study (2-15 Years of Age) Adult StudiesVehicle Ointment N = 116 PROTOPIC Ointment 0.03% N = 117VehicleOintment N = 212 PROTOPICOintment 0.03% N = 211PROTOPIC Ointment 0.1% N = 209100% 4 (3%) 14 (12%) 2 (1%) 21 (10%) 20 (10%) >/=90% 8 (7%) 42 (36%) 14 (7%) 58 (28%) 77 (37%) >/=75% 18 (16%) 65 (56%) 30 (14%) 97 (46%) 117 (56%) >/=50%31 (27%)85 (73%)42 (20%)130 (62%)152 (73%)0.03% and 41% of those treated with PROTOPIC Ointment 0.1%, regressed from this state of improvement at 2 weeks after end-of-treatment. In pediatric patients who had achieved >/= 90% improvement, 54% of those treated with PROTOPIC Ointment 0.03% regressed from this state of improvement at 2 weeks after end-of-treatment. Because patients were not followed for longer than 2 weeks after end-of-treatment, it is not known how many additional patients regressed at periods longer than 2 weeks after cessation of therapy.In both PROTOPIC Ointment treatment groups in adults and in the PROTOPIC Ointment 0.03% treatment group in pediatric patients, a significantly greater improvement compared to vehicle (p < 0.001) was observed in the secondary efficacy endpoints of percent body surface area involved, patient evaluation of pruritus, erythema, edema, excoriation, oozing, scaling, and lichenification. The following two graphs depict the time course of improvement in the percent body surface area affected in adult and in pediatric patients as a result of treatment.The following two graphs depict the time course of improvement in erythema in adult and in pediatric patients as a result of treatment.The time course of improvement in the remaining secondary efficacy variables was similar to that of erythema, with improvement in lichenification slightly slower.A total of 571 patients applied PROTOPIC Ointment 0.1% in long-term adult and pediatric safety studies for up to one year. In the adult study, 246 patients were evaluated for at least 6 months and 68 patients for 12 months. In the pediatric study, 219 patients were evaluated for at least 6 months and 180 patients for 12 months. On average, patients received treatment for 87% of study days.INDICATIONS AND USAGEPROTOPIC Ointment, both 0.03% and 0.1% for adults, and only 0.03% for children aged 2 to 15 years, is indicated for short-term and intermittent long-term therapy in the treatment of patients with moderate to severe atopic dermatitis in whom the use of alternative, conventional therapies are deemed inadvisable because of potential risks, or in the treatment of patients who are not adequately responsive to or are intolerant of alternative, conventional therapies.CONTRAINDICATIONSPROTOPIC Ointment is contraindicated in patients with a history of hypersensitivity to tacrolimus or any other component of the preparation.PRECAUTIONSGeneralStudies have not evaluated the safety and efficacy of PROTOPIC Ointment in the treatment of clinically infected atopic dermatitis. Before commencing treatment with PROTOPIC Ointment, clinical infections at treatment sites should be cleared.While patients with atopic dermatitis are predisposed to superficial skin infections including eczema herpeticum (Kaposi's varicelliform eruption), treatment with PROTOPIC Ointment may be associated with an increased risk of varicella zoster virus infection (chicken pox or shingles), herpes simplex virus infection, or eczema herpeticum. In the presence of these infections, the balance of risks and benefits associated with PROTOPIC Ointment use should be evaluated.In clinical studies, 33 cases of lymphadenopathy (0.8%) were reported and were usually related to infections (particularly of the skin) and noted to resolve upon appropriate antibiotic therapy. Of these 33 cases, the majority had either a clear etiology or were known to resolve. Transplant patients receiving immunosuppressive regimens (e.g., systemic tacrolimus) are at increased risk for developing lymphoma; therefore, patients who receive PROTOPIC Ointment and who develop lymphadenopathy should have the etiology of their lymphadenopathy investigated. In the absence of a clear etiology for the lymphadenopathy, or in the presence of acute infectious mononucleosis, discontinuation of PROTOPIC Ointment should be considered. Patients who develop lymphadenopathy should be monitored to ensure that the lymphadenopathy resolves.The enhancement of ultraviolet carcinogenicity is not necessarily dependent on phototoxic mechanisms. Despite the absence of observed phototoxicity in humans (see ADVERSE REACTIONS ), PROTOPIC Ointment shortened the time to skin tumor formation in an animal photocarcinogenicity study (see Carcinogenesis, Mutagenesis, Impairment of Fertility ). Therefore, it is prudent for patients to minimize or avoid natural or artificial sunlight exposure.The use of PROTOPIC Ointment may cause local symptoms such as skin burning (burning sensation, stinging, soreness) or pruritus. Localized symptoms are most common during the first few days of PROTOPIC Ointment application and typically improve as the lesions of atopic dermatitis heal. With PROTOPIC Ointment 0.1%, 90% of the skin burning events had a duration between 2 minutes and 3 hours (median 15 minutes). Ninety percent of the pruritus events had a duration between 3 minutes and 10 hours (median 20 minutes).The use of PROTOPIC Ointment in patients with Netherton's Syndrome is not recommended due to the potential for increased systemic absorption of tacrolimus. The safety of PROTOPIC Ointment has not been established in patients with generalized erythroderma.Information for Patients(See patient package insert)Patients using PROTOPIC Ointment should receive the following information and instructions:1.Patients should use PROTOPIC Ointment as directed by the physician. PROTOPIC Ointment is for external use only. As withany topical medication, patients or caregivers should wash hands after application if hands are not an area for treatment.2.Patients should minimize or avoid exposure to natural or artificial sunlight (tanning beds or UVA/B treatment) while usingPROTOPIC Ointment.3.Patients should not use this medication for any disorder other than that for which it was prescribed.4.Patients should report any signs of adverse reactions to their physician.5.Before applying PROTOPIC Ointment after a bath or shower, be sure your skin is completely dry.Drug InteractionsFormal topical drug interaction studies with PROTOPIC Ointment have not been conducted. Based on its minimal extent of absorption, interactions of PROTOPIC Ointment with systemically administered drugs are unlikely to occur but cannot be ruled out. The concomitant administration of known CYP3A4 inhibitors in patients with widespread and/or erythrodermic disease should be done with caution. Some examples of such drugs are erythromycin, itraconazole, ketoconazole, fluconazole, calcium channel blockers and cimetidine.Carcinogenesis, Mutagenesis, Impairment of FertilityNo evidence of genotoxicity was seen in bacterial ( Salmonella and E. coli ) or mammalian (Chinese hamster lung-derived cells) in vitro assays of mutagenicity, the in vitro CHO/HGPRT assay of mutagenicity, or in vivo clastogenicity assays performed in mice. Tacrolimus did not cause unscheduled DNA synthesis in rodent hepatocytes.Oral (feed) carcinogenicity studies have been carried out with systemically administered tacrolimus in male and female rats and mice. In the 80-week mouse study and in the 104-week rat study no relationship of tumor incidence to tacrolimus dosage was found at daily doses up to 3 mg/kg [9X the Maximum Recommended Human Dose (MRHD) based on AUC comparisons] and 5 mg/kg (3X the MRHD based on AUC comparisons), respectively.A 104-week dermal carcinogenicity study was performed in mice with tacrolimus ointment (0.03%-3%), equivalent to tacrolimus doses of 1.1-118 mg/kg/day or 3.3-354 mg/m 2 /day. In the study, the incidence of skin tumors was minimal and the topical application of tacrolimus was not associated with skin tumor formation under ambient room lighting. However, a statistically significant elevation in the incidence of pleomorphic lymphoma in high dose male (25/50) and female animals (27/50) and in the incidence of undifferentiated lymphoma in high dose female animals (13/50) was noted in the mouse dermal carcinogenicity study. Lymphomas were noted in the mouse dermal carcinogenicity study at a daily dose of 3.5 mg/kg (0.1% tacrolimus ointment) (26X MRHD based on AUC comparisons). No drug-related tumors were noted in the mouse dermal carcinogenicity study at a daily dose of 1.1 mg/kg (0.03% tacrolimus ointment) (10X MRHD based on AUC comparisons).In a 52-week photocarcinogenicity study, the median time to onset of skin tumor formation was decreased in hairless mice following chronic topical dosing with concurrent exposure to UV radiation (40 weeks of treatment followed by 12 weeks of observation) with tacrolimus ointment at >/=0.1% tacrolimus.Reproductive toxicology studies were not performed with topical tacrolimus. In studies of oral tacrolimus no impairment of fertility was seen in male and female rats. Tacrolimus, given orally at 1.0 mg/kg (0.12X MRHD based on body surface area [BSA]) to male and female rats, prior to and during mating, as well as to dams during gestation and lactation, was associated with embryolethality and with adverse effects on female reproduction. Effects on female reproductive function (parturition) and embryolethal effects were indicated by a higher rate of pre-implantation loss and increased numbers of undelivered and nonviable pups. When given at 3.2mg/kg (0.43X MRHD based on BSA), tacrolimus was associated with maternal and paternal toxicity as well as reproductive toxicity including marked adverse effects on estrus cycles, parturition, pup viability, and pup malformations.PregnancyTeratogenic Effects: Pregnancy Category CThere are no adequate and well-controlled studies of topically administered tacrolimus in pregnant women. The experience with PROTOPIC Ointment when used by pregnant women is too limited to permit assessment of the safety of its use during pregnancy. Reproduction studies were carried out with systemically administered tacrolimus in rats and rabbits. Adverse effects on the fetus were observed mainly at oral dose levels that were toxic to dams. Tacrolimus at oral doses of 0.32 and 1.0 mg/kg (0.04X-0.12X MRHD based on BSA) during organogenesis in rabbits was associated with maternal toxicity as well as an increase in incidence of abortions. At the higher dose only, an increased incidence of malformations and developmental variations was also seen. Tacrolimus, at oral doses of 3.2 mg/kg during organogenesis in rats, was associated with maternal toxicity and caused an increase in late resorptions, decreased numbers of live births, and decreased pup weight and viability. Tacrolimus, given orally at 1.0 and 3.2 mg/kg (0.04X-0.12X MRHD based on BSA) to pregnant rats after organogenesis and during lactation, was associated with reduced pup weights.No reduction in male or female fertility was evident.There are no adequate and well-controlled studies of systemically administered tacrolimus in pregnant women. Tacrolimus is transferred across the placenta. The use of systemically administered tacrolimus during pregnancy has been associated with neonatal hyperkalemia and renal dysfunction. PROTOPIC Ointment should be used during pregnancy only if the potential benefit to the mother justifies a potential risk to the fetus.Nursing MothersAlthough systemic absorption of tacrolimus following topical applications of PROTOPIC Ointment is minimal relative to systemic administration, it is known that tacrolimus is excreted in human milk. Because of the potential for serious adverse reactions in nursing infants from tacrolimus, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.Pediatric UsePROTOPIC Ointment 0.03% may be used in pediatric patients 2 years of age and older. Two phase 3 pediatric studies were conducted involving 606 patients 2-15 years of age: one 12-week randomized vehicle-controlled study and one open-label, 1 year, long-term safety study. Three hundred and thirty (330) of these patients were 2 to 6 years of age.The most common adverse events associated with PROTOPIC Ointment application in pediatric patients were skin burning and pruritus (see ADVERSE REACTIONS). In addition to skin burning and pruritus, the less common events (<5%) of varicella zoster (mostly children pox), and vesiculobullous rash were more frequent in patients treated with PROTOPIC Ointment 0.03% compared to vehicle. In the long-term 1 year safety study involving 255 pediatric patients using PROTOPIC Ointment, the incidence of adverse events, including infections, did not increase with increased duration of study drug exposure or amount of ointment used. In 491 pediatric patients treated with PROTOPIC Ointment, 3(0.6%) developed eczema herpeticum. Since the safety and efficacy of PROTOPIC Ointment have not been established in pediatric patients below 2 years of age, its use in this age group is not recommended.Geriatric UseTwenty-five (25) patients >/= 65 years old received PROTOPIC Ointment in phase 3 studies. The adverse event profile for these patients was consistent with that for other adult patients.ADVERSE REACTIONSNo phototoxicity and no photoallergenicity was detected in clinical studies of 12 and 216 normal volunteers, respectively. One out of 198 normal volunteers showed evidence of sensitization in a contact sensitization study.In three randomized vehicle-controlled studies and two long-term safety studies, 655 and 571 patients respectively, were treated with PROTOPIC Ointment.The following table depicts the adjusted incidence of adverse events pooled across the 3 identically designed 12 week studies for patients in vehicle, PROTOPIC Ointment 0.03%, and PROTOPIC Ointment 0.1% treatment groups, and the unadjusted incidence of adverse events in two one year long-term safety studies, regardless of relationship to study drug.Phase 3 Studies12-Week Adjusted Incidence Rate (%) Tacrolimius Ointment Incidence(%)Adult Pediatric Adult PediatricVehicle n=2120.03%TacrolimusOintmentn=2100.1%TacrolimusOintmentn=209Vehiclen=1160.03%TacrolimusOintmentn=118 n=316 n=255Skin Burning †26 46 58 29 43 47 26 Pruritus †37 46 46 27 41 25 25 Flu-like symptoms †19 23 31 25 28 22 35 Allergic Reaction 8 12 6 8 4 22 15 Skin Erythema 20 25 28 13 12 12 9 Headache †11 20 19 8 5 10 18 Skin Infection 11 12 5 14 10 11 11 Fever 4 4 1 13 21 2 18 Infection 1 1 2 9 7 14 8 Cough Increased 2 1 1 14 18 3 15 Asthma 4 6 4 6 6 5 16 Herpes Simplex 4 4 4 2 0 12 5 Eczema Herpeticum 0 1 1 0 2 2 0 Pharyngitis 3 3 4 11 6 5 10 Accidental Injury 4 3 6 3 6 4 12 Pustular Rash 2 3 4 3 2 6 8 Folliculitis † 1 6 4 0 2 11 2 Rhinitis 4 3 2 2 6 5 5 Otitis Media 4 0 1 6 12 1 7 Sinusitis † 1 4 2 8 3 3 7 Diarrhea 3 3 4 2 5 4 6 Urticaria 3 3 6 1 1 5 5 Lack of Drug Effect 1 1 0 1 1 10 2 Bronchitis 0 2 2 3 3 3 6 Vomiting 0 1 1 7 6 1 5 Maculopapular Rash 2 2 2 3 0 4 3 Rash † 1 5 2 4 2 2 5 Abdominal Pain 3 1 1 2 3 1 5 Fungal Dermatitis 0 2 1 3 0 2 6 Gastroenteritis 1 2 2 3 0 4 2 Alcohol Intolerance †0 3 7 0 0 6 0 Acne † 2 4 7 1 0 2 4 Sunburn 1 2 1 0 0 4 4 Skin Disorder 2 2 1 1 4 1 4 Conjunctivitis 0 2 2 2 1 4 2 Pain 1 2 1 0 1 4 3 Vesiculobullous Rash† 3 3 2 0 4 2 2 Lymphadenopathy 2 2 1 0 3 2 3 Nausea 4 3 2 0 1 1 2 Skin Tingling † 2 3 8 1 2 2 1 Face Edema 2 2 1 2 1 3 1 Dyspepsia † 1 1 4 0 0 1 4Other adverse events which occurred at an incidence greater than or equal to 1% in any clinical study include: alopecia, ALT or AST increased, anaphylactoid reaction, angina pectoris, angioedema, anorexia, anxiety, arrhythmia, arthralgia, arthritis, bilirubinemia, breast pain, cellulitis, cerebrovascular accident, cheilitis, chills, constipation, creatinine increased, dehydration, depression, dizziness, dyspnea, ear pain, ecchymosis, edema, epistaxis, exacerbation of untreated area, eye disorder, eye pain, furunculosis, gastritis, hemia, hyperglycemia, hypertension, hypoglycemia, hypoxia, laryngitis, leukocytosis, leukopenia, liver function tests abnormal, lung disorder, malaise, migraine, neck pain, neuritis, palpitations, paresthesia, peripheral vascular disorder, photosensitivity reaction, procedural complication, routine procedure, skin discoloration, sweating, taste perversion, tooth disorder, unintended pregnancy, vaginal moniliasis, vasodilatation, and vertigo. OVERDOSAGEPROTOPIC Ointment is not for oral use. Oral ingestion of PROTOPIC Ointment may lead to adverse effects associated with systemic administration of tacrolimus. If oral ingestion occurs, medical advice should be sought. DOSAGE AND ADMINISTRATION ADULTPROTOPIC Ointment 0.03% and 0.1%Apply a thin layer of PROTOPIC Ointment 0.03% or 0.1% to the affected skin areas twice daily and rub in gently and completely. Treatment should be continued for one week after clearing of signs and symptoms of atopic dermatitis.The safety of PROTOPIC Ointment under occlusion which may promote systemic exposure, has not been evaluated. PROTOPIC Ointment 0.03% and 0.1% should not be used with occlusive dressings. PEDIATRICPROTOPIC Ointment 0.03%Apply a thin layer of PROTOPIC Ointment 0.03% to the affected skin areas twice daily and rub in gently and completely. Treatment should be continued for one week after clearing of signs and symptoms of atopic dermatitis. The safety of PROTOPIC Ointment under occlusion, which may promote systemic exposure, has not been evaluated. PROTOPIC Ointment 0.03% should not be used with occlusive dressings.Dry Skin 7 3 3 0 1 0 1 Hyperesthesia † 1 3 7 0 0 3 0 Skin Neoplasm Benign ‡‡ 1 1 1 0 0 2 3 Back Pain † 0 2 2 1 1 3 1 Peripheral Edema 2 4 3 0 0 2 1 Varicella Zoster/ Herpes Zoster ‡ 0 1 0 0 5 1 3 Contact Dermatitis 1 3 3 3 4 1 1 Asthenia 1 2 3 0 0 2 1 Pneumonia 0 1 1 2 0 1 2 Eczema 2 2 2 0 0 3 0 Insomnia 3 4 3 1 1 1 0 Exfoliative Dermatitis 3 3 1 0 0 0 2 Dysmenorrhea 2 4 4 0 0 0 2 Periodontal Abscess 1 0 1 0 0 3 0 Myalgia † 0 3 2 0 0 1 0 Cyst †13† May be reasonably associated with the use of this drug product.‡ Four cases of chicken pox in the pediatric 12-week study, 1 case of "zoster of the lip" in the adult 12-week study; 7 cases ofchicken pox and 1 case of shingles in the open-label pediatric study; 2 cases of herpes zoster in the open-label adult study.‡‡ Generally "warts".Patient Information AboutProtopic® (tacrolimus) OintmentRead this important information before you start using PROTOPIC [pro-TOP-ik] Ointment and each time you refill your prescription. There may be new information. This summary is not meant to take the place of your doctor's advice.What is PROTOPIC?PROTOPIC Ointment is a prescription medicine that is used to treat eczema (atopic dermatitis). It is for adults and children age 2 years and older. You can use PROTOPIC for short or intermittent long periods of treatment. Intermittent means starting and stopping repeatedly, as directed by your doctor. You can use it on all affected areas of your skin, including your face and neck.Who should not use PROTOPIC?Do not use PROTOPIC if you arel breastfeedingl allergic to PROTOPIC Ointment or any of its ingredients. The active ingredient is tacrolimus. Ask your doctor or pharmacist about the inactive ingredients.Before you start using PROTOPIC, tell your doctor if you are:l using any other prescription medicines, non-prescription (over-the-counter) medicines, or supplementsl receiving any form of light therapy (phototherapy, UVA or UVB) on your skinl using any other type of skin productl pregnant or planning to become pregnantHow do I use PROTOPIC?Use PROTOPIC only to treat eczema that has been diagnosed by a doctor.l Wash your hands before using PROTOPIC.l Apply a thin layer of PROTOPIC to all skin areas that your doctor has diagnosed as eczema. Try to cover the affected areas completely. Most people find that a pea-sized amount squeezed from the tube covers an area about the size of a two-inchcircle (approximately the size of a silver dollar).l Apply the ointment twice a day, about 12 hours apart.l Before applying PROTOPIC Ointment after a bath or shower, be sure your skin is completely dryl Do not cover the skin being treated with bandages, dressings or wraps. Unless otherwise instructed by your doctor, do not apply another type of skin product on top ofPROTOPIC Ointment. However, you can wear normal clothingl Do not bathe, shower or swim right after applyingPROTOPIC. This could wash off the ointment.l If you are a caregiver applying PROTOPIC Ointment to a patient, or if you are a patient who is not treating your hands, wash your hands with soap and water after applying PROTOPIC. This should remove any ointment left on the hands.l Use PROTOPIC only on your skin. Do not swallowPROTOPIC.Because 2 strengths of PROTOPIC are available for adult patients, your doctor will decide what strength ofPROTOPIC Ointment is best for you.Many people notice that their skin starts to improve after the first few weeks of treatment. Even though your skin looks and feels better, it is important to keep usingPROTOPIC as instructed by your doctor.If you do not notice an improvement in your eczema or if your eczema gets worse within the first few weeks of treatment, tell your doctor.What Should I Avoid While Using PROTOPIC?l Avoid sunlight and sun lamps, tanning beds, and treatment with UVA or UVB light. If you need to be outdoors after applying PROTOPIC, wear loose fitting clothing that protects the treated area from the sun. In addition, ask your doctor what other type of protection from the sun you should use.l Check with your doctor or pharmacist before you¡start taking any new medicines while using PROTOPIC¡start using any other ointment, lotions, or creams on you skinWhat Are The Possible Side Effects of PROTOPIC?。
[他克莫司软膏的作用]他克莫司软膏多久用一次
![[他克莫司软膏的作用]他克莫司软膏多久用一次](https://img.taocdn.com/s3/m/41bc16fd541810a6f524ccbff121dd36a32dc498.png)
[他克莫司软膏的作用]他克莫司软膏多久用一次篇一: 他克莫司软膏多久用一次他克莫司软膏多久用一次?他克莫司软膏是用于治疗皮炎、银屑病、湿疹等皮肤病的进口免疫调节剂,可作为短期或间歇性长期治疗药物,接下来我们就马上来看看他克莫司软膏多久用一次吧。
他克莫司软膏适用于因潜在危险而不宜使用传统疗法、或对传统疗法反应不充分、或无法耐受传统疗法的中到重度特应性皮炎患者,作为短期或间歇性长期治疗。
他克莫司软膏有0.03%*10g 和0.1%*10g 两种规格,0.03%和0.1%浓度的他克莫司软膏均可用于成人,但只有0.03%浓度的他克莫司软膏可用于2岁及以上的儿童。
那么,他克莫司软膏多久用一次呢?他克莫司软膏的使用方法介绍如下:如想了解更多他克莫司软膏的相关评论,欢迎点击-comment/1、成人:0.03%和0.1%他克莫司软膏在患处皮肤涂上一薄层本品,轻轻擦匀,并完全覆盖,一天两次,持续至特应性皮炎症状和体征消失后一周。
2、儿童:0.03%他克莫司软膏在患处皮肤涂上一薄层本品,轻轻擦匀,并完全覆盖,一天两次,持续至特应性皮炎症状和体征消失后一周。
3、他克莫司软膏是一种霜剂型的外用药,药膏为无色,油腻状,外形有点像一支牙膏,使用的时候先用刀具在胶袋的前端开一个小口,使用后应放在避光和温度适宜的地方。
4、他克莫司软膏每天使用两次,间隔为12小时一次,一般使用过的朋友都是在晚上睡觉前和早上起床后使用。
有的朋友会在使用前沐浴,以清洁皮肤。
当然,沐浴并不是必须的。
壹药网温馨提示:相信通过本文的阐述后,大家对他克莫司软膏多久用一次这个问题已经有答案了吧,建议大家在用药前先仔细阅读药品说明书或先咨询专业医师或药师的意见,以保证用药安全。
如需购买他克莫司软膏,可登录.html篇二: 他克莫司软膏使用注意事项他克莫司软膏是一种常见的用于治疗皮肤性炎症的药膏,治疗效果非常明显,适用性也非常不错。
对于常患有皮肤癣、湿疹的患者来说,非常值得推荐。
他克莫司软膏

他克莫司软膏【药品名称】通用名称:他克莫司软膏英文名称:T acrolimus Ointment【成份】本品主要成分及其化学名称为:他克莫司。
【适应症】本品适用于因潜在危险而不宜使用传统疗法、或对传统疗法反应不充分、或无法耐受传统疗法的中到重度特应性皮炎患者,作为短期或间歇性长期治疗。
0.03%和0.1%浓度...【用法用量】成人:0.03%和0.1%他克莫司软膏,在患处皮肤涂上一薄层本品,轻轻擦匀,并完全覆盖,一天两次,持续至特应性皮炎症状和体征消失后一周。
封包疗法可能会促进全身性吸收,其安全性未进行过评价。
本品不应采用封包敷料外用。
儿童:0.03%他克莫司软膏,在患处皮肤涂上一薄层本品,轻轻擦匀,并完全覆盖,一天两次,持续至特应性皮炎症状和体征消失后一周。
封包疗法可能会促进全身性吸收,其安全性未进行过评价。
本品不应采用封包敷料外用。
【不良反应】1.在分别有12例和216例健康志愿者参加的临床研究中,未发现药物具有光毒性和光致敏性。
(光毒性是指强烈的阳光中的紫外线本身就能够造成严重的日光性皮炎,甚至皮肤癌。
许多药物对人体不会造成伤害,但在阳光中的紫外线的作用下,渗入人体皮肤蛋白质中的这些药物便会发生化学反应,从而引发皮肤过敏症。
强烈的阳光可使药物活化,直接破坏或杀死皮肤细胞,使暴露在光线下的皮肤在日晒后的几分钟或几小时内产生轻度的光毒性反应,其症状类似于日晒斑或日光性皮炎。
而且引发皮肤癌的风险也比常人更大。
)在对198例健康志愿者进行的接触致敏研【禁忌】对他克莫司或制剂中任何其他成分有过敏史的患者禁用本品。
【注意事项】一般注意事项:1.本品在临床上对感染性特应性皮炎的安全性和有效性未进行过评价。
在开始使用本品治疗前,应首先清除治疗部位的感染灶。
2.特应性皮炎患者易患浅表皮肤感染,包括疱疹性湿疹(卡波济水痘样疹),使用本品治疗可能会增加带状疱疹病毒感染(水痘或带状疱疹)、(水痘是由水痘带状疱疹病毒初次感染引起的急性传染病。
普特彼(他克莫司软膏)简要说明书

【普特彼药品名称】
通用名:
他xx软膏
商品名:
普特彼
xx:
TacrolimusOintment
汉语拼音:
TaKeMoSiRuanGao
【普特彼成份】
普特彼主要成份为:
他xx。
【普特彼性状】
普特彼为白色至淡黄色软膏。
【普特彼处方组成】
每克普特彼含他克莫司0.03或0.1(w/w),软膏基质为矿物油、石蜡、碳酸丙烯酯、白凡士林和白蜡。
使用商品时,请仔细阅读说明书,并按说明书使用;如药品,请在医师指导下服用。
【普特彼有效期】
30个月
【普特彼贮藏】
室温25℃保存;允许的温度范围是15-30℃。
【普特彼批准文号】
国内分装批准文号:
国药准字J20148
【普特彼生产企业】
国内分装企业名称:
xxxx制药(xx)有限公司
【
商品图片信息展示仅供参考,最终包装以商品实物为准。欢迎纠错!
说明书内容仅供查阅参考,最终以商品包装内说明书为准。欢迎纠错!
3.患者不应将普特彼用于处方以外的疾病。
4.患者应向医生报告不良反应的症状。
5.沐浴或淋浴后应等皮肤完全干燥后再应用普特彼。
【普特彼药物相互作用】
对普特彼局部应用的药物相互作用未进行过研究。由于吸收量极少,普特彼不太可能与全身性给药的药物发生相互作用,但是也不能完全排除。皮炎较广泛的患者和/或红皮病患者合用已知的CYP3A4抑制剂时应当谨慎,这些药物的例子包括红霉素、伊曲康唑、酮康唑、氟康唑、钙通道阻滞剂和西米替丁等。
【普特彼禁忌】
对他克莫司或制剂中任何其他成分有过敏史的患者禁用普特彼。
【普特彼注意事项】
一般注意事项
他克莫司软膏说明书

快易捷医药网【通用名】他克莫司软膏【商品名】普特彼【英文名】英文名:Tacrolimus Ointment【汉语拼音】Ta Ke Mo Si Ruan Gao本品主要成分及其化学名称为:他克莫司,【性状】本品为白色至淡黄色软膏。
【处方组成】每克本品含他克莫司0.03%或0.1%(w/w),软膏基质为矿物油、石蜡、碳酸丙烯酯、白凡士林和白蜡。
【适应症】本品适用于因潜在危险而不宜使用传统疗法、或对传统疗法反应不充分、或无法耐受传统疗法的中到重度特应性皮炎患者,作为短期或间歇性长期治疗。
0.03%和0.1%浓度的本品均可用于成人,但只有0.03%浓度的本品可用于2岁及以上的儿童。
【用法与用量】成人0.03%和0.1%他克莫司软膏在患处皮肤涂上一薄层本品,轻轻擦匀,并完全覆盖,一天两次,持续至特应性皮炎症状和体征消失后一周。
封包疗法可能会促进全身性吸收,其安全性未进行过评价。
本品不应采用封包敷料外用。
儿童0.03%他克莫司软膏在患处皮肤涂上一薄层本品,轻轻擦匀,并完全覆盖,一天两次,持续至特应性皮炎症状和体征消失后一周。
封包疗法可能会促进全身性吸收,其安全性未进行过评价。
本品不应采用封包敷料外用。
【禁忌症】对他克莫司或制剂中任何其他成分有过敏史的患者禁用本品。
【注意事项】外用本品可能会引起局部症状,如皮肤烧灼感(灼热感、刺痛、疼痛)或瘙痒。
局部症状最常见于使用本品的最初几天,通常会随着特应性皮炎受累皮肤好转而消失。
应用0.1%浓度的本品治疗时,90%的皮肤烧灼感持续时间介于2分钟至3小时(中位时间为15分钟)之间,90%的瘙痒症状持续时间介于3分钟至10小时(中位时间为20分钟)之间。
不推荐使用本品治疗Netherton综合征患者,因为可能会增加他克莫司的全身性吸收。
本品对弥漫性红皮病患者治疗的安全性尚未建立。
【患者须知】使用本品的患者应接受下列信息和指导:1.患者应在医生的指导下使用本品。
本品仅供外用。
他克莫司

FK506的临床应用与血药浓度检测一、概述FK506(商品名为tacrolimus,普乐可复)是从土壤真菌中提取的一种大环内酯类抗生素,常温下呈白色结晶或晶状粉末。
分子式C44H69NO12.H2O,分子量为822.05道尔顿。
因该药在水中的溶解度低,制造时与水溶性多聚物(羟丙基甲基纤维素)制成固体扩散剂,口服吸收具有较好的稳定性,半衰期 5-8小时(1-5)。
该药1989年开始用于临床,1995年经FDA获准后在美国正式使用。
FK 506 与FK 506受体结合后抑制了磷酸酶的活性,从而抑制核调蛋白和 T细胞活化因子(如白介素-2)、原癌基因和细胞因子相应受体(如白介素-2受体)的表达。
在体外试验中,FK 506抑制混合淋巴细胞反应的作用强度比环孢素强一百倍(6、7)。
1996年在西班牙举办的世界移植大会中, Hoof f 等报告提示 FK506 能明显减低急性排斥反应,耐激素排斥反应,以及慢性排斥反应的发生率。
应用 FK 506 除震颤发生率较高外,未见发生多毛并发症,齿龈增生、痤疮和心律失常发生率也明显低于环孢素组。
Woodle等报告应用 FK506治疗移植肾顽固性急性排斥反应,认为FK506 能有效逆转难治的急性排斥反应,有低排斥反应发生率和较好的人肾成活率。
根据美国匹兹堡大学报告,在使用环孢素无效的援救疗法中,肝移植患者应用FK506 有87%的成功率,而肾移植患者有74%成功率(8、9)。
二、药代动力学FK506的吸收在个体之间相差较大,而且受食物的影响,其生物利用度的变化也较大,在5%-67%(平均20%)之间。
达到血高峰浓度需要0.5-6小时(平均1.5小时)。
FK506在体内分布广泛,进入体内后大部分分布于血液之外的组织中,半衰期4-41小时(平均5-8小时)。
口服后达到稳态血浓度一般要3天(10)。
药物的吸收与影响因素FK506是一种难溶于水的药物,不易通过胃肠道吸收。
常用羟脯氨酸甲基纤维素制备成固体分散状以供临床使用。
他克莫司胶囊说明书(英文)

month 1-3: 7-20 ng/mL month 4-12: 5-15 ng/mL
In combination with MMF/IL-2 receptor antagonist Adult Liver transplant Pediatric Liver transplant Adult Heart transplant
Careful and frequent monitoring of tacrolimus trough concentrations is recommended; Black patients may require higher doses in order to achieve comparable trough concentrations (2.1)
0.1 mg/kg/day
0.10-0.15 mg/kg/day 0.15-0.20 mg/kg/day 0.075 mg/kg/day
month 1-12: 4-11 ng/mL
month 1-12: 5-20 ng/mL month 1-12: 5-20 ng/mL
month 1-3: 10-20 ng/mL month ≥4: 5-15 ng/mL
Prograf is a calcineurin-inhibitor immunosuppressant indicated for
Prophylaxis of organ rejection in patients receiving allogeneic liver, kidney or heart transplants (1.1, 1.2, 1.3)
Only physicians experienced in immunosuppressive
普乐可复(Tacrolimus Capsules)说明书.pdf_1693991539.01903

核准日期:2007年05月24日修改日期:2011年11月05日2014年01月02日2015年04月21日2017年02月24日2019年01月17日2020年01月13日2020年08月17日2022年03月19日他克莫司胶囊说明书请仔细阅读说明书并在医师指导下使用由于免疫抑制,发生淋巴瘤和其他恶性肿瘤,尤其是皮肤癌的风险增加;对细菌、病毒、真菌和原虫感染包括机会性感染在内的易感性增加。
本品应由有免疫抑制治疗和器官移植病人管理经验的医师处方。
服用本品的患者应由配备足够实验室设备和医护人员的医疗机构进行随访。
负责维持治疗的医师应掌握进行随访所需的全部信息。
【药品名称】通用名称:他克莫司胶囊商品名称:普乐可复英文名称:Tacrolimus Capsules汉语拼音:Takemosi Jiaonang【成份】化学名称:[3S-[3R*[E(1S*,3S*,4S*)],4S*,5R*,8S*,9E,12R*,14R*,15S*,16R*,18S*,19S*,26aR*]]-5,6,8,11,12,13,14,15,16,17,18,19,24,25,26,26a-六-癸氢-5,19-二羟基-3-[2-(4-羟-3-甲氧环己基)-1-甲基乙烯基]-14,16-二甲氧-4,10,12,18-四甲基-8-(2-丙烯基)-15,19-环氧-3H-吡啶并[2,1-c][1,4]氧杂氮杂环二十三碳烯-1,7,20,21(4H,23H)-四酮,一水合物。
化学结构式:分子式:C44H69NO12·H2O分子量:822.03辅料:羟丙基甲基纤维素、交联羧甲基纤维素钠、一水乳糖、硬脂酸镁【性状】0.5mg:浅黄色硬质胶囊,内容物为白色粉末。
1mg:白色硬质胶囊,内容物为白色粉末。
【适应症】预防肝脏或肾脏移植术后的移植物排斥反应。
治疗肝脏或肾脏移植术后应用其他免疫抑制药物无法控制的移植物排斥反应。
【规格】0.5mg;1mg。
他克莫司 用法

他克莫司用法他克莫司用法1. 什么是他克莫司?他克莫司(Tacrolimus)是一种免疫抑制剂,主要用于预防和治疗器官移植后的排斥反应。
它属于钙调神经素-肾素II(calcineurin-inhibitor)抑制剂药物,通过抑制T细胞活性来达到免疫抑制的效果。
2. 他克莫司的使用方法•擦剂:他克莫司擦剂是用于治疗皮肤炎症的外用药物,一般适用于湿疹、银屑病等炎症性皮肤病。
使用时,将适量擦剂涂抹在患处,轻轻按摩至完全吸收。
避免接触到眼睛和开放性伤口。
•口服胶囊:他克莫司口服胶囊主要用于器官移植后的免疫抑制治疗。
一般情况下,应在餐前1小时或餐后2小时内口服,可以与面包、苹果酱或砂糖混合食用。
剂量根据患者情况进行调整,通常从较低剂量开始,逐渐增加至维持剂量。
•静脉注射:他克莫司静脉注射剂一般用于急性移植排斥反应的治疗。
由专业医生在医院内通过静脉注射给药,剂量和使用频率根据患者具体情况来决定。
3. 注意事项•使用他克莫司时,患者应按照医生的指导进行用药,不可擅自调整剂量或停药。
•在使用他克莫司擦剂时,避免与眼睛接触,以免引起眼部不适或损伤。
•患者在口服他克莫司胶囊时,应与医生密切合作,定期复查药物血浓度,以确保药物在有效范围内。
•他克莫司可能会导致免疫系统抑制,增加感染的风险。
患者在使用期间应注意避免接触病菌,保持良好的个人卫生。
•和其他药物相比,他克莫司与某些药物可能存在相互作用,因此在用药前应告知医生已经使用的其他药物、保健品或补充剂。
4. 副作用他克莫司的使用可能会引起一些副作用,常见的包括:•恶心、呕吐、腹泻等消化系统不适;•高血压、头痛等心血管系统症状;•感染、瘙痒、皮疹等过敏反应。
如果出现上述或其他不适症状,患者应立即告知医生,并根据医生的建议采取相应的处理措施。
结论他克莫司是一种重要的免疫抑制剂,可以有效预防和治疗器官移植后的排斥反应。
在使用他克莫司时,患者应遵循医生的建议,并注意药物的使用方法、注意事项和副作用。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
xxxxxxTacrolimus Capsules0-1200-720-inf Due to intersubject variability in tacrolimus pharmacokinetics, individualization of dosing regimen is necessary for optimal therapy. (See DOSAGE AND ADMINISTRATION ). Pharmacokinetic data indicate that whole blood concentrations rather than plasma concentrations serve as the more appropriate sampling compartment to describe tacrolimus pharmacokinetics. AbsorptionAbsorption of tacrolimus from the gastrointestinal tract after oral administration is incomplete and variable. The absolute bioavailability of tacrolimus was 17± 10% in adult kidney transplant patients (N=26), 22 ± 6% in adult liver transplant patients (N=17) and 18 ± 5% in healthy volunteers (N=16).A single dose study conducted in 32 healthy volunteers established the bioequivalence of the 1 mg and 5 mg capsules. Another single dose study in 32 healthy volunteers established the bioequivalence of the 0.5 mg and 1 mg capsules. Tacrolimus maximum blood concentrations (C max ) and area under the curve (AUC) appeared to increase in a dose-proportional fashion in 18 fasted healthy volunteers receiving a single oral dose of 3, 7, and 10 mg.In 18 kidney transplant patients, tacrolimus trough concentrations from 3 to 30 ng/mL measured at 10 to 12 hours post-dose (C min ) correlated well with the AUC (correlation coefficient 0.93). In 24 liver transplant patients over a concentration range of 10 to 60 ng/mL, the correlation coefficient was 0.94. Food EffectsThe rate and extent of tacrolimus absorption were greatest under fasted conditions. The presence and composition of food decreased both the rate and extent of tacrolimus absorption when administered to 15 healthy volunteers.The effect was most pronounced with a high-fat meal (848 kcal, 46% fat): mean AUC and C max were decreased 37% and 77%, respectively; T max was lengthened 5-fold. A high-carbohydrate meal (668 kcal, 85% carbohydrate) decreased mean AUC and mean C max by 28% and 65%, respectively.In healthy volunteers (N=16), the time of the meal also affected tacrolimus bioavailability. When given immediately following the meal, mean C max was reduced 71%, and mean AUC was reduced 39%, relative to the fasted condition. When administered 1.5 hours following the meal, mean C max was reduced 63%, and mean AUC was reduced 39%, relative to the fasted condition.In 11 liver transplant patients, tacrolimus capsules administered 15 minutes after a high fat (400 kcal, 34% fat) breakfast, resulted in decreased AUC (27 ± 18%) and C max (50 ± 19%), as compared to a fasted state. DistributionThe plasma protein binding of tacrolimus is approximately 99% and is independent of concentration over a range of 5-50 ng/mL. Tacrolimus is bound mainly to albumin and alpha-1-acid glycoprotein, and has a high level of association with erythrocytes. The distribution of tacrolimus between whole blood and plasma depends on several factors, such as hematocrit, temperature at the time of plasma separation, drug concentration, and plasma protein concentration. In a U.S. study, the ratio of whole blood concentration to plasma concentration averaged 35 (range 12 to 67). MetabolismTacrolimus is extensively metabolized by the mixed-function oxidase system, primarily the cytochrome P-450 system (CYP3A). A metabolic pathway leading to the formation of 8 possible metabolites has been proposed. Demethylation and hydroxylation were identified as the primary mechanisms of biotransformation in vitro . The major metabolite identified in incubations with human liver microsomes is 13-demethyl tacrolimus. In in vitro studies, a 31-demethyl metabolite has been reported to have the same activity as tacrolimus. ExcretionThe mean clearance following IV administration of tacrolimus is 0.040, 0.083, and 0.053, in healthy volunteers, adult kidney transplant patients, adult liver transplant patients, respectively. In man, less than 1% of the dose administered is excreted unchanged in urine.In a mass balance study of IV administered radiolabeled tacrolimus to 6 healthy volunteers, the mean recovery of radiolabel was 77.8±12.7%. Fecal elimination accounted for 92.4±1% and the elimination half-life based on radioactivity was 48.1±15.9 hours whereas it was 43.5±11.6 hours based on tacrolimus concentrations. The mean clearance of radiolabel was 0.029±0.015 L/hr/kg and clearance of tacrolimus was 0.029±0.009 L/hr/kg. When administered PO, the mean recovery of the radiolabel was 94.9±30.7%. Fecal elimination accounted for 92.6±30.7%, urinary elimination accounted for 2.3±1.1% and the elimination half-life based on radioactivity was 31.9±10.5 hours whereas it was 48.4± 12.3 hours based on tacrolimus concentrations. The mean clearance of radiolabel was 0.226±0.116 L/hr/kg and clearance of tacrolimus 0.172± 0.088 L/hr/kg. Special Populations PediatricPharmacokinetics of tacrolimus have been studied in liver transplantation patients, 0.7 to 13.2 years of age. Following oral administration to 9 patients, mean AUC and C max were 337 ± 167 ng±hr/mL and 48.4 ± 27.9 ng/mL, respectively. The absolute bioavailability was 31 ± 24%.Whole blood trough concentrations from 31 patients less than 12 years old showed that pediatric patients needed higher doses than adults to achieve similar tacrolimus trough concentrations. (See DOSAGE AND ADMINISTRATION ).Renal and Hepatic InsufficiencyThe mean pharmacokinetic parameters for tacrolimus following single administrations to patients with renal and hepatic impairment are given in the following table. Population(No. of Patients) Dose AUC 0 - t (ng·hr/mL) t ½ (hr) V (L/kg) CI(L/hr/kg) RenalImpairment (n=12)0.02 mg/kg/4hrIV 393 ± 123 (t = 60 hr) 26.3 ± 9.2 1.07 ±0.20 0.038 ± 0.014 Mild Hepatic Impairment (n=6)0.02 mg/kg/4hrIV 367 ± 107 (t = 72 hr) 60.6 ± 43.8 Range: 27.8-141 3.1±1.6 0.042 ± 0.02 7.7 mg PO488 ± 320 (t = 72 hr) 66.1 ± 44.8 Range: 29.5-138 3.7 ± 4.7* 0.034 ± 0.019* Severe Hepatic Impairment (n=6, IV)0.02 mg/kg/4hr IV (n=2) 762 ± 204 (t = 120 hr) 198 ± 158 Range: 81-4363.9 ± 10.017 ± 0.013(n=5, PO)†0.01 mg/kg/8hr IV (n=4) 8 mg PO (n=1) 289 ± 117 (t = 144 hr)658 (t =120 hr) 119 ± 35 Range: 85-1783.1 ± 3.4*0.016 ± 0.011*5 mg PO (n=4) 4 mg PO (n=1)533 ± 156 (t = 144 hr)*corrected for bioavailability; †1 patient did not receive the PO doseRenal InsufficiencyTacrolimus pharmacokinetics following a single IV administration were determined in 12 patients (7 not on dialysis and 5 on dialysis, serum creatinine of 3.9 ± 1.6 and 12 ± 2.4 mg/dL, respectively) prior to their kidney transplant. The pharmacokinetic parameters obtained were similar for both groups.The mean clearance of tacrolimus in patients with renal dysfunction was similar to that in normal volunteers (see previous table). Hepatic InsufficiencyTacrolimus pharmacokinetics have been determined in six patients with mild hepatic dysfunction (mean Pugh score: 6.2) following single IV and oral administrations. The mean clearance of tacrolimus in patients with mild hepatic dysfunction was not substantially different from that in normal volunteers (see previous table). Tacrolimus pharmacokinetics were studied in 6 patients with severe hepatic dysfunction (mean Pugh score: >10). The mean clearance was substantially lower in patients with severe hepatic dysfunction, irrespective of the route of administration. RaceA formal study to evaluate the pharmacokinetic disposition of tacrolimus in Black transplant patients has not been conducted. However, a retrospective comparison of Black and Caucasian kidney transplant patients indicated that Black patients required higher tacrolimus doses to attain similar trough concentrations. (See DOSAGE AND ADMINISTRATION ). GenderA formal study to evaluate the effect of gender on tacrolimus pharmacokinetics has not been conducted, however, there was no difference in dosing by gender in the kidney transplant trial. A retrospective comparison of pharmacokinetics in healthy volunteers, and in kidney and liver transplant patients indicated no gender-based differences.CLINICAL STUDIES Liver TransplantationThe safety and efficacy of tacrolimus-based immunosuppression following orthotopic liver transplantation were assessed in two prospective, randomized, non-blinded multicenter studies. The active control groups were treated with a cyclosporine-based immunosuppressive regimen. Both studies used concomitant adrenal corticosteroids as part of the immunosuppressive regimens. These studies were designed to evaluate whether the two regimens were therapeutically equivalent, with patient and graft survival at 12 months following transplantation as the primary endpoints. The tacrolimus-based immunosuppressive regimen was found to be equivalent to the cyclosporine-based immunosuppressive regimens.In one trial, 529 patients were enrolled at 12 clinical sites in the United States; prior to surgery, 263 were randomized to the tacrolimus-based immunosuppressive regimen and 266 to a cyclosporine-based immunosuppressive regimen (CBIR). In 10 of the 12 sites, the same CBIR protocol was used, while 2 sites used different control protocols. This trial excluded patients with renal dysfunction, fulminant hepatic failure with Stage IV encephalopathy, and cancers; pediatric patients (≤ 12 years old) were allowed.In the second trial, 545 patients were enrolled at 8 clinical sites in Europe; prior to surgery, 270 were randomized to the tacrolimus-based immunosuppressive regimen and 275 to CBIR. In this study, eachcenter used its local standard CBIR protocol in the active-control arm. This trial excluded pediatric patients, but did allow enrollment of subjects with renal dysfunction, fulminant hepatic failure in Stage IV encephalopathy, and cancers other than primary hepatic with metastases.One-year patient survival and graft survival in the tacrolimus -based treatment groups were equivalent to those in the CBIR treatment groups in both studies. The overall 1-year patient survival (CBIR and tacrolimusbased treatment groups combined) was 88% in the U.S. study and 78% in the European study. The overall 1-year graft survival (CBIR and tacrolimus-based treatment groups combined) was 81% in the U.S. study and 73% in the European study. In both studies, the median time to convert from IV to oral tacrolimus capsules dosing was 2 days.Because of the nature of the study design, comparisons of differences in secondary endpoints, such as incidence of acute rejection, refractory rejection or use of OKT3 for steroid-resistant rejection, could not be reliably made.Kidney Transplantation Tacrolimus/azathioprineTacrolimus-based immunosuppression in conjunction with azathioprine and corticosteroids following kidney transplantation was assessed in a Phase 3 randomized, multicenter, non-blinded, prospective study. There were 412 kidney transplant patients enrolled at 19 clinical sites in the United States. Study therapy was initiated when renal function was stable as indicated by a serum creatinine ≤ 4 mg/dL (median of 4 days after transplantation, range 1 to 14 days). Patients less than 6 years of age were excluded.There were 205 patients randomized to tacrolimus-based immunosuppression and 207 patients were randomized to cyclosporine-based immunosuppression. All patients received prophylactic induction therapy consisting of an antilymphocyte antibody preparation, corticosteroids and azathioprine. Overall 1 year patient and graft survival was 96.1% and 89.6%, respectively and was equivalent between treatment arms. Because of the nature of the study design, comparisons of differences in secondary endpoints, such as incidence of acute rejection, refractory rejection or use of OKT3 for steroid-resistant rejection, could not be reliably made.Tacrolimus/mycophenolate mofetil(MMF)Tacrolimus-based immunosuppression in conjunction with MMF, corticosteroids, and induction has been studied. In a randomized, open-label, multi-center trial (Study 1), 1589 kidney transplant patients received tacrolimus (Group C, n=401), sirolimus (Group D, n=399), or one of two cyclosporine regimens (Group A, n=390 and Group B, n=399) in combination with MMF and corticosteroids; all patients, except those in one of the two cyclosporine groups, also received induction with daclizumab. The study was conducted outside the United States; the study population was 93% Caucasian. In this study, mortality at 12 months in patients receiving tacrolimus/MMF was similar (2.7%) compared to patients receiving cyclosporine/MMF (3.3% and 1.8%) or sirolimus/MMF (3 %). Patients in the tacrolimus group exhibited higher estimated creatinine clearance rates (eCL cr ) using the Cockcroft-Gault formula (Table 1) and experienced fewer efficacy failures, defined as biopsy proven acute rejection (BPAR), graft loss, death, and/or lost to follow-up (Table 2) in comparison to each of the other three groups. Patients randomized to tacrolimus/MMF were more likely to develop diarrhea and diabetes after the transplantation and experienced similar rates of infections compared to patients randomized to either cyclosporine/MMF regimen (see ADVERSE REACTIONS ).Table 1: Estimated Creatinine Clearance at 12 Months in Study 1 GroupeCLcr [mL/min] at Month 12a N MEAN SD MEDIAN Treatment Differencewith Group C (99.2%CI b )(A) CsA/MMF/CS390 56.5 25.8 56.9 -8.6 (-13.7,-3.7) (B) CsA/MMF/CS/Daclizumab 399 58.9 25.6 60.9 -6.2 (-11.2,-1.2) (C) Tac/MMF/CS/Daclizumab 401 65.1 27.4 66.2 -(D) Siro/MMF/CS/Daclizumab 399 56.2 27.4 57.3 -8.9 (-14.1, -3.9) Total158959.226.860.5Key: CsA=Cyclosporine, CS=Corticosteroids, Tac=Tacrolimus, Siro=Sirolimusa) All death/graft loss (n=41,27, 23 and 42 in Groups A, B, C and D) and patients whose last recorded creatinine values were prior to month 3 visit (n=10, 9, 7 and 9 in Groups A, B, C and D) were inputed with GFR of 10 mL/min; a subject’s last observed creatinine value from month 3 on was used for the remainder of subjects with missing creatinine at month 12 (n=11, 12, 15 and 19 for Groups A, B, C and D). Weight was also imputed in the calculation of estimated GFR, if missing b) Adjusted for multiple (6) pairwise comparisons using Bonferroni corrections.Table 2: Incidence of BPAR, Graft Loss, Death or Loss to Follow-up at 12 Months in Study 1ABCDN=390N=399N=401 N=399Overall Failure141 (36.2%) 126(31.6%) 82 (20.4%) 185 (46.4%) Components of efficacy failure BPAR113(29%) 106 (26.6%) 60 (15%) 152(38.1%) Graft loss excluding death 28 (7.2%) 20 (5%) 12 (3%) 30 (7.5%) Mortality13 (3.3%) 7(1.8%) 11(2.7%) 12 (3%) Lost to follow-up5(1.3%) 7(1.8%)5 (1.3%)6(1.5%) Treatment Difference of efficacy failure compared to 15.8% (7.1%, 11.2% (2.7%, -26% (17.2%, Group C (99.2% CI a )24.3%)19.5%)34.7%)Group A =CsA/MMF/CS, B =CsA/MMF/CS/Daclizumab, C=Tac/MMF/CS/Daclizumab, and D=Siro/MMF/CS/Daclizumab a) Adjusted for multiple (6) pairwise comparisons using Bonferroni corrections.The protocol-specified target tacrolimus trough concentrations (C ,Tac) were 3-7 ng/mL; however, the trough observed median C ,Tac approximated 7 ng/mL throughout the 12 month study (Table 3).troughs Table 3: Tacrolimus Whole Blood Trough Concentrations (Study 1) TimeMedian (P10-P90 a ) tacrolimus whole blood trough concentrations (ng/mL) Day 30 (N=366) 6.9 (4.4-11.3) Day90 (N=351) 6.8 (4.1-10.7) Day 180 (N=355) 6.5 (4 - 9.6) Day 365 (N=346)6.5 (3.8 -10)a) Range of C trough , Tac that excludes lowest 10% and highest 10% of C trough , TacThe protocol-specified target cyclosporine trough concentrations (C trough ,CsA) for Group B were 50-100ng/mL; however, the observed median C trougs , CsA approximated 100 ng/mL throughout the 12 month study. The protocol-specified target C trougs ,CsA for Group A were 150-300 ng/mL for the first 3 months and 100-200 ng/mL from month 4 to month 12; the observed median C trougs , CsA approximated 225 ng/mL for the first 3 months and 140 ng/mL from month 4 to month 12.While patients in all groups started MMF at 1g BID, the MMF dose was reduced to <2 g/day in 63% of patients in the tacrolimus treatment arm by month 12 (Table 4); approximately 50% of these MMF dose reductions were due to adverse events. By comparison, the MMF dose was reduced to <2 g/day in 49% and 45% of patients in the two cyclosporine arms (Group A and Group B, respectively), by month 12 and approximately 40% of MMF dose reductions were due to adverse events. Table 4: MMF Dose Over Time in tacrolimus/MMF (Group C) (Study 1) Time period (Days) Time-averaged MMF dose (g/day)a <2 2 >20-30 (N=364) 37% 60% 2% 0-90 (N=373) 47% 51% 2% 0-180 (N=377) 56% 42% 2% 0-365 (N=380)63% 36%1%Time-averaged MMF dose = (total MMF dose)/(duration of treatment)a) Percentage of patients for each time-averaged MMF dose range during various treatment periods. Two g/day of time-averaged MMF dose means that MMF dose was not reduced in those patients during the treatment periods.In a second randomized, open-label, multi-center trial (Study 2), 424 kidney transplant patients received tacrolimus (n=212) or cyclosporine (n=212) in combination with MMF 1 gram BID, basiliximab induction, and corticosteroids. In this study, the rate for the combined endpoint of biopsy proven acute rejection, graft failure, death, and/or lost to follow-up at 12 months in the tacrolimus/MMF group was similar to the rate in the cyclosporine/MMF group. There was, however, an imbalance in mortality at 12 months in those patients receiving tacrolimus/MMF (4.2%) compared to those receiving cyclosporine/MMF (2.4%), including cases attributed to overimmunosuppression (Table 5).Table 5: Incidence of BPAR, Graft Loss, Death or Loss to Follow-up at 12 Months in Study 2Tacrolimus/MMF (n=212) Cyclosporine/MMF (n=212) Overall Failure32(15.1%) 36 (17%) Components of efficacy failure BPAR16 (7.5%) 29 (13.7%) Graft loss excluding death 6 (2.8%) 4(1.9%) Mortality9 (4.2%) 5 (2.4%) Lost to follow-up4(1.9%) 1 (0.5%) Treatment Difference of efficacy failure compared to tacrolimus/MMF group (95% CI a )- 1.9% (-5.2%, 9%)a) 95% confidence interval calculated using Fisher’s Exact TestThe protocol-specified target tacrolimus whole blood trough concentrations (C trough ,Tac) in Study 2 were 716 ng/mL for the first three months and 5-15 ng/mL thereafter. The observed median C troughs ,Tac approximated 10 ng/mL during the first three months and 8 ng/mL from month 4 to month 12 (Table 6). Table 6: Tacrolimus Whole Blood Trough Concentrations (Study 2) TimeMedian (P10-P90a ) tacrolimus whole blood trough concentrations ng/mLDay 30 (N=174) 10.5 (6.3 -16.8) Day 60 (N=179) 9.2(5.9-15.3) Day 120 (N=176) 8.3 (4.6 -13.3) Day 180(N=171) 7.8 (5.5 -13.2) Day 365 (N=178)7.1(4.2-12.4)a) Range of C troughs , Tac that excludes lowest 10% and highest 10% of C troughs TacThe protocol-specified target cyclosporine whole blood concentrations (Ctr o ugh,CsA) were 125 to 400 ng/ mL for the first three months, and 100 to 300 ng/mL thereafter. The observed median Ctroughs, CsA approximated 280 ng/mL during the first three months and 190 ng/mL from month 4 to month 12.Patients in both groups started MMF at 1g BID. The MMF dose was reduced to <2 g/day by month 12 in 62% of patients in the tacrolimus/MMF group (Table 7) and in 47% of patients in the cyclosporine/MMF group. Approximately 63% and 55% of these MMF dose reductions were because of adverse events in the tacrolimus/MMF group and the cyclosporine/MMF group, respectively. Table 7: MMF Dose Over Time in the tacrolimus/MMF group (Study 2) Time period (Days) Time-averaged MMF dose (g/day)a <2 2 >20-30(N=212) 25% 69% 6% 0-90 (N=212) 41% 53% 6% 0-180 (N=212) 52% 41% 7% 0-365 (N=212)62% 34%4%Time-averaged MMF dose=(total MMF dose)/(duration of treatment)a) Percentage of patients for each time-averaged MMF dose range during various treatment periods. Two g/day of time-averaged MMF dose means that MMF dose was not reduced in those patients during the treatment periods.INDICATIONS AND USAGETacrolimus capsules are indicated for the prophylaxis of organ rejection in patients receiving allogeneic liver, or kidney transplants. It is recommended that tacrolimus be used concomitantly with adrenal corticosteroids. In kidney transplant recipients, it is recommended that tacrolimus be used in conjunction with azathioprine or mycophenolate mofetil (MMF). The safety and efficacy of the use of tacrolimus with sirolimus has not been established (see CLINICAL STUDIES ).CONTRAINDICATIONSTacrolimus capsules are contraindicated in patients with a hypersensitivity to tacrolimus.WARNINGS(See boxed WARNING .)Post-Transplant Diabetes MellitusInsulin-dependent post-transplant diabetes mellitus (PTDM) was reported in 20% of tacrolimus-treated kidney transplant patients without pretransplant history of diabetes mellitus in the Phase III study (See Tables Below). The median time to onset of PTDM was 68 days. Insulin dependence was reversible in 15% of these PTDM patients at one year and in 50% at 2 years post transplant. Black and Hispanic kidney transplant patients were at an increased risk of development of PTDM.Incidence of Post Transplant Diabetes Mellitus and Insulin Use at 2 Years in Kidney Transplant Recipients in the Phase III studyStatus of PTDM*Tacrolimus CBIR Patients without pretransplant history of diabetes mellitus. 151151New onset PTDM*, 1st Year30/151 (20%) 6/151 (4%) Still insulin dependent at one year in those without prior history of diabetes.25/151 (17%) 5/151 (3%) New onset PTDM* post 1 year 1Patients with PTDM* at 2 years16/151 (11%)5/151 (3%)* use of insulin for 30 or more consecutive days, with <5 day gap, without a prior history of insulin dependent diabetes mellitus or non insulin dependent diabetes mellitus.Development of Post Transplant Diabetes Mellitus by Race and by Treatment Group during First Year Post Kidney Transplantation in the Phase III study TacrolimusCBIRPatient Race No. of Patients at Risk Patients Who Developed PTDM* No. of PatientsAt Risk Patients WhoDeveloped PTDM* Black 41 15 (37%) 36 3 (8%) Hispanic 17 5 (29%) 18 1 (6%) Caucasian 82 10 (12%) 87 1 (1%) Other 11 0 (0%) 10 1 (10%) Total151 30 (20%)1516 (4%)*use of insulin for 30 or more consecutive days, with <5 day gap, without a prior history of insulin dependent diabetes mellitus or non insulin dependent diabetes mellitus.Insulin-dependent post-transplant diabetes mellitus was reported in 18% and 11% of tacrolimus-treated liver transplant patients and was reversible in 45% and 31% of these patients at 1 year post transplant, in the U.S. and European randomized studies, respectively (See Table below). Hyperglycemia was associated with the use of tacrolimus in 47% and 33% of liver transplant recipients in the U.S. and European randomized studies, respectively, and may require treatment (see ADVERSE REACTIONS )Incidence of Post Transplant Diabetes Mellitus and Insulin Use at 1 Year in Liver Transplant Recipients Status of PTDM*U.S. StudyEuropean StudyTacrolimus CBIR Tacrolimus CBIR Patients at risk** 239 236 239 249 New Onset PTDM*42(18%) 30(13%) 26(11%) 12(5%) Patients still on insulin at 1 year23(10%) 19(8%)18(8%) 6 (2%)* use of insulin for 30 or more consecutive days, with < 5 day gap, without a prior history of insulin dependent diabetesmellitus or non insulin dependent diabetes mellitus** Patients without pretransplant history of diabetes mellitusNephrotoxicityTacrolimus can cause nephrotoxicity, particularly when used in high doses. Nephrotoxicity was reported in approximately 52% of kidney transplantation patients and in 40% and 36% of liver transplantation patients receiving tacrolimus in the U.S. and European randomized trials, respectively (see ADVERSE REACTIONS ). More overt nephrotoxicity is seen early after transplantation, characterized by increasing serum creatinine and a decrease in urine output. Patients with impaired renal function should be monitored closely as the dosage of tacrolimus may need to be reduced. In patients with persistent elevations of serum creatinine who are unresponsive to dosage adjustments, consideration should be given to changing to another immunosuppressive therapy. Care should be taken in using tacrolimus with other nephrotoxic drugs. In particular, to avoid excess nephrotoxicity, tacrolimus should not be used simultaneously with cyclosporine. tacrolimus or cyclosporine should be discontinued at least 24 hours prior to initiating the other. In the presence of elevated tacrolimus or cyclosporine concentrations, dosing with the other drug usually should be further delayed. HyperkalemiaMild to severe hyperkalemia was reported in 31% of kidney transplant recipients and in 45% and 13% of liver transplant recipients treated with tacrolimus capsules in the U.S. and European randomized trials, respectively, and may require treatment (see ADVERSE REACTIONS ). Serum potassium levels should be monitored and potassium-sparing diuretics should not be used during tacrolimus capsules therapy (see PRECAUTIONS ). NeurotoxicityTacrolimus capsules can cause neurotoxicity, particularly when used in high doses. Neurotoxicity, including tremor, headache, and other changes in motor function, mental status, and sensory function were reported in approximately 55% of liver transplant recipients in the two randomized studies. Tremor occurred more often in tacrolimus capsules-treated kidney transplant patients (54%) compared to cyclosporine-treated patients. The incidence of other neurological events in kidney transplant patients was similar in the two treatment groups (see ADVERSE REACTI ONS ). Tremor and headache have been associated with high whole-blood concentrations of tacrolimus and may respond to dosage adjustment. Seizures have occurred in adult and pediatric patients receiving tacrolimus capsules (see ADVERSE REACTIONS ). Coma and delirium also have been associated with high plasma concentrations of tacrolimus.Patients treated with tacrolimus have been reported to develop posterior reversible encephalopathy syndrome (PRES). Symptoms indicating PRES include headache, altered mental status, seizures, visual disturbances and hypertension. Diagnosis may be confirmed by radiological procedure. If PRES is suspected or diagnosed, blood pressure control should be maintained and immediate reduction of immunosuppression is advised. This syndrome is characterized by reversal of symptoms upon reduction or discontinuation of immunosuppression.Malignacy and Lymphoproliferative DisordersAs in patients receiving other immunosuppressants, patients receiving tacrolimus capsules are at increased risk of developing lymphomas and other malignancies, particularly of the skin. The risk appears to be related to the intensity and duration of immunosuppression rather than to the use of any specific agent. A lymphoproliferative disorder (LPD) related to Epstein-Barr Virus (EBV) infection has been reported in immunosuppressed organ transplant recipients. The risk of LPD appears greatest in young children who are at risk for primary EBV infection while immunosuppressed or who are switched to tacrolimus capsules following long-term immunosuppression therapy. Because of the danger of oversuppression of the immune system which can increase susceptibility to infection, combination immunosuppressant therapy should be used with caution. Latent Viral InfectionsImmunosuppressed patients are at increased risk for opportunistic infections, including latent viral infections. These include BK virus associated nephropathy and JC virus associated progressive multifocal leukoencephalopathy (PML) which have been observed in patients receiving tacrolimus. These infections may lead to serious, including fatal, outcomes.PRECAUTIONS GeneralHypertension is a common adverse effect of tacrolimus therapy (see ADVERSE REACTIONS ). Mild or moderate hypertension is more frequently reported than severe hypertension. Antihypertensive therapy may be required; the control of blood pressure can be accomplished with any of the common antihypertensive agents. Since tacrolimus may cause hyperkalemia, potassium-sparing diuretics should be avoided. While calcium-channel blocking agents can be effective in treating tacrolimus-associated hypertension, care should be taken since interference with tacrolimus metabolism may require a dosage reduction (see Drug Interactions ).Renally and Hepatically Impaired PatientsFor patients with renal insufficiency some evidence suggests that lower doses should be used (see CLINICAL PHARMACOLOGY and DOSAGE AND ADMINISTRATION ).The use of tacrolimus capsules in liver transplant recipients experiencing post-transplant hepatic impairment may be associated with increased risk of developing renal insufficiency related to high whole-blood levels of tacrolimus. These patients should be monitored closely and dosage adjustments should be considered. Some evidence suggests that lower doses should be used in these patients (see DOSAGE AND ADMINISTRATION ). Myocardial HypertrophyMyocardial hypertrophy has been reported in association with the administration of tacrolimus capsules, and is generally manifested by echocardiographically demonstrated concentric increases in left ventricular posterior wall and interventricular septum thickness. Hypertrophy has been observed in infants, children and adults. This condition appears reversible in most cases following dose reduction or discontinuance of therapy. In a group of 20 patients with pre- and post-treatment echocardiograms who showed evidence of myocardial hypertrophy, mean tacrolimus whole blood concentrations during the period prior to diagnosis of myocardial hypertrophy ranged from 11 to 53 ng/mL in infants (N=10, age 0.4 to 2 years), 4 to 46 ng/ mL in children (N=7, age 2 to 15 years) and 11 to 24 ng/mL in adults (N=3, age 37 to 53 years).In patients who develop renal failure or clinical manifestations of ventricular dysfunction while receiving tacrolimus therapy, echocardiographic evaluation should be considered. If myocardial hypertrophy is diagnosed, dosage reduction or discontinuation of tacrolimus should be considered. Information for PatientsPatients should be informed of the need for repeated appropriate laboratory tests while they are receiving tacrolimus capsules. They should be given complete dosage instructions, advised of the potential risks during pregnancy, and informed of the increased risk of neoplasia. Patients should be informed that changes in dosage should not be undertaken without first consulting their physician.Patients should be informed that tacrolimus capsules can cause diabetes mellitus and should be advised of the need to see their physician if they develop frequent urination, increased thirst or hunger.As with other immunosuppressive agents, owing to the potential risk of malignant skin changes, exposure to sunlight and ultraviolet (UV) light should be limited by wearing protective clothing and using a sunscreen with a high protection factor. Laboratory TestsSerum creatinine, potassium, and fasting glucose should be assessed regularly. Routine monitoring of metabolic and hematologic systems should be performed as clinically warranted. Drug InteractionsDue to the potential for additive or synergistic impairment of renal function, care should be taken when administering tacrolimus capsules with drugs that may be associated with renal dysfunction. These include, but are not limited to, aminoglycosides, amphotericin B, and cisplatin. Initial clinical experience with the co-administration of tacrolimus and cyclosporine resulted in additive/synergistic nephrotoxicity. Patients switched from cyclosporine to tacrolimus should receive the first tacrolimus capsules dose no sooner than 24 hours after the last cyclosporine dose. Dosing may be further delayed in the presence of elevated cyclosporine levels.Drugs that May Alter Tacrolimus ConcentrationsSince tacrolimus is metabolized mainly by the CYP3A enzyme systems, substances known to inhibit these enzymes may decrease the metabolism or increase bioavailability of tacrolimus as indicated by increased whole blood or plasma concentrations. Drugs known to induce these enzyme systems may result in an increased metabolism of tacrolimus or decreased bioavailability as indicated by decreased whole blood or plasma concentrations. Monitoring of blood concentrations and appropriate dosage adjustments are essential when such drugs are used concomitantly.*Drugs That May Increase Tacrolimus Blood Concentrations Calcium Channel Blockers Antifungal Agents Macrolide Antibiotics diltiazem clotrimazole clarithromycin nicardipine fluconazole erythromycin nifedipine itraconazole troleandomycinverapamilketoconazole** voriconazoleGastrointestinal Prokinetic AgentsOther Drugs cisapride bromocriptine metoclopramidechloramphenicol cimetidine cyclosporine danazol ethinyl estradiol methylprednisolone lansoprazole*** omeprazole protease inhibitors nefazodonemagnesium-aluminum-hydroxide* This table is not all inclusive**In a study of 6 normal volunteers, a significant increase in tacrolimus oral bioavailability (14±5% vs. 30±8%) was observed with concomitant ketoconazole administration (200 mg). The apparent oral clearance of tacrolimus during ketoconazole administration was significantly decreased compared to tacrolimus alone (0.430±0.129 L/hr/kg vs. 0.148±0.043 L/hr/kg). Overall, IV clearance of tacrolimus was not significantly changed by ketoconazole co-administration, although it was highly variable between patients.*** Lansoprazole (CYP2C19, CYP3A4 substrate) may potentially inhibit CYP3A4-mediated metabolism of tacrolimus and thereby substantially increase tacrolimus whole blood concentrations, especially in transplant patients who are intermediate or poor CYP2C19 metabolizers, as compared to those patients who are efficient CYP2C19 metabolizers.*Drugs That May Decrease Tacrolimus Blood ConcentrationsAnticonvulsants Antimicrobialscarbamazepine rifabutin phenobarbital caspofungin phenytoinrifampin Herbal Preparations Other Drugs St. John’s Wort sirolimus*This table is not all inclusive.St. John’s Wort (Hypericum perforatum) induces CYP3A4 and P-glycoprotein. Since tacrolimus is a substrate for CYP3A4, there is the potential that the use of St. John’s Wort in patients receiving tacrolimus capsules could result in reduced tacrolimus levels.In a single-dose crossover study in healthy volunteers, co-administration of tacrolimus and magnesiumaluminum-hydroxide resulted in a 21% increase in the mean tacrolimus AUC and a 10% decrease in the mean tacrolimus C max relative to tacrolimus administration alone.In a study of 6 normal volunteers, a significant decrease in tacrolimus oral bioavailability (14 ± 6% vs. 7 ± 3%) was observed with concomitant rifampin administration (600 mg). In addition, there was a significant increase in tacrolimus clearance (0.036 ± 0.008 L/hr/kg vs. 0.053 ± 0.01 L/hr/kg) with concomitant rifampin administration.Interaction studies with drugs used in HIV therapy have not been conducted. However, care should be exercised when drugs that are nephrotoxic (e.g., ganciclovir) or that are metabolized by CYP3A (e.g., nelfinavir, ritonavir) are administered concomitantly with tacrolimus. Based on a clinical study of 5 liver transplant recipients, co-administration of tacrolimus with nelfinavir increased blood concentrations of tacrolimus significantly and, as a result, a reduction in the tacrolimus dose by an average of 16-fold was needed to maintain mean trough tacrolimus blood concentrations of 9.7 ng/mL. Thus, frequent monitoring of tacrolimus blood concentrations and appropriate dosage adjustments are essential when nelfinavir is used concomitantly. Tacrolimus may affect the pharmacokinetics of other drugs (e.g., phenytoin) and increase their concentration. Grapefruit juice affects CYP3A-mediated metabolism and should be avoided (see DOSAGE AND ADMINISTRATION ).Following co-administration of tacrolimus and sirolimus (2 or 5 mg/day) in stable renal transplant patients, mean tacrolimus AUC 0-12 and C min decreased approximately by 30% relative to tacrolimus alone. Mean tacrolimus AUC 0-12 and C min following co-administration of 1 mg/day of sirolimus decreased approximately 3% and 11%, respectively. The safety and efficacy of tacrolimus used in combination with sirolimus for the prevention of graft rejection has not been established and is not recommended. Other Drug InteractionsImmunosuppressants may affect vaccination. Therefore, during treatment with tacrolimus capsules, vaccination may be less effective. The use of live vaccines should be avoided; live vaccines may include, but are not limited to measles, mumps, rubella, oral polio, BCG, yellow fever, and TY 21a typhoid.1At a given MMF dose, mycophenolic acid (MPA) exposure is higher with tacrolimus co-administration than with cyclosporine co-administration due to the differences in the interruption of the enterohepatic recirculation of MPA. Clinicians should be aware that there is also a potential for increased MPA exposure after crossover from cyclosporine to tacrolimus in patients concomitantly receiving MMF or MPA. Carcinogenesis, Mutagenesis, Impairment of FertilityAn increased incidence of malignancy is a recognized complication of immunosuppression in recipients of organ transplants. The most common forms of neoplasms are non-Hodgkin’s lymphomas and carcinomas of the skin. As with other immunosuppressive therapies, the risk of malignancies in tacrolimus recipients may be higher than in the normal, healthy population. Lymphoproliferative disorders associated with Epstein-Barr Virus infection have been seen. It has been reported that reduction or discontinuation of immunosuppression may cause the lesions to regress.No evidence of genotoxicity was seen in bacterial (Salmonella and E. coli) or mammalian (Chinese hamster lung-derived cells) in vitro assays of mutagenicity, the in vitro CHO/HGPRT assay of mutagenicity, or in vivo clastogenicity assays performed in mice; tacrolimus did not cause unscheduled DNA synthesis in rodent hepatocytes.Carcinogenicity studies were carried out in male and female rats and mice. In the 80-week mouse study and in the 104-week rat study no relationship of tumor incidence to tacrolimus dosage was found. The highest doses used in the mouse and rat studies were 0.8 - 2.5 times (mice) and 3.5 - 7.1 times (rats) the recommended clinical dose range of 0.1 - 0.2 mg/kg/day when corrected for body surface area.No impairment of fertility was demonstrated in studies of male and female rats. Tacrolimus, given orally at 1 mg/kg (0.7 - 1.4X the recommended clinical dose range of 0.1 - 0.2 mg/kg/day based on body surface area corrections) to male and female rats, prior to and during mating, as well as to dams during gestation and lactation, was associated with embryolethality and with adverse effects on female reproduction. Effects on female reproductive function (parturition) and embryolethal effects were indicated by a higher rate of pre-implantation loss and increased numbers of undelivered and nonviable pups. When given at 3.2 mg/kg (2.3 - 4.6X the recommended clinical dose range based on body surface area correction), tacrolimus was associated with maternal and paternal toxicity as well as reproductive toxicity including marked adverse effects on estrus cycles, parturition, pup viability, and pup malformations.。