间位取代卟啉及其锌卟啉的电子吸收光谱
一种烟酸取代的尾式卟啉及其锌、锰配合物的合成与表征

t e d t n te p rn  ̄ e r te r t a. h a a i h a e f e s a e h oei 1 l s c
・ ,
・
《 >
>c o a o N
分
—
S h me e s n h ss r u e o i a i c d h i d p r h rn c e l Th y t e i o t f n c c d a i a l o p y i e l a d a d i o lx s i n g n t c mp e e s
1实验 酸 钠 的 合 成 . 3烟 2 取 2 g烟 酸 溶 于 2 0 mL
i§ 一 r 三
元素分析通过 P ri— lme20 自动元 热水中, ekn Ee r4C 滴加氢 氧化钠 溶液( 1 ∞ 素 分 析 仪 测 量 ;红 外 光谱 通 过 Ni lt5 C t l ) P c e P o / 调 H至 8~ , 浴蒸 o oL 9水  ̄ -R 红 外光谱仪 测量( B TI K r压 片)紫外可 见 干 。 ; 1 . 烟 酸 尾 式 卟 啉 的 .4 2 光 谱在 Si a z V一 4 波谱 仪上 测量 ;H hm du U 20 I N MR 在 V r n U l - 0 5 0 MH ) ai nt 5 0 f0 z波谱 仪 上 合 成 a y 一 测 量 (D 1为溶 剂 )尾 式 5 (~烟 酸 酰 氧 基 乙 C C ; 一4 在 装 有 回 流 冷 凝 管 的 Fg 2 i UV- s s e t Vi p c r o a f Fg 1 H i 1 NM R s e tu f 。 p cr m o 氧基) 苯基 一 ,1,1一三 苯 基 卟 啉 (E 5 0 5 N 及 10 mL圆 底 烧 瓶 中加 入 6 0 0 N P P () E T P nh E T P a,N P P Z () NEPT P P g烟 酸 钠 和 01 o 单 溴 . mm l 3 其 Mn n配 合 物 (E T P n E T P C) ,z N P P Z ,N P P Mn 1 的 m a d N P P Mn I(C n E T P C ) 合 成 路 线 如 S hme 1 示 。 ce 所 代 卟 啉 醚 ,并 加 人 2 0 mL D , 浴 7 加热 搅拌反 MF水 5 1 . 验 过程 2实 ; 珏 1 . 5 f一羟 基 ) 基 一 O1,O . 1 -4 2 苯 1,52 一三 苯 基 应, 薄层 层析法检测反应 并用 进程 , 反应 基本 完全, 待 停止 卟啉 (— pH 的合 成 3 参考 文献【】 1, 并做 了适 当改进 , 成 了 5 反应, 合 一 冷却, 加入 6 L饱 和 0m 食盐水使产物 析出,过滤, 分 ( 4 ~羟基) 苯基 一 Ol,0 1 , 2 一三苯基 卟啉 。 5 干 将 63 .6 g(00 o)苯甲醛 、. O.2 别 用蒸 馏 水 和 甲醇洗 涤 , . m1 6 24 g 0 4 mo r 基 苯 甲醛 和 2 0 m 1 羟 ) 0 L丙 酸 加 入 到 50 燥, 0 粗产 品以 A2, 固定相 , l 为 0 mL三颈瓶中, 在磁力搅拌下加热 回流, 体系控制 氯 仿 为 洗 脱 液进 行 柱 层 析 分 F g I s e tu o i 3 R p c r m f 收集第 二 红色 带, 去溶 蒸 在微沸状态. 用滴液漏斗缓慢滴加 5 I新蒸 离 , . m 6 NE P P 吡咯与 2 L丙酸的混合溶液,0 m n加完, 0m 3 i 滴 剂,用氯仿 一甲醇重结 晶, 得 定 v— i方 即烟酸尾式卟啉, 产率 9 % 搅 拌 至 回流 , 时取 样 用 u Vs 法 观 测 卟 啉 0 加 的过程 中,溶液颜色 由橙黄色逐渐 变为棕黑 紫红色 晶体化合物, 配体 向金属卟啉配合物转化的紫外可见光谱吸 色. 继续搅拌 回流 4 n然后蒸去大部分丙酸, 以上 。 0mi, 主要特征为 Q带吸收峰数 目的减 少和峰 1. . 5烟酸尾式 卟啉过渡金属 配合物的合 收峰 ( 2 冷却到室温, 在搅拌 下加入 8 L乙醇 , Om 放置过 位 的移 动 ) 至 最 大 吸收 峰 不 再 变化 ,停 止 加 ,直 夜. 抽滤, 并用 乙醇洗涤晶体 , 真空干燥, 得深紫色 成 冷却, 大量饱 和食盐水, 加人 待产物充分析 出 N P P Z : 10 E T P n 在 0 mL三颈 瓶 中 , 8 热, 将 O mg 晶体. 将粗产 品溶 于氯 仿中, 以未经处理 的氯仿 过滤, 分别用水 和甲醇洗两次, 干燥得粗产 品. P P ,. n 1力 人 0 mL DMF中,Ⅱ 后, 力热 fR作淋洗剂, A) 中性 Al 作为 固定相, 2 0] 进行柱层 NE T P02 gZ C2 Ⅱ 2 粗 产 品采 用 柱 层 析 法分 离 , 以 f 转 6 下 8页 )
计算阅读文章记录

5.30 金属卟啉配合物的分子识别研究进展.刘海洋,计亮年.无机化学学报1997介绍了金属卟啉配合物在分子形状与大小识别、官能团识别和手性识别方面。
生物大分子之间的专一结合现象称为分子识别。
分子识别在酶促反应,免疫反应和蛋白质的生物合成等生命化学过程中有重要意义。
配体上带有某种官能团、特定空间构象的配合物与有机分子和生物分子之间也存在的不同专一结合的现象就是配合物的分子识别。
在生物体系中,卟啉的合成以甘氨酸和琥珀酸辅酶A为基本原料在在酶的作用下经多步反应得与完成的。
目前利用金属卟啉配合物已实现对如下生物机能的模拟:1血红蛋白和肌红蛋白模型-对氧分子的可逆结合。
细胞色素P450及过氧化氢酶模型-温和条件下催化氧化还原反应。
3光合作用前期光化学系统模型-光能的转换。
金属卟啉配合物为主体分子(host molecules)有其独特的优点。
首先,卟啉环具有刚性结构,周边官能团的方向和位置可以较好地得到控制,使之与客体分子(guest molecule)之间有最佳的相互作用;其次,卟啉分子有较大的表面,对金属卟啉分子轴向配体周围的空间容积和相互作用方向的控制余地较大;再次,金属卟啉配合物具有多样性(即在下篇文章中描述具有四个meso位和八个β位,可以立体分子设计)。
于此,金属卟啉配合物可以对多种有机和生物分子进行识别,金属卟啉配合物的分子识别研究具有广阔前景。
1 金属卟啉配合物的分子识别1.1分子形状与大小识别一般铁卟啉与氧分子作用通过下述途径而生成不能可逆载氧的μ-O二聚体:为阻止反应2的发生,人们合成了栅栏型(临乙酰胺)、盖帽型(临-OOCO-四取代苯)和吊带型卟啉,利用适当的空间结构阻止μ-O二聚体形成,实现对氧分子的选择性可逆结合。
生越久靖通过控制吊带卟啉中吊带与卟啉平面所形成空穴的大小,使之与苄基吡啶生成不同的配体作用模式(中心离子Fe与吡啶中N作用)。
举例四苯基卟啉铁(III)为催化剂均相催化甾醇底物侧链双键,保留环内双键。
卟啉与金属卟啉化合物_图文

命名与结构
相互关系
卟吩(porphine) 卟啉的骨架
中位碳或外环碳被取代
卟啉(porphyrin)
与金属离子结合
金属卟啉(metalloporphyrin)
命名与结构
3
5
7
4
6
2
8
外环碳
1 20
9 10
19 18
11
中位(meso)碳
12
16
14
17
15
13
IUPAC编号法
卟吩(porphq3
八乙基卟啉铂(PtOEP)与TPP相比具有更 高的光致发光量子效率,在利用单线态能量 同时还利用了三线态能量发光,使器件的内 量子效率理论上突破了25%的极限。将其掺 杂于PNP中可使发光效率达到29%。
(a)PtOE
吉林大学的研究小组也在这方面开展了 一些工作 ,他们把四苯基羰基钌掺杂Alq3以 及把四苯基卟啉铂(PtTPP)掺杂双(酚基吡啶) 铍(BePP2),利用主客体的能量转移获得了纯 红光器件。
A paradigm
2.在生物化学方面的应用
由于卟啉在生物体内起着及其重要的作 用,是血红素、细胞色素和叶绿素等生物大 分子的核心部分,故可以用作生物体内氧化 过程的模型,而其中以模拟单加氧酶P-450 、血红蛋白及肌红蛋白最引人注目。
关于模拟单加氧酶P-450
在具多转化底物能力的血红素蛋白中, 细 胞色素P-450意义重大, 它能催化各种有机物 和分子氧之间称之为混合功能氧化的化学反应 ,但由于它们的分子量巨大, 很难研究其催化 反应的详细机制。同时由于它们不稳定, 制备 很困难。由于铁卟啉配合物和P-450有类似的 结构性质, 人们利用它去对P-450进行模拟。 一种由咪唑的铁卟啉络合物和亚甲基丙烯酸共 价结合的模拟体系,如下所示:
两种高分子化锌卟啉络合物与特丁津相互作用的光谱性能研究

dition, both the Soret and Q absorption bands of ZnPP鄄PGMA exhibited red shift in the electronic rescence quenching for ZnHPP鄄PGMA was a little less than ZnPP鄄PGMA. It is because the hydrogen terbuthylazine. 摇 bonding between ZnHPP鄄PGMA and terbuthylazine led to the weaker axial coordination. Moreover, the fluorescence quenching of ZnHPP鄄PGMA was strengthed with the increasing concentration of
characterized by nuclear magnetic resonance ( 1 H鄄NMR) spectroscopy. The spectroscopic properties spectroscopy. The axial coordination reaction between two kinds of Zn porphyrin鄄functionalized
Spectroscopic Properties of Two Kinds of Zn Porphyrin鄄functionalized Polymer and Their Coordination Products with Terbuthylazine
YU Long1 , WANG Rui鄄xin1* , GAO Bao鄄jiao1 , GENG Tian鄄qi2 , CHEN Mei鄄jun2 ,
咪唑修饰的卟啉及其锌、铜配合物的合成和非线性光学性质

n c a man t eo a c u l r g e crsn n e( e i H
) ut vo t ibe( V— s se t s p , o r r r s r i rr (T I ) , l ail — s l U Vi p c oc y F ui a f m f e P —R r evi ) r o e tn o na d
I i a o e a d I sZi , ppe m p e e m d z l n t nc Co rCo lx s
ZHAN G a — o g Xi o H n J AO I Zhi Y AN e — e W iW i RU AN e J a W n—u n ZHU ZhiAn - g
咪 唑修 饰 的卟 啉及 其锌 、 配合物 的合成 和 非线 性光 学性 质 铜
张晓红 矫 志 闰ຫໍສະໝຸດ 伟 阮文娟 30 7 ) 0 0 1
朱志 昂
( 南开大学化学学 院, 天津
摘 要 : 合 成 了一种 新型咪 唑修 饰 的卟啉() 1及其锌 、 配合物 (、)通 过核磁共振 氢谱( MR 、 铜 2 3, N )紫外一 H 可见 ( V Vi 光谱 、 U—s ) 傅里 叶变换 红9 (rI ) bF — 光谱 、 R 电喷雾质谱 (S— ) E I 及元素分析等 多种谱学 方法对其结构进行表 MS 征. 卟啉环流效应对侧链咪唑芳 环的影响导致咪唑环上三个氢原子的化学 位移 向高 场移动, 且卟啉的紫外一 可见 光谱 的 S rt oe 带发生裂分 . 用分子模拟方法得到的 自由卟啉最低能量构象与光谱 分析结果一致, 采 即侧链咪唑环 位于卟啉环上方. 同时, 利用 z扫描技术对 卟啉及其锌 、 一 铜配合物的三阶非线性光学性质进行 了研究, 结果 表明: 卟啉及其锌、 铜配合物均具有很强的反饱 和吸收性质, 且铜卟啉的非线性光学性质强于锌 卟啉 的.
四苯基卟啉及其金属配合物的制备及光谱测定
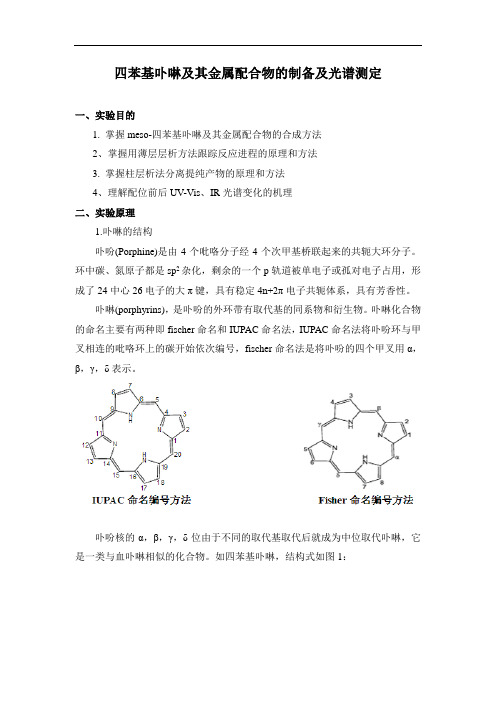
四苯基卟啉及其金属配合物的制备及光谱测定一、实验目的1. 掌握meso-四苯基卟啉及其金属配合物的合成方法2、掌握用薄层层析方法跟踪反应进程的原理和方法3. 掌握柱层析法分离提纯产物的原理和方法4、理解配位前后UV-Vis、IR光谱变化的机理二、实验原理1.卟啉的结构卟吩(Porphine)是由4个吡咯分子经4个次甲基桥联起来的共轭大环分子。
环中碳、氮原子都是sp2杂化,剩余的一个p轨道被单电子或孤对电子占用,形成了24中心26电子的大π键,具有稳定4n+2π电子共轭体系,具有芳香性。
卟啉(porphyrins),是卟吩的外环带有取代基的同系物和衍生物。
卟啉化合物的命名主要有两种即fischer命名和IUPAC命名法,IUPAC命名法将卟吩环与甲叉相连的吡咯环上的碳开始依次编号,fischer命名法是将卟吩的四个甲叉用α,β,γ,δ表示。
卟吩核的α,β,γ,δ位由于不同的取代基取代后就成为中位取代卟啉,它是一类与血卟啉相似的化合物。
如四苯基卟啉,结构式如图1:卟啉环中心的氢原子电离后,形成的空腔可以与金属离子配位形成金属卟啉配合物。
周期表中几乎所有金属元素都可以和卟啉类大环配位,金属卟啉也广泛存在于自然界。
例如动物体内的血红素是含铁卟啉化合物,血蓝素是铜卟啉化合物,植物体内的叶绿素是含镁的卟啉化合物,维生素B12是含钴的卟啉化合物。
卟啉化合物由于其母体卟吩具有刚性为主兼有柔性的大环共轭结构,因而具稳定性好,光谱响应宽,对金属离子络合能力强的特性。
卟啉化合物巨大的应用前景激起了化学家和生物学家对卟啉化学极大的兴趣和研究热情。
人们相信卟啉化合物在医学、仿生学、材料化学、药物化学、电化学、光物理与化学、分析化学、功能分子的设计、合成及应用研究等各个领域都有很大应用前景。
2、中位取代卟啉的一般光谱特征红外光谱(1)卟啉化合物的的红外光谱特征峰为在1590-1300 cm-1C=N伸缩振动峰,在1000 cm-1左右的卟啉骨架振动峰,在3550-3300 cm-1的N-H伸缩振动峰和在970-960 cm-1的N-H面内变形峰。
卟啉类光电功能材料的研究进展

圈l 胡萝h素(c)一卟啭(P)一醌(Q)三元化台物一醌(Qn)的结构 式
F【g 1 Structure of carot叶porphyri卅quinone tmd
2模拟生物光合作用中心的光致电荷转移和 能量转移
绿色植物光合作用中心是一个多步电荷转移体系.而这个 体系是由一系列按能量和空间排列的分子组成.具有相当高的 光致电荷转移和分离效率.在这种意义上来说,自然界的植物光 合作用中心是一种在分子水平上的高效率的光伏器件.因此模 拟生物光合成中心的光致电子转移和能量转移对太阳能的光电 转换和收集具有重要的理论和应用价值.其中卟啉化合物构成 叶绿素等生物大分子的核心部分.参与植物光合作用等一系列 重要过程。从80年代初开始.人们设计和合成了许多古有胡萝 h素、醌等官能团的卟啉类超分子体系来模拟和了解研究光合 作用中心的光致电子转移和能量转移等过程,并取得了很大的 进展…”
%—-磷, 圈3 胡萝h案(c)一卟啉锌(P^)一卟啉(PB)壕醌(瓯)苯醌 (Qn)五元化合物的结构式
Flg 3 StlucTure of carotene-Zn porphy叶porphyrin_naphthoqui—
none_benzoquinone pentad
图4 胡萝p素(c)一卟啉(I’H)一富勒烯(c。。)三元化奇物的结构 武
枣 攀 基。 丞凡Ⅳ 。
DⅥ 斜双,
陌6分予开关nA-D的结构
F19 6 Struct山_e of I)一A D m01ecular swltch
waⅢlewskl等Ⅲ3台成了结构复杂的电子给体一受体一受体一
给体【D。IA。一A。一Dz)型分于阵列(图7a),利用其中一对光生离
子对(Di—A.。)所产生的本征电场来影响和控制另一对给体一受
微量元素检测的方法学分析

微量元素检测的方法学分析准确检测微量元素在人体中的含量是任何理论研究与临床应用的前提和基础,如果没有准确地检测,根本谈不上研究与应用。
虽然从20世纪70年代就开始了微量元素研究,但它毕竟是一个新兴学科,检测微量元素的手段还比较陈旧和落后,无论从采样到测试前处理到测试直到结果分析都需专业人士来操作,步骤相当复杂,污染严重,且出结果时间长。
这也正是医院在人体微量元素检测方面无法普及的重要原因之一。
随着医疗水平的不断提高,微量元素与人体健康的关系得到了充分的认识,人们更加关心如何补充微量元素,如何排除有害元素。
微量元素在人体内是一个平衡过程,微量元素的缺乏和过量都会对人体产生不良影响。
因此如何准确快速、方便地检测人体微量元素含量就成为医务工作者亟须解决的课题。
目前我国的各级医疗保健单位,尤其是妇幼保健单位、儿童医院、综合医院等,已经将人体元素(铅、锌、铜、钙、镁、铁等)检测作为常规项目。
如何选择一种适合的仪器,是医院管理者在采购过程中面临的首要问题。
出于对病人健康的高度责任感和可能出现医患纠纷的自我保护,选择一种能够准确而且规范的测量仪器最为重要;其次应考虑操作流程简便性、设备使用安全性和稳定性;还要考虑受检者经济承担能力和受影响程度,满足其希望能够又准又快又便宜地完成检测的要求;最后,也要考虑到仪器利用率高,保证投资收益。
下面就微量元素检测的方法学做一介绍一传统的微量元素检测的方法目前可用于人体微量元素检测的方法有:同位素稀释质谱法、分子光谱法、原子发射光谱法、原子吸收光谱法、X射线荧光光谱分析法、中子活化分析法、生化法、电化学分析法等。
但在临床医学上广泛应用的方法主要为生化法、电化学分析法、原子吸收光谱法这几种。
下面简单介绍一下生化法、电化学分析法这两种检验方法的主要特点:1 生化法(锌原卟啉法、双硫腙法、其它比色法等)的特点:用血量较大需要前处理,操作复杂,澄清血清耗时长检测血清,而血清受近期饮食等因素影响极大,从而使数据缺乏客观准确性试剂成本较高检测元素种类受限制灵敏度达不到临床检测的要求重复性差2 电化学分析法的特点:(目前尚有部分基层医院和非正规医疗机构采用,常称之为电位溶出法或溶出伏安法等)仪器价格较低可以用于痕量的测量,但误差较大测定多种元素时,重复性差对环境和实验人员污染严重很难将保养到最佳条件前处理极其繁杂耗时整个实验很难控制,结果非常不稳定虽然上述的两种方法均可以在临床测定人体微量元素中应用,但由于其自身的种种弊端,已基本被现代更先进、更准确的方法所取代。
紫外——可见光谱法在卟啉类化合物结构分析中的应用

紫外-可见光谱法在卟啉类化合物结构表征中的应用摘要:简述了紫外-可见光谱分析的基本原理,及其在有机化化学中的应用;结合卟啉、金属卟啉的吸收特点,对紫外-可见光谱在其结构表征中的应用作了归纳性的总结。
关键词:紫外-可见光谱法;应用;卟啉;金属卟啉;结构表征1 紫外-可见吸收光谱分析基本原理紫外光谱(UV)是指波长在200~400nm;可见光谱则是波长在400~800nm的电磁波吸收光谱。
相应于上述波长的能量范围约在670~314kJ/mol和314~155kJ/mol。
因此,它们是属于π电子(成键的或孤对的电子)跃迁。
所以,不是所有的有机化合物,都能给出它们的吸收光谱,而主要是对具有共轭双键结构的化合物和芳香族化合物才能给出光谱。
如果用紫外和可见光照射含有共轭的不饱和化合物溶液,可以看到一部分光线被吸收了,吸收光线的多少,取决于入射光的波长和化合物的结构。
如果以波长为横坐标,以紫外、可见光线的吸收强度(有时也称消光系数或摩尔吸收度)为纵坐标作图,就得到紫外或可见光谱图。
同一种物质对不同波长的光吸收不同;不同浓度的同一种物质,其吸收曲线形状相似、λmax不变,只是吸光度大小不同;而对于不同物质,它们的吸收曲线形状和λmax均不同。
当外层电子吸收紫外或可见辐射后,就从基态向激发态(反键轨道)跃迁。
主要有四种跃迁形式,如图1。
所需能量ΔΕ大小顺序为:n→π*< π→π*< n→σ*< σ→σ*。
吸收带是指吸收峰在光谱中的波带位置,根据电子及分子轨道理论,有机化合物紫外-可见光区的吸收带有四种类型:R吸收带——由化合物中的n→π*跃迁产生的吸收带。
其强度小,ε<100;λmax位于较长波长处,>270nm;K吸收带——由共轭体系中π→π*跃迁产生的吸收带。
其强度大,ε>104;λmax比R带的短,一般>200nm;B 吸收带——由苯环本身振动及闭合环状共轭双键π→π*跃迁产生的吸收带。
锌卟啉-富勒烯配合物的光谱和光伏性质研究

O C H 3 ) Z n P ・ C 6 。 i s o b t a i n e d,e s p e c i a l l y W i n t h e O 2 / H 2 O r e d o x c o u p l e , a n d t h e g r e a t e s t v a l u e o f p h o t o v o h a i e p o t e n t i a l i s 2 1 5 m V .
卟啉锌化合物结构特点

卟啉锌化合物结构特点
卟啉锌是一种化学物质,其结构特点如下:
1. 中心原子:卟啉锌的中心原子是锌(Zn),其价态为+2。
2. 配体:卟啉锌的配体是卟啉,它是一种由四个吡咯环通过共轭的甲烯基相连形成的环状化合物。
3. 空间构型:由于四个吡咯环的相对位置,卟啉锌呈现出一个中心对称的结构,即它具有四个对称的配位点。
4. 键合方式:在卟啉锌中,锌原子与四个氮原子(来自四个吡咯环)通过配位键结合,形成五元环状结构。
这四个配位键在同一平面上,形成了一个平面四边形结构。
5. 稳定性:由于五元环的稳定性以及中心原子与配体的强配位相互作用,卟啉锌在常温常压下稳定。
6. 光学和电子性质:卟啉锌具有特定的吸收光谱和发射光谱,可在特定波长下吸收或发射光。
此外,其电子结构和化学性质也与其它金属卟啉有所不同。
如需获取更多详细信息,建议查阅相关文献或咨询化学领域专业人士。
苯基卟啉衍生物及其锌、锰配合物的合成、光物理性能及电化学性质

p e yp r h r d r a ie ; meal p r h rn o l x s p o o h se p o et s e e to h mia h n l op y n e v t s i i v tl o y i c mp e e ; h tp y ia o p l rp ri ; lc rc e c l e
V0 . 0 No 3 13 . S p. 0 1 e 2 1
苯 基 卟啉 衍 生物 及 其 锌 、 配 合 物 的合 成 、 锰 光 物 理 性 能 及 电化 学 性 质
陈 连清, 徐华诚, 森, 程国 曹蒂薇
( 中南 民族大学 化学与材料科学学院 , 武汉 4 0 7 ) 30 4 摘 要 为寻求更好的苯基卟啉类光电材料 , 以吡咯和取代苯 甲醛为原料 , 通过改进 的 Adr le 法合成 了 3种苯 基卟
苯基卟啉化合物由于其母体卟吩具有刚性为主
兼有 柔性 的大 环共轭 结构 , 具有 一定芳 香性 , 稳定 性 好, 光谱 响应 宽 , 金 属 离 子络 合 能 力 强 , 对 这些 性 质 使 得苯基 卟 啉具 有 广 泛 的用 途 J .苯基 卟啉 具 有 分子 晶格 , 子 间作 用 能低 , 收光 后 引 起 分 子 激 分 吸 发 , 发后 的卟 啉化合 物 易 形 成 带 电荷 的卟 啉 自由 激
啉衍生物及其锌 、 锰配合物 , 目标化合物 进行 了 F — MS X S和元 素分析等表 征 , 测试 了化 合物 的紫外、 对 TI R、 、 P 并 荧 光光谱 , 探讨 了其 光物理性 能和电化学性质.结果表 明 : 通过改进的合成 方法 , 苯基卟啉衍生物及其锌 、 锰配合物合 成简单 、 收率高且其光电性 能 良好 .
电化学文献报告

电化学测量原理课程报告题目:中央取代卟啉的电化学行为:阳离子自由基对半波氧化电势分裂的作用专业:分析化学班级:2016级2班日期:2016年11月23日湖南大学化学化工学院中央取代卟啉的电化学行为:阳离子自由基对半波氧化电势分裂的作用摘要:在这项研究中,通过循环伏安法(CV)和密度泛函理论(DFT)检测游离碱和锌内消旋卟啉的电化学行为。
结果表明,四苯基卟啉(H2TPP)及其锌配合物(ZnTPP)的两个氧化态(ΔE=第二个E1 / 2-第一个E1 / 2)的半波氧化电位分裂高于卟啉及其锌与内消旋取代的五元杂环的络合物。
对于内消旋卟啉和它们各自的锌络合物,ΔE值遵循TPP> T(3'-噻吩基)P> T(3'-呋喃基)P> T(2'-噻吩基)P的趋势。
通过采用DFT计算,发现ΔE值的趋势与阳离子基团的最高自旋密度(HSD)分布和HOMO-LUMO能隙以及中心卟啉和内消旋取代环之间的π共轭的趋势一致。
此外,它们表现出卟啉环与内消旋取代环之间更好的共振,如从卟啉和它们的锌配合物与苯基环到五元杂环。
计算结果和实验结果之间的良好一致性表明阳离子自由基,特别是它们的自旋密度分布,确实在中波卟啉及其锌络合物的半波氧化势分裂中起重要作用。
1 研究目的:由于卟啉类化合物在催化,电子传递系统[1]和光电器件中[2]的广泛应用,及其在电化学[3]与已经引起了很多关注。
卟啉和金属卟啉包含广泛的共轭π环系统,其电子转移行为取决于电子结构的离域程度,π系统离域越高,由于电子转移时结构的微小变化,电子的吸收或释放越容易[4]。
可逆电子转移反应中的结构变化的程度可以通过第一和第二氧化态之间的半波氧化电势差(ΔE= E2 – E1)来检查[5]。
循环伏安法(CV)是研究新系统的化学行为的流行方法[5],并且密度函数理论(DFT)是计算化合物的电子状态的有前途的方法[6],本文采用CV和DFT 计算的组合来研究卟啉及其金属配合物与内消旋取代的苯基和五元环(图2)的电化学行为,同时探索了阳离子自由基最高自旋密度效应对化学性质的影响。
紫外——可见光谱法在卟啉类化合物结构分析中的应用
紫外-可见光谱法在卟啉类化合物结构表征中的应用摘要:简述了紫外-可见光谱分析的基本原理,及其在有机化化学中的应用;结合卟啉、金属卟啉的吸收特点,对紫外-可见光谱在其结构表征中的应用作了归纳性的总结。
关键词:紫外-可见光谱法;应用;卟啉;金属卟啉;结构表征1 紫外-可见吸收光谱分析基本原理紫外光谱(UV)是指波长在200~400nm;可见光谱则是波长在400~800nm的电磁波吸收光谱。
相应于上述波长的能量范围约在670~314kJ/mol和314~155kJ/mol。
因此,它们是属于π电子(成键的或孤对的电子)跃迁。
所以,不是所有的有机化合物,都能给出它们的吸收光谱,而主要是对具有共轭双键结构的化合物和芳香族化合物才能给出光谱。
如果用紫外和可见光照射含有共轭的不饱和化合物溶液,可以看到一部分光线被吸收了,吸收光线的多少,取决于入射光的波长和化合物的结构。
如果以波长为横坐标,以紫外、可见光线的吸收强度(有时也称消光系数或摩尔吸收度)为纵坐标作图,就得到紫外或可见光谱图。
同一种物质对不同波长的光吸收不同;不同浓度的同一种物质,其吸收曲线形状相似、λmax不变,只是吸光度大小不同;而对于不同物质,它们的吸收曲线形状和λmax均不同。
当外层电子吸收紫外或可见辐射后,就从基态向激发态(反键轨道)跃迁。
主要有四种跃迁形式,如图1。
所需能量ΔΕ大小顺序为:n→π*< π→π*< n→σ*< σ→σ*。
吸收带是指吸收峰在光谱中的波带位置,根据电子及分子轨道理论,有机化合物紫外-可见光区的吸收带有四种类型:R吸收带——由化合物中的n→π*跃迁产生的吸收带。
其强度小,ε<100;λmax位于较长波长处,>270nm;K吸收带——由共轭体系中π→π*跃迁产生的吸收带。
其强度大,ε>104;λmax比R带的短,一般>200nm;B 吸收带——由苯环本身振动及闭合环状共轭双键π→π*跃迁产生的吸收带。
卟啉型染料分子

卟啉型染料分子卟啉是由4 个吡咯环通过亚甲基相连形成的具有18 电子体系的共轭大环化合物, 其分子配位性能突出, 周期表上几乎所有的金属原子都能和中心的氮原子配位形成金属卟啉配合物。
在卟啉分子周围, 有两类取代位置, 分别为间位(meso)和β位, 可以通过化学方法引入不同的取代基。
卟啉化合物具有良好的光、热和化学稳定性, 在可见光区有很强的特征电子吸收光谱。
近年来, 利用卟啉及其配合物独特的电子结构和光电性能, 设计合成光电功能材料和器件已成为国际上十分活跃的研究领域。
在获取能源方面, 大自然选择了卟啉配合物。
光合作用中, 卟啉衍生物叶绿素是光能转换的反应中心。
能够将太阳能转化成化学能, 关键是叶绿素分子受光激发产生的电荷分离态寿命可长达1s,这是电荷有效输出的重要前提。
实验表明, 太阳能电池中, 不论电子注入TiO2 的效率还是速度, 卟啉的表现都不逊于多吡啶钌类化合物。
导带电子和卟啉激发态的复合速率约几个微秒, 这段时间足够电解质中的电子回传到卟啉基态上, 完成染料的还原。
这些结果表明, 卟啉有望成良好的太阳能电池光敏染料。
3.1 卟啉单分子作为DSSC 的吸光染料卟啉化合物无论是单分子还是聚合物, 在各种染料太阳能电池中都有应用, 特别是用卟啉作为光敏剂的敏化纳米晶太阳能电池性能突出。
目前, 研究最多的间位-四(对羧基苯基)卟啉(TCPP)及其金属配合物(M-TCPP),分子激发态寿命较长(>1 ns),HOMO 和LUMO 能级高低合适, 是较为理想的DSSC 染料候选化合物。
Gr"tzel 和Fox 等都报道了Zn-TCPP 敏化纳米TiO2 光电池IPCE(B 带)=42%,但没有报道η值;Boschloo 和Goossens 报道了它的光电转化效率η为1.1%,(IPCE(B 带)=40%)[。
间位- 四对苯磺酸基卟啉锌(Zn-TSPP)染料敏化纳米晶TiO2电池的IPCE 高达99.4%, 大幅超过多吡啶钌类DSSC [33]。
卟啉吸收光谱

卟啉吸收光谱卟啉是一种重要的生物分子,它在生物体内扮演着重要的生理功能,比如光合作用、呼吸作用等等。
卟啉分子的特殊结构决定了它有很强的吸光能力,因此也被广泛应用于光谱学中,成为研究生物分子结构和相互作用的工具之一。
本文主要介绍卟啉吸收光谱的原理以及在分析生物分子中所起的作用。
一、卟啉分子的结构和电子结构卟啉分子是一种环状大分子,由四个吡咯环组成,它的化学式为C34H36N4。
卟啉分子中含有22个.pi.电子,这些电子能产生吸收光谱,因此使得卟啉分子在可见光区和紫外光区具有很强的吸收能力,这也是卟啉成为研究生物分子的一个理由。
此外,卟啉分子中的.nbond.键和.cbeta.原子还具有很强的带电性,也是卟啉分子体系中重要的结构基础。
二、卟啉吸收光谱的原理卟啉分子中的22个.pi.电子的共振吸收是卟啉吸收光谱的主要原理。
在卟啉分子中,这些.pi.电子的状态可以被用Huckel理论描述,Huckel理论是一种将分子中.pi.电子抽象为组成部分的计算方法。
在这个理论框架下,卟啉分子中的电子运动可以被表示为具有简约的线性组合基原子轨道(LCAO)的线性方程组。
这个方程组运用分子轨道理论中的简化原理,可以将分子中的电子运动形象地表达出来。
卟啉分子中的可见光和紫外光区的波长都恰好能够与22个.pi.电子的吸收相匹配。
由于电子的共振吸收会导致分子吸收和轨道的变化,因此在分子的吸收光谱中就能够观察到许多特殊的吸收峰。
这些吸收峰的位置和大小反映了分子内部的电子状态,因此可以用来分析分子的结构和相互作用。
三、卟啉在生物体系中的应用卟啉作为一种重要的生物分子,在生物体系中也有广泛应用。
它在生物体内参与了光合作用、呼吸作用等等生理活动,因此也成为生物体系中的重要研究对象。
通过对卟啉分子的吸收光谱分析,研究人员可以了解卟啉分子在生物体系中的特殊作用,比如它的电子传输功能和光合成功能等等。
此外,卟啉分子还是许多生物分子的组成部分,比如血红素中就含有卟啉分子。
卟啉配位键

卟啉配位键卟啉配位键是指卟啉分子通过氮原子与金属离子形成配位键的化学现象。
卟啉是一种含有四个吡咯环的杂环化合物,具有非常特殊的性质和结构。
其配位键的形成对于理解卟啉及其衍生物的结构与功能具有重要意义。
本文将分为三个部分介绍卟啉配位键的形成、性质及其在生物体中的应用。
第一部分:卟啉配位键的形成卟啉分子具有24个π电子,其中20个属于共轭π电子体系。
通过提供第四周期元素的金属中心,卟啉分子可以形成中央金属离子和卟啉配合物。
卟啉的吡咯环中的氮原子作为配体与金属离子形成靠近垂直的平面的配位键。
这种配位键的形成与卟啉分子的结构和电子结构有关。
第二部分:卟啉配位键的性质卟啉配位键形成后,可改变其光谱性质和化学性质。
例如,卟啉配合物的紫外-可见吸收光谱显示不同的吸收峰和颜色。
这是因为金属中心对卟啉的π电子造成局域扭曲,引起共振结构的改变。
此外,金属离子还可以通过与卟啉配位键的构型改变来调控卟啉分子的电子密度和反应性。
卟啉配合物还可以通过配合键与溶剂分子进行相互作用,改变其性质和活性。
第三部分:卟啉配位键在生物体中的应用卟啉配合物在生物体中具有广泛的应用。
其中最为著名的就是血红素和叶绿素,它们分别与细胞色素和叶绿体相关蛋白质结合,参与氧气的运输和光合作用。
血红素的配位键通过与铁离子形成,使氧气能够与血红素结合,从而实现氧气的输送。
叶绿素的配位键通过与镁离子形成,实现光合作用的光能吸收和电子转移。
此外,卟啉配合物还在催化、电化学和生物传感等领域有着广泛的应用。
综上所述,卟啉配位键是一种通过氮原子与金属离子形成的化学现象。
卟啉配位键的形成受到卟啉分子结构和电子结构的影响,改变了卟啉分子的光谱性质和化学性质。
在生物体中,卟啉配合物通过配位键参与氧气的运输、光合作用等重要生物过程。
这些研究对于理解卟啉及其衍生物的结构与功能,以及在生物体系中的应用具有重要意义。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
[Article]物理化学学报(Wuli Huaxue Xuebao )Acta Phys.⁃Chim.Sin .2012,28(5),1085-1093May Received:December 12,2011;Revised:February 29,2012;Published on Web:March 2,2012.∗Corresponding author.Email:qibinchen@;Tel/Fax:+86-21-64252767.The project was supported by the National Natural Science Foundation of China (20806025,21103047)and National 111Project of China ʹs Higher Education (B08021).国家自然科学基金(20806025,21103047)和中国高校111计划(B08021)资助项目ⒸEditorial office of Acta Physico ⁃Chimica Sinicadoi:10.3866/PKU.WHXB201203024间位取代卟啉及其锌卟啉的电子吸收光谱曹振锋陈启斌*卢运祥刘洪来胡英(华东理工大学,结构可控先进功能材料及其制备教育部重点实验室,上海200237)摘要:取代的卟啉类衍生物在气敏传感器方面具有广泛的应用前景.本文采用了密度泛函理论(DFT)和含时密度泛函理论(TD-DFT)研究了四种不同取代基的卟啉衍生物(meso 位四硝基苯基/四氨基苯基卟啉(NO 2PP,NH 2PP)及其相应的锌金属卟啉衍生物(NO 2ZnPP,NH 2ZnPP))的紫外和近紫外光谱特征.利用两种不同的交换相关泛函(广义梯度近似泛函(PBE)和杂化密度泛函(B3LYP))优化了上述四种物质的结构,并应用TD-DFT 计算了相应的电子激发能量和振动强度.结果表明,取代卟啉的吸收光谱与大量的电子跃迁有关;与B3LYP 泛函预测的光谱相比,PBE 泛函所得B 带以及Q 带的波长位置与实验值更为接近.另外,计算所得硝基取代基卟啉的B 带相对于氨基取代基卟啉的B 带发生了红移,这与实验现象也保持一致.由于卟啉衍生物的三重激发态在电子转移中有很重要的应用,因此在PBE/6-31G(d )水平上计算了四种物质的最低三重激发态能量,分别为1.426、1.469、1.608和1.581eV.关键词:含时密度泛函理论;电子吸收光谱;电子跃迁;红移;三重激发态中图分类号:O641Electronic Absorption Spectra of Meso -SubstitutedPorphyrins and Their Zinc DerivativesCAO Zhen-FengCHEN Qi-Bin *LU Yun-XiangLIU Hong-LaiHU Ying(Key Laboratory for Advanced Material and Development of Chemistry,East China University of Science and Technology,Shanghai 200237,P .R.China )Abstract:Meso -substituted porphyrin derivatives have demonstrated great potential as sensing materials for toxic gas detection.In this paper,density functional theory (DFT)and its time-dependent DFT approach (TD-DFT)were employed to investigate the ultraviolet-visible (UV-Vis)or the near-ultraviolet-visible (near-UV-Vis)absorption spectra of meso -tetra (o -nitrophenyl/o -aminophenyl)porphyrins (NO 2PP,NH 2PP)and their corresponding zinc derivatives,NO 2ZnPP and NH 2ZnPP.The geometry optimizations for these four molecules were obtained from two different exchange-correlation functionals,the generalized-gradient approximation functional PBE (Perdew-Burke-Ernzerhof)and the hybrid functional B3LYP (Becke,three-parameter,Lee-Yang-Parr).The excitation energies and oscillation strengths were obtained from TD-DFT calculations.Calculations show that the optical absorptions are associated with numerous electronic transitions.In addition,the PBE-predicted wavelengths of the B and Q bands are more consistent with experiment than those predicted by B3LYP.The B band of NO 2-substituted derivative exhibits a bathochromic shift different from that of NH 2-containing material,also consistent with experimental results.In addition,at the PBE/6-31G(d )level of theory,the calculated energies of the lowest triplet excited states of NO 2PP,NH 2PP,NO 2ZnPP,and NH 2ZnPP are 1.426,1.469,1.608,and 1.581eV,respectively.1085Acta Phys.⁃Chim.Sin.2012V ol.28Key Words:TD-DFT;Electronic absorption spectrum;Electronic transition;Bathochromic shift;Triplet state1IntroductionPorphyrins and related metalloporphyrins have been the sub-jects of numerous experimental and theoretical studies for de-cades.1-12Due to their remarkable photochemical,electrochemi-cal,and biochemical properties,13-15there has been renewed in-terests in the use of these compounds in some fields including catalysts,optical machinery,solar cells,and non-linear optics. Exploiting their interesting properties,porphyrin films have been scrutinized for possible applications as sensitive,selec-tive,and stable active layers of transducers in gas sensor,act-ing as an electron donating or accepting unit during the process of electron transfer when interacting with the analytes,which could lead to pronounced variation of the B-bands and Q-bands in the absorption spectra,as supported by many spectroscopic experiments.16,17Chen et al.18have recently reported a new chlo-rophyll f(C55H70O6N4Mg)by replacing the methyl group at C2 position with formyl group and discovered that oxygenic photo-synthesis can be extended further into the infrared region, which clearly verifies the importance of the substituent.This implies that besides the analyte,the different substitutional(the electron donating and/or withdrawing)groups of the porphyrin derivatives likely have a significant effect on their electron transfer and transition.On the other hand,compared with free-base porphyrins,metalloporphyrins play much more important roles in biological systems such as oxygen storage and transfer, metabolism,photosynthesis and chemical sensors,and thus have more artificial applications.19Additionally,Richardson et al.20have synthesized different metalloporphyrins to detect var-ious kinds of toxic gas by in situ UV-Vis measurements.Be-cause of all those important applications,understanding the de-tailed knowledge of the structure,electronic and molecular ex-citations as well as orientation of adequate transition moments is of urgent necessity to interpret energy or electron transfer processes.In particular,considering the crucial role of the sub-stituent and the complex metal atom in the properties of por-phyrin derivatives described herein,it is indispensable to sys-tematically study these effects on the electronic properties and the optical absorption of these molecules.In order to explore the effect of the substituents and the cen-tral metal atom on the absorption spectra,meso-tetra(o-amino-phenyl)porphyrin(NH2PP),meso-tetra(o-nitrophenyl)porphy-rin(NO2PP),and the corresponding zinc derivatives(NH2ZnPP, NO2ZnPP)were synthesized in our laboratory.21Here,―NH2and ―NO2are the most common and simple electron donating and withdrawing groups,respectively,which could clearly influ-ence the electron distribution of the porphyrin ring,leading to the variation of the absorption spectra.To our knowledge,up to the present,these four porphyrin derivatives have not been well investigated.As a whole,their experimental absorption spectra seem to be similar to those of porphine(H2P)and zinc porphine(ZnP);22,23namely,free-base porphyrins have a strong B-band in the near-UV region and four weak Q-bands in the visible region,while metalloporphyrins have a B-band and two Q-bands due to their higher symmetry.The four Q peaks are la-beled as Q0x,Q1x,Q0y,Q1y,where Q0x and Q0y peaks are generally in-terpreted as pure electronic transition,24,25while Q0and Q1 peaks always present in the metalloporphyrins(Q1is vibronic). Generally,the absorption spectra of porphine and metal-por-phine are qualitatively interpreted on the basis of Goutermanʹs four-orbital model,24-26in which the spectra mostly depend on the electron transition between two highest occupied orbitals and two lowest unoccupied orbitals.However,with the addi-tion of various substituents and the decrease of the symmetry of molecules,strong mixing of configurations should occur and thus more excited states are responsible for the absorption spectra.As a result,the detailed absorption spectra of the por-phyrin derivatives should differ from one another,which need to be analyzed carefully.During the past decades,some theoretical investigations have appeared to study the excitation spectra of porphyrin de-rivatives by means of different level of theories,for examples, the symmetry-adapted-cluster configuration-interaction(SAC-CI) calculations on excited states of chlorophyll and pheophytin,4 the multiconfiguration second-order perturbation theory (CASPT2)on the excitation energy of N2(NH)2.27However,the calculated spectrum results were not in well consistency with the experimental observations.Recently,the emergence of the DFT and TD-DFT has provided new impetus in the field of computational chemistry.21,28Taking electron correlation into ac-count in an implicit and expedient manner,DFT and TD-DFT calculations on the molecular structure and excited state of por-phyrins have proved to be useful and suitable.22,29In this work, with the purpose of studying the effects of the substitute group and zinc atom on the geometries and the excited electronic tran-sition of porphyrins,we used DFT and TD-DFT with two dif-ferent functionals(PBE and B3LYP)to optimize the geome-tries of the four porphyrin derivatives and calculate the elec-tronic transitions which are relevant to absorption spectra, moreover,comparing with the experimental results.The ener-gies of the triplet excited state with respect to the ground states were also evaluated owing to its relevance to the molecular electronics.2Theoretical and experimental procedures The synthesis of NO2PP/NH2PP and the corresponding metal-loporphyrins NO2ZnPP/NH2ZnPP was carried out according to our previous work21and the general procedure.30UV-Vis spec-tra were recorded on a Shimadzu UV-2450spectrometer1086CAO Zhen-Feng et al.:Electronic Absorption Spectra of Meso-Substituted Porphyrins and Their Zinc Derivatives No.5(Japan).The samples were firstly dissolved in dichlorometh-ane(Chemical Reagent)and a quartz cuvette of1.0cm pathlength containing dichloromethane was used as reference whenmeasuring UV-Vis spectra of dichloromethane solution.The molecular structures of four porphyrin derivatives werefirstly optimized via two different DFT functionals:the gener-alized-gradient approximation(GGA)exchange-correlationfunctionals PBE31-33and the hybrid functional B3LYP.34,35Theformer is a reformulation of the first-principles GGA function-al PW91(more simple but improved over PW91),whereas thelatter involves the semiempirical parameters used in DFT cal-culations.22,36,37Since these two functionals have been success-fully used in the TD-DFT calculations of absorption spectra ofporphine and metal porphine,22,28,29,36,37they were also applied in this work to calculate the excitation transition energies and os-cillator strengths based on the optimized structures.The first step of the TD-DFT calculation employed a self-consistent ground state Kohn-Sham(KS)computation.The second step consisted of solving the central equation of the TD-DFT re-sponse theory by using the adiabatic local density approxima-tion(ALDA)for the functional derivatives of the exchange-cor-relation potential.38The split-valence basis set6-31G(d)was used throughout the calculations.For each molecule,a structure optimization was performed firstly,followed by a frequency calculation to confirm that the optimized structure was indeed a minimum (with no imaginary frequencies).The TD-DFT was then em-ployed to evaluate the excitation energy to predict the absorp-tion spectra.All the calculations in this work were performed using the Gaussian03package.393Results and discussion3.1Optimized ground-state geometriesThe structural formula and optimized structures with PBE functional used for four porphyrin derivatives are displayed in Fig.1and Fig.2.For simplicity,we employ numbers1,2,3, and4to represent four derivatives NO2PP,NH2PP,NO2ZnPP, and NH2ZnPP,respectively.Some selected structural parame-ters of these derivatives predicted with two different function-als are listed in Tables1and2.For comparison,the experimen-tal parameters for H2TPP and ZnTPP are also listed.40-42Accord-ing to the present calculations,H2P and ZnP show D2h and D4h symmetries,respectively.However,with the addition of the NO2and NH2substituents,the symmetry of the studied deriva-tives reduces to C2v symmetry.It is known that different me-so-substituents and their orientations would produce different symmetries for porphyrin skeleton.Moreover,it is found that the D2v symmetries is the most stable geometry for these four derivatives.As can be seen from the data in Tables1and2,the car-bon-carbon bond lengths of the derivatives under study are lo-cated between those of the carbon-carbon single and double bonds,indicating that the conjugation exists in porphyrins rings,as well as between porphyrins ring and phenyl group. Generally speaking,the length of carbon-carbon single bond is 0.154nm and the length of the carbon-carbon double bond is 0.133nm.Thus,the porphyrin molecules are in the conjuga-tion condition because the carbon-carbon length is between 0.133and0.154nm according to our calculation results.Previ-ous studies on the meso-phenylporphyrins have revealed that the meso-phenyl substitution and the central metal would cause more or less the out-of-plane distortion.42,43The NCαC m Cαdihe-dral angle is often used to evaluate the extent of out-of-porphy-rin distortion.From Tables1and2,it is obvious that the NCαC m Cαangles(such as N19C24C23C3,N4C3C23C24)are very small(<3°),indicating that the meso-phenyl substitutions and zinc metal affect the out-of-distortion to a relatively less de-gree because of the suited size of zinc ion within the porphyrin core.For the in-plane-distortion,the largest distortion usually occurs around the C m atoms,which can be shown clearly by the alteration of the C m―Cαbond lengths and by comparing the distances of two opposite C m atoms of the derivatives with those of ZnP and H2P.The C m―Cαbond length and C m―C m in-ter-atomic distance tend to decrease,especially in zinc deriva-tives.For example,the C m―C m distance in1/PBE is shown to be0.694nm,whereas in3/PBE the distance shortens to0.692 nm.Moreover,it can be found that the geometry of the por-phine ring remains almost unchanged with the addition of NO2 and NH2groups.On the different behavior of the two function-als,the B3LYP functional yields slightly smaller values com-pared with those by PBE.The most remarkable difference of the two different functionals can be manifested by the C2C1C6C5 dihedral angle:B3LYP predicts much larger values compared with that by PBE,especially in NH2-substituted porphyrins. 3.2Electronic structuresAs mentioned above,due to the reduction of the symmetry of the porphyrins by adding meso-phenyl substitutents,the Gautermanʹs four-orbital theory is not suitable for calculating the electronic transition and predicting the absorption spectra. More frontier orbitals are involved and more mixing of config-urations should be taken into consideration.The energies of some frontier MOs of four porphyrin derivatives are shown in Figs.3and4.The highest occupied molecular orbital(HOMO) 1Structures and nomenclature of the fourporphyrin derivatives1087Acta Phys.⁃Chim.Sin .2012V ol.28energy is used as a reference here,that is,the HOMO energy is set to zero.From Figs.3and 4,it can be seen that for 1,2,3and 4,PBE functional yields the HOMO-LUMO (the lowest unoccupied molecular orbital)energy gaps to be 1.618,1.694,1.673,and 1.736eV ,respectively,while larger energy gaps (2.705,2.767,2.837,and 2.899eV)are obtained with the hybrid functional B3LYP.Furthermore,it is found that the orbital energies be-tween LUMO and LUMO+5of 1/PBE are almost identical and the maximum differences are smaller than 0.25eV ,whereas the energy gap between HOMO and HOMO -3appears larger,especially from HOMO -1to HOMO -2.However,opposite trends are observed for 2/PBE:the orbital energies of LUMO+2and LUMO+3are much larger than those of LUMO and LUMO+1,while the orbital energy values from HOMO -1to HOMO -5are essentially degenerate.These tendencies are al-so shown in metalloporphyrins (3/PBE and 4/PBE).Generally,with the addition of substituents (NO 2,NH 2)and metal zinc,the geometries of the considered derivatives remain almost un-changed (vide supra ),while the electronic structures are shown to be distinctly different as shown in Fig.5.This can be as-cribed to the fact that NO 2is an electron-withdrawing groupTable 1Some calculated bond lengths and bond angles in free-base porphyrins 1,2using two different functionals B3LYP and PBEL is bond length,A is bond angle,and D is dihedral angle.aData are taken from X-ray crystallography.40,41Parameter L (N —H)/nm L (N —C α)/nm L (C α—C β)/nm L (C β—C β)/nm L (C m —C α)/nm L (C β—H)/nm L (C m —C m )/nm L (C1—C6)/nm A (H —N —C α)/(°)A (C α—N —C α)/(°)A (N —C α—C β)/(°)A (C α—C β—C β)/(°)A (C α—C m —C α)/(°)D (N 4C 3C 23C 24)/(°)D (N 19C 24C 23C 3)/(°)D (N 8C 7C 6C 5)/(°)D (N 4C 5C 6C 7)/(°)D (C 2C 1C 6C 5)(°)106.3-108.11/B3LYP 0.1020.1370.143-0.1460.135-0.1370.140-0.1410.1080.6910.150124.7105.4-110.6106.6-110.0125.9-1.5240.018-0.0181.52478.641/PBE 0.1030.137-0.1380.144-0.1460.136-0.1380.141-0.1420.1090.6940.150124.7104.8-110.5106.8-111.5126.0-2.0590.476-0.4762.05976.07106.1-108.02/B3LYP 0.1020.137-0.1380.144-0.1460.135-0.1370.1410.1080.6940.150124.6105.2-110.7106.4-111.1106.3-108.2125.1-1.5221.281-1.2811.52279.702/PBE 0.1030.1380.144-0.1460.136-0.1380.141-0.1420.1090.6960.150124.6104.7-110.6106.6-111.5125.1-2.3541.956-1.9562.35474.24106.2-108.1H 2TPP a 0.0930.1370.1430.1350.140125.2125.6Fig.2Optimized structures of the four porphyrin derivatives with PBE functional(a)1/PBE,(b)2/PBE,(c)3/PBE,(d)4/PBE.1,2,3,4represent NO 2PP,NH 2PP,NO 2ZnPP,and NH 2ZnPP,respectively.The letter after “/”refers tothe generalized-gradient approximation functional PBE.1088CAO Zhen-Feng et al.:Electronic Absorption Spectra of Meso-Substituted Porphyrins and Their Zinc Derivatives No.5and NH2is an electron-donating group.Apparently,the elec-tron is well delocalized around the porphyrin ring of these de-rivatives and their HOMO shapes are relatively similar to each other.However,significant discrepancies are observed for the LUMOs:the central electrons of NO2-substitued derivatives are prone to move to NO2group,while in NH2-substituted de-rivatives these electrons are rearranged regularly around the porphyrin ring.In addition,due to the full shell of d orbital in zinc metal,there are also some differences in the distribution of the electron around the porphyrin ring of zinc porphyrins in comparison to free-base porphyrins.In summary,these dispari-ties should affect the electronic transition and absorption spec-tra of these molecules to a large degree,as shown in this work.3.3UV-Vis absorption spectraThe experimental absorption spectra of four porphyrin deriv-atives were measured in dichloromethane solution.3.3.1NO2PP(1)and NH2PP(2)According to the above discussions,new orbital interactions are expected to occur for porphyrin derivatives when the ortho positions are substituted by NO2and/or NH2group.As a conse-quence,the electronic structures and the Q/B-bands in the UV-Vis absorption spectrum should be changed.The wave-lengths and oscillator strengths of TD-DFT/PBE-calculated ex-citation of NO2PP and NH2PP,which can be assigned to the Q and B bands,are complied in Table3.The experimental wave-lengths are also listed.Due to more one-electron transitions involved,more excita-tions are obtained from the TD-DFT calculations,referring to Supporting Information.These excitations are probably related to the lower wavelength absorption bands of porphyrins,al-though they have slightly larger oscillator strengths than ex-pected.The calculated and the corresponding experimental ab-sorption spectra of NO2PP and NH2PP are presented in Fig.6 with a suitable broadening of Gaussian lineshape.3Calculated energies of the frontier molecular orbitals offree-base porphyrins1,2using the HOMO energy as areference4Calculated energies of the frontier molecular orbitals ofmetalloporphyrins3,4using the HOMO energy as a reference Table2Some calculated bond lengths and bond angles in metalloporphyrins3,4using two different functionals B3LYP and PBEa Data are taken from literature42.L(N—Cα)/nmL(Cα—Cβ)/nmL(Cβ—Cβ)/nmL(C m—Cα)/nmL(Cβ—H)/nmL(C m—C m)/nmL(C1—C6)/nmA(N—Zn—N)/(°)A(Zn—N—Cα)/(°)A(Cα—N—Cα)/(°)A(Cα—Cβ—Cβ)/(°)A(Cα—C m—Cα)/(°)D(N19C24C23C3)/(°)D(N4C3C23C24)/(°)D(N4C5C6C7)/(°)D(N8C7C6C5)/(°)D(C2C1C6C5)/(°)0.1380.144-0.1450.1360.1400.1080.6900.15089.8126.6-126.8106.4-106.5106.9125.40.627-2.1582.158-0.62777.460.1380.1450.1370.1410.1090.6920.15089.8126.9-127.1105.8-105.9106.8125.30.056-2.6242.624-0.05675.360.1380.1450.1360.1410.1080.6920.15090.0126.7-126.8106.4-106.6107.0124.61.174-1.2281.228-1.17482.440.1390.1450.1370.1410.1090.6950.15090.0126.8-127.0105.8-106.1106.9124.42.138-2.6032.603-2.13873.57a0.1380.1440.1351.400106.6107.41089Acta Phys.⁃Chim.Sin .2012V ol.28As evident from Table 3and Fig.6,the calculated electronicexcitations of Q bands are at 629.8and 586.8nm for 1/PBE and 665.8and 560.1nm for 2/PBE,which agree well with ex-perimental wavelengths (650.8and 551.2nm for 1,649.4and 550.1nm for 2).Calculations also show that there are several excitations between 500and 700nm assigned to Q band;how-ever,their oscillator strengths are very small (<10-3),and no ex-perimental absorptions could be related to these excitations.Fur-thermore,Q bands of 1/PBE originate from only two HOMOs (HOMO and HOMO -1)and more unoccupied MOs,because its HOMO and HOMO -1are identical and the unoccupied MOs are almost degenerate (see above).On the contrary,Q bands of 2/PBE result only from two LUMOs (LUMO and LUMO+1)and more occupied MOs.As far as the B band con-cerned,more electronic excitations are also obtained in the wavelength of 400-450nm (see Supporting Information).TheFig.5HOMO and LUMO isoamplitude surfaces of four porphyrin derivatives with PBE functionalTable 3TD-DFT/PBE-calculated and experimental wavelength (λ),and the corresponding one-electron transition andoscillator strengths of free-based porphyrins 1,2H:HOMO;L:LUMO;Abs:absorbanceMolecule NO 2PPNH 2PPBand Q0xQ 0yBQ 0x Q 0yBTD-DFT/PBEλ/nm 629.8586.8438.3665.1560.1408.7Excitation energy/eV1.972.112.831.892.213.03One-electron transitionH -1→L;H -1→L+5;H →L+1;H →L+4H →L+4;H -1→L;H -1→L+5H -3→L+4;H -1→L;H -1→L+5;H →L+4;H →L+9H -1→L+1;H -4→L;H →L H -5→L+1;H -4→L;H →L H -11→L+2;H -9→L;H -7→L+1;H -5→L;H -3→L+1Oscillator strength0.0170.0510.2540.0280.0160.247Experimentλ/nm 650.8551.2421.2649.4550.8419.2Excitation energy/eV 1.912.252.941.912.252.95Abs0.0330.0861.0020.0440.1050.896Fig.6Experimental and calculated absorption spectra of NO 2PP and NH 2PP1090CAO Zhen-Feng et al.:Electronic Absorption Spectra of Meso-Substituted Porphyrins and Their Zinc Derivatives No.5large number of excitations in the B band region can be attribut-ed to more one-electron transitions taking place between many occupied and unoccupied molecular orbitals.The addition of the NO2and NH2groups reduces the symmetry of the mole-cules,and therefore,more electronic excitations are allowed. In addition,Fig.6shows that the B band of NO2PP is more bathochromic than that of NH2PP in the calculated spectra, which is also observed in the experimental spectra.The red-shift can be explained by the characteristics of the NO2 group,which is electron withdrawing group and makes the electron of porphyrin ring delocalized well,leading to the slightly smaller gap between HOMO and LUMO than that of NH2PP(see Fig.3).The TD-DFT/B3LYP calculated results of the excitations are given in parison of Q and B bands in Tables3and 4reveals that the B3LYP absorption bands are all shifted to-ward lower wavelengths compared to those of PBE.This might be due to the sensitivity of the TD-DFT calculations to the as-ymptotic decay of the exchange-correlation potential,which in turn impacts the quality of the orbital energy differences that enter the TD-DFT formalism as a first approximation to the ex-citation energy.38,44It can be deduced from comparing with Fig.3that for1/B3LYP and2/B3LYP,the gap between the or-bital energies are2.705and2.767eV,respectively,which are about1eV larger than those obtained by PBE functional.Al-though as anticipated,hybrid functionals,which in general ex-hibit an improved asymptotic decay of the exchange-correla-tion potential over GGA functionals,would yield more accu-rate excitation energies.However,the hybrid functionals are improved by mixing of exact Hartree-fock exchange with the semilocal functionals,and the optimum amount of mixing is far from universal,45whereas no semiempirical parameters are involved for the PBE functional.From the reality of calcula-tions,the PBE functional is shown to be more accurate for pre-dicting the absorption spectra than B3LYP functional,and this is also true for the calculation of the metalloporphyrinʹs spectra (vide infra).3.3.2NO2ZnPP(3)and NH2ZnPP(4)The calculated and experimental spectra of NO2ZnPP and NH2ZnPP are presented in Fig.7with a suitable broadening of Gaussian line shape.Examination of this figure discloses that PBE-calculated wavelengths of Q bands for3/PBE and4/PBE are at582.9and562.9nm,respectively,with oscillator strengths of0.028and0.018,which coincides well with the ex-perimental values(601.8and599.2nm).In particular,in the absorption spectrum of NH2ZnPP,a peak lies at637nm with relatively strong oscillator strength of0.025,which may also be assigned to Q band.In addition,there are many other excita-tions with low oscillator strengths within the region of Q band, which can not be observed in experimental spectra.PBE-calcu-lated electronic excitations at416.4and415.1nm with maxi-mum oscillator strength of0.673and0.504for NO2ZnPP and NH2ZnPP,respectively,are assigned to B band,again in accor-dance with the experimental spectra in which the B band lo-cates at424.5and422.0nm,respectively.As shown in Fig.7(c,d),in comparison with the spectra cal-culated with PBE functional,the B3LYP-calculated spectra are not well in agreement with experimental spectra.Besides, there are two peaks between450and550nm which could be assigned to Q bands.This can be mainly attributed to the fact that the gap between the HOMO and LUMO calculated by B3LYP functional is1eV larger than that of PBE. Furthermore,similar to NO2PP and NH2PP,the red-shift is al-so observed in the absorption spectrum of NO2ZnPP with NH2ZnPP.In the experimental absorption spectra,the red-shift is about2nm for NO2PP and NH2PP and2.5nm for NO2ZnPP and NH2ZnPP.In the PBE-calculated absorption spectra,the difference between the B bands is30and1.3nm,respectively. The calculated red-shift follows the same trend as that revealed by the experiment,although there appear some disparities.The red-shift can be ascribed to the addition of the metal zinc in the center of the porphyrin ring and the meso-subsitituted groups, which influence the electron distribution of the porphyrins ring.3.4Triplet statesAs a potential material in optoelectronic field,such as organ-ic photovoltaic,organic field effect transistor,optical limiting, and artificial photosynthesis mimic,46,47porphyrin derivatives are usually investigated as an electron donor(in donor-accep-tor systems)to mimic the multi-step electron-transfer process. Generally speaking,the electron firstly transits from ground state to singlet state,then relaxes to triplet state,and eventual-ly,the excitation energy of triplet state transfers to the accep-Table4TD-DFT/B3LYP-calculated and experimental wavelength,and the corresponding one-electron transition andoscillator strengths of free-based porphyrins1,2MoleculeNO2PP NH2PP Q0xQ0yBQ0xQ0yBTD-DFT/B3LYPλ/nm580.2542.8377.1569.2533.5373.4Excitation energy/eV2.142.283.292.182.323.32One-electron transitionH-1→L+1;H→LH-1→L;H→L+1H-3→L+1;H-1→L;H-1→L+5;H→L+1H-1→L;H→L+1H-1→L+1;H→LH-7→L;H-5→L;H-3→L+1;H-1→L+1;H→LOscillator strength0.0120.0261.2410.0040.0230.949Experimentλ/nm650.8551.2421.2649.4550.8419.2Excitation energy/eV1.912.252.941.882.212.96Abs0.0330.0861.0020.0440.1050.8961091。