菲布力说明书
非布丙醇胶丸

非布丙醇胶丸
【药品名称】
通用名称:非布丙醇胶丸
英文名称:Febuprol Soft Capsules
【成份】
本品主要成分及其化学名称:本品的主要成分为非布丙醇,其化学名称为苯丁氧丙醇。
【适应症】
用于治疗胆囊炎、胆石症。
【用法用量】
口服:每次200mg胶丸,每日3次,儿童口服:2~4mg/kg,每日3次。
饭前服用。
【不良反应】
偶有胃部不适,用药初期会发生腹泻。
【禁忌】
肝功能不全或胆道梗阻的患者禁用、胃肠肿瘤、消化性溃疡肠道急性炎症禁用。
【注意事项】
用药初期会发生腹泻,此时应减量或停药数日,重新用药时由低剂量开始逐渐增加至所需剂量。
【特殊人群用药】
儿童注意事项:
儿童用药应根据体重年龄酌情减量。
妊娠与哺乳期注意事项:
孕妇尤其妊娠前三月及哺乳期妇女禁用。
老人注意事项:
老年患者慎用。
【药物相互作用】
尚不明确。
【药理作用】
本品有利胆作用,动物实验证明,无论肝实质是否损伤,均可使胆汁分泌增加,此外尚具有松弛胆管平滑肌及奥狄括约肌、降低血中胆固醇的作用。
【贮藏】
避光,密闭凉处保存。
有效期二年。
【有效期】
24个月
【批准文号】
国药准字H21022923
【说明书修订日期】
核准日期:2007年04月02日
【生产企业】
企业名称:沈阳同联药业有限公司
生产地址:沈阳市沈北新区金星街55号。
Philips HeartStart FR2-Series Defibrillator 说明书

Philips HeartStart FR2-Series DefibrillatorSupplies and accessoriesProducts and services,maximizing defibrillator performanceQuick Reference GuideItem #M3860-97800The Quick Reference Guide is a briefinstruction guide for defibrillator operators.Its short captions and straightforward drawings break down each step of the defibrillation process.Carry CasesThere are four carry cases available for the HeartStart FR2-Series Defibrillator:the Standard Carry Case,either semi-rigid cordora or vinyl;the Temperature Control Carry Case;and the hard-shell waterproof case.FR2-Series Carry CaseSemi-rigidItem #M3868AA red cordura,semi-rigid carrying case(M3868A) holds the device as well as its main accessories and supplies.VinylItem #M3869AAn easy-to-clean vinyl carrying case (M3869A) is also available.Both the cordura and vinyl carrying cases are intended for use in environments where the defibrillator is protected from moisture and harsh treatment.Dimensions:9.5" (24 cm) w X 8.5" (21 cm) h X 4.8" (12 cm) dFast Response KitItem #68-pchatThe Fast Response Kit contains tools and supplies typically needed for patient care and personal protection:2 pairs of hypoallergenic nitrile gloves,a pocket breathing mask,paramedic scissors,a chest hair razor,and a large extra-absorbent paper towel.These items are housed in a zippered pouch which attaches securely to the handle of either the semi-rigid or vinyl carry case.Dimensions:9.5" (24 cm) w X 5.5" (14 cm) hTemperature Control Case Item #989803133171With the T emperature Control Carrying Case (989803133171),you can safely store your HeartStart FR2-Series Defibrillator in the trunk or storage compartment of a vehicle during extreme winter and summer temperatures.It provides protection from temperatures as low as –20°F (-29ºC) and as high as 140°F(60ºC),perfect for utility trucks,police cruisers and other vehicles in which heating or air conditioning isn’t always available.The case’s thermostat-regulated heater can be powered by a 12-volt vehicle battery.The built-in cut-off module turns off the heater before it depletes the battery.And a thick foam insulation layer slows the heating effect of hot summer days.Dimensions:9.5" (24 cm) w X 8.5" (21 cm) h X 3.5" (9 cm) dHard-shell Carry Case Item #YCOur waterproof carry case made of hard-shell plastic (product code YC)is suited for more rigorous use,particularly in wet outdoor settings.It can also accommodate the contents of the Fast Response Kit.Dimensions:13.5" (34 cm) w X 12" (30 cm) h X 6" (15 cm) d*The Americans with Disabilities Act requires that objects not protrude more than 4 inches into foot traffic areas of openaisles and walkways unless the object’s bottom edge is no higher than 27 inches from the ground.Premium Surface Mounted Cabinet Item #PFE7024DDimensions:16" (41 cm) w X 22.5" (57 cm) h X 6" (15 cm) dPremium Semi-recessed Cabinet Item #PFE7023DDimensions:Recessed Compartment –14" (36 cm) w X 22" (56 cm) h X 6" (15 cm) d Footprint on wall –16.5" (42 cm) w X 24.5" (62 cm) h X 2.5" (6 cm) dBasic Surface Mounted Cabinet Item #989803136531Dimensions:16.5" (42 cm) w X 15" (38 cm) h X 6" (15 cm) dWall Mounting SolutionsPhilips Wall Mount Bracket and Defibrillator Cabinets let you strategically place defibrillators for fast access and response.Wall Mount Bracket Item #M3857ADimensions:10.6" (270 mm) w X 8" (204 mm) h X 5" (126 mm) d Weight:19 ounces (.5 kg)Item #M3859A Secure-pull SealWall Mount BracketThe Wall Mount Bracket (M3857A) is designed specifically for housing a Philips HeartStart Defibrillator and its accessories.Thedefibrillator’s carry case can be tethered to the Wall Mount Bracket with a breakaway Secure-Pull Seal (M3859A),to discourage tampering.A broken seal indicates that the defibrillator has been removed from the Wall Mount and accessories may need to be replenished.Defibrillator CabinetsT o mobilize an emergency medical response or deter AED theft,Philips offers 3 different battery-operated,alarmed wall cabinets.The basic cabinet has a simple audible alarm.Also available are two premium cabinets:a wall surface mounted cabinet and a semi-recessed cabinet* that is inserted into a wall cut-out for a less obtrusive look.The premium cabinets feature combination audible and flashing light alarms.They are made of sturdy heavy-gauge steel,and are large enough to accommodate additional medical supplies,such as oxygen.Y ou can also connect the premium cabinets’alarms to your internal security system so that a more coordinated emergency response can be mobilized centrally.AED Wall Sign Item #M3858AAn AED Wall Sign (M3858A) hanging above a Wall Mount Bracket or Defibrillator Cabinet gives even greater visibility to the defibrillator.Dimensions:7" (18 cm) w X 10" (25.5 cm) hPads and AdaptersHeartStart Adult AED Defibrillator Pads Item # DP2/DP6HeartStart Adult AED Defibrillator Pads (DP2 and DP6),recommended for patients 8years and older,or above 55 pounds (25kg),are optimized for use with Philips FR2-series defibrillators.Constructed of a thin flexible conductor sandwichedbetween a protective polymer backing and a hydro-gel adhesive,these pads withstand the rigors of CPR and harsh conditions.Soft,flexible and oval-shaped,HeartStart Adult AED Pads conform to body contours for ample surface contact and adhesion.For smooth handoff to advanced life support (ALS) personnel,the pads connector is compatible with the hands-free cable of HeartStart manual defibrillators which have advanced capabilities.Once attached,these multifunction electrode pads can provide ECG monitoring,external pacing,and synchronized cardioversion,in addition to defibrillation.FR2 Infant/Child Reduced-Energy Defibrillator Pads Item # M3870AHeartStart FR2 Infant/Child Defibrillator Pads (M3870A) are for treating infants and children under 8 years of age or 55 pounds with an FR2-series defibrillator.These special infant/child pads contain electronics that attenuate,or reduce,the energy of the defibrillator’s shock from 150to 50 Joules,a more appropriate dosage of SMART Biphasic therapy for infants and small children.1And the FR2-seriesdefibrillator can evaluate children’s heart rhythms with outstanding accuracy.2The pads’ design was carefully thought out to ensure that,even for the most inexperienced user under the most stressful circumstances,it is instantly obvious that these pads are for children and infants only.This helps ensure that the correct pads are chosen when you need to be prepared to treat both adults and children.The packaging,graphics,and pink teddy bear-shaped connector clearly communicate pediatric use.1.T ang,et al.Pediatric Fixed Energy Biphasic Waveform Defibrillation Using a Standard AED and Special Pediatric Electrodes.Supplement to Circulation,Vol 102,No 18,October 31,2000,II-437.2.Cecchin,et al.Is Arrhythmia Detection by Automatic External Defibrillator Accurate for Children?Sensitivity and Specificity of an AED Algorithm in 696 Pediatric Arrhythmias.Circulation 2001;103:2483-2488,May 22,2001.HeartStart AdaptersHeartStart Adult AED Defibrillator Pads may also be used with defibrillators from other manufacturers,using Philips HeartStart Adapters,whether defibrillators are biphasic or monophasic,manual or automated.The adapters allow ALScaregivers to connect the pads to their manual defibrillator.Adapters cannot be used with FR2 Infant/Child Defibrillator Pads.Permanent ConfigurationRemovable ConfigurationAdapter Model 05-10200Fits Philips hands-free cables M3507A or M1750A/B for connection to Hewlett-Packard CodeMaster 100,XE,XL,and XL+;PhilipsHeartstream/HeartStart XL and XLT;HeartStart MRx and Laerdal Heartstart 4000 defibrillator/monitorsAdapter Model 05-10000Removable adapter for Medtronic Physio-Control Quik-Combo LifePak 9,10C,11,12,20,and 500 defibrillatorsAdapter Model 05-10100Removable adapter for Zoll 1200,1400,1600 and M-Series defibrillatorsFR2-Series Standard Battery Item #M3863AFR2-series defibrillators come with adisposable lithium manganese dioxide battery (M3863A),which has a shelf life of 5 years,plus typical standby life of 5 years (4 years minimum).**U.S.airline customers should order the FAA-certified version of this battery (989803136291).Power SuppliesFR2+ Rechargeable Battery Item #M3848AIn hospital and EMS environments,where the useful life of a defibrillator’s disposable battery can be consumed in a relatively short period of time,the FR2+ Rechargeable Battery(M3848A) provides a cost-effective option for frequent-use applications,such as back-to-back sudden cardiac arrest responses and monitoring patients during transport.This Lithium-Ion battery fully charges(using the Charger M3849A) in just 3 hours to provide 100 shocks (typical) or 5 hours (typical) of ECG display time.The “fuel gauge”on the FR2+ displays the battery’s remaining power.Under normal conditions,the FR2+Rechargeable Battery withstands 300 charge-discharge cycles or 2.5 years of use.** Use of the defibrillator,additional battery insertion tests,or exposure to temperature extremes may shorten the battery life.*** FR2+ ECG screen is not suitable for diagnostic and ST -segment interpretation.FR2+ ECG Assessment ModuleIdeal where manual defibrillators and monitors are impractical,the Philips FR2+ ECGAssessment Module enables the professional responder to use the HeartStart FR2+Defibrillator with ECG to assess the heart rhythms of conscious and/or breathing patients who may be in cardiac distress.With this reusable unit,caregivers knowledgeable in ECG rhythms can use the defibrillator’s display to view the Lead II ECG rhythm of a patient who may be experiencing dizziness,chestdiscomfort,or becomes unconscious but is still breathing.***If arrhythmias are observed or the patient’s status changes,the responder can proactively determine next steps.And if the patient goes into cardiac arrest,the responder can react immediately by switching to defibrillation pads.The FR2+ ECG Assessment Module,compatible with most standard snapmonitoring electrodes,including M2202A (radioluscent foam),is available in two configurations:Item #M3873AAAMI color convention with red,white and black leadwire connections.Item #M3874AIEC color convention with green,red and yellow leadwire connections.Training SolutionsTraining and Administration Pack When equipped with the rechargeable training and administration pack (M3864A),the FR2-series defibrillator acts as a trainer.Its shock delivery capability is disabled while you train with nine realistic scenarios.And whenconnected to a simulator or a special training manikin capable of producing an ECG rhythm,the FR2+ with training and administration pack will produce realistic responses to various heart rhythms.In addition to training,the pack allows you to reconfigure the defibrillator’s behavior to your specific cardiac arrest protocol.*AED Trainer 2For training many responders simultaneously,HeartStart AED T rainer 2 (M3752A) is a more flexible and economical solution.AED T rainer 2 helps your responders learn to use FR2-series defibrillators in simulated sudden cardiac arrest episodes for an extremely realistic training experience.AED T rainer 2looks and behaves like the FR2-series defibrillators,but cannot deliver an actual defibrillation shock.It is pre-configured with 10 realistic training scenarios,developed in accordance with internationally recognized emergencyresponder training programs.Voice prompts in various languages and additional customscenarios can be configured using the optional programming kit (M3754A).An optional remote control (M3753A) lets the instructor adjust the T rainer’s volume,select a scenario,pause and then resume the scenario in progress to give instruction,and override the scenario to test how students respond to a variety of situations they may encounter.The AED T rainer 2 comes equipped with one set of adult-size reusable training pads (07-10900),a quick reference instruction card,a user guide,and a carrying case.HeartStart AED Trainer 2Item # M3752ARemote Control Item # M3753ATraining and Administration Pack with ChargerItem # M3864A with M3855AProgramming Kit Item # M3754AHeartStart Training PadsItem #07-10900*Changes to default values should be done only by authorized personnel under the oversight of a medical professional.FR2 infant/Child Training Pads Item # M3871AAED Little Anne Training System Also available is the all-in-one AED Little Anne T raining System (M3756A),for added realism.In addition to the AED T rainer 2, quick reference card,user guide,and carrying case,this total training solution also includes the remote control,special training pads (M3755A) and a Laerdal AED Little Anne training manikin.The T rainer,AED Little Anne manikin,and special training pads work together to provide feedback on the quality of pads placement.Instructor's Training Toolkit The training toolkit (M5066-89100) includes instructional aids,such as a videotape and CD with a PowerPoint presentation,for teaching groups of people to operate the HeartStart FR2-series defibrillator.Data Collection and StorageData Card and TrayItem #M3854AThe data card (M3854A) holds approximately 8 hours of incident and ECG information,or one hour with voice recording.One card can hold data from multiple cases.A flash data card reader enables data transfer from the card to a personal computer for use with HeartStart Event Review data management software.Event ReviewItem #M3834A (single PC)or 989803141811 (organization-wide)Event Review allows you to download patient data from your defibrillator,and view it on your PC screen,annotate it with your comments,and add basic response and patient status information. Y ou can save the case to a file or to a database,allowing ad hoc case queries and case reports.It is available with single PC pricing or unlimited organization-wide pricing.Event Review ProItem #861276 Opt A01Event Review Pro is our comprehensive case management tool for the most demanding data managers and medical directors,with even more detailed data entry screens to record every aspect of the response, including detailed response times,interventions,and patient observations.In addition to the individual case reports,you get Utstein reporting and graphical summaries of your system’s overall response times to help you manage your service levels more efficiently.Philips Medical Systems is part of Royal Philips Electronics/heartstart*******************Philips Medical Systems3000 Minuteman RoadAndover,MA 01810-1085+1800 934-7372©Koninklijke Philips ElectronicsN.V.2005.All rights reserved.Reproduction in whole or in partis prohibited without prior writtenconsent of the copyright holder.Philips Medical Systems NorthAmerica Corporation reservesthe right to make changes inspecifications or to discontinue anyproduct at any time without noticeor obligation and will not be liablefor any consequences resultingfrom the use of this publication.Philips is a registered trademark ofKoninklijke Philips Electronics N.V.Printed in The Netherlands.4522 962 06871/861 * AUG 2005HeartStart EssentialsPhilips helps implement a comprehensive program with most everything you and your medical director will need to help save a life from sudden cardiac arrest.We’ll get you started with pre-implementation consulting and site assessments.We can help manage your everyday needs including medical direction from a licensed physician,web-based program management,data management software,and responder and instructor training.Philips offers ongoing support with a five-year warranty,optional extended warranty and continued customer support from supplies ordering to technical assistance.Philips Medical SuppliesPhilips is committed to producing and supporting the finest quality medical equipmentand supplies.Our supplies are thoughtfully designed,tested and manufactured todeliver reliable and accurate results from your HeartStart Defibrillators.For a completelist of supplies,please visit .。
非布司他021856lbl

HIGHLIGHTS OF PRESCRIBING INFORMATIONThese HIGHLIGHTS do not include all the information needed to use ULORIC safely and effectively. See full prescribing information for ULORIC.ULORIC (febuxostat) tablet for oral useInitial U.S. Approval: 2009----------------------------INDICATIONS AND USAGE--------------------------- ULORIC is a xanthine oxidase (XO) inhibitor indicated for the chronic management of hyperuricemia in patients with gout. (1)ULORIC is not recommended for the treatment of asymptomatic hyperuricemia. (1)------------------------DOSAGE AND ADMINISTRATION---------------------- • ULORIC is recommended at 40 mg or 80 mg once daily. The recommended starting dose of ULORIC is 40 mg once daily. For patients who do not achieve a serum uric acid (sUA) less than 6 mg per dL after 2 weeks with 40 mg, ULORIC 80 mg is recommended.(2.1)• ULORIC can be administered without regard to food or antacid use.(2.1)• No dose adjustment is necessary when administering ULORIC to patients with mild to moderate renal or hepatic impairment. (2.2)--------------------DOSAGE FORMS AND STRENGTHS---------------------- Tablet: 40 mg, 80 mg. (3)------------------------------CONTRAINDICATIONS------------------------------- ULORIC is contraindicated in patients being treated with azathioprine, mercaptopurine, or theophylline. (4)-------------------------WARNINGS AND PRECAUTIONS---------------------- • Gout Flare: An increase in gout flares is frequently observed during initiation of anti-hyperuricemic agents, including ULORIC. If a gout flare occurs during treatment, ULORIC need not be discontinued.Prophylactic therapy (i.e., non-steroidal anti-inflammatory drug(NSAID) or colchicine upon initiation of treatment) may be beneficial for up to six months. (2.4, 5.1) • Cardiovascular Events: A higher rate of cardiovascular thromboembolic events was observed in patients treated withULORIC than allopurinol in clinical trials. Monitor for signs andsymptoms of MI and stroke. (5.2)• Liver Enzyme Elevation: Transaminase elevations have been observed in ULORIC-treated patients. Monitor liver function testsperiodically. (5.3)------------------------------ADVERSE REACTIONS------------------------------- Adverse reactions occurring in at least 1% of ULORIC-treated patients, and, at least 0.5% greater than placebo, are liver function abnormalities, nausea, arthralgia, and rash. (6.1)To report SUSPECTED ADVERSE REACTIONS, contact Takeda Pharmaceuticals at 1.877.825.3327 or FDA at 1.800.FDA.1088 or /medwatch.-------------------------------DRUG INTERACTIONS------------------------------ Concomitant administration of ULORIC with XO substrate drugs, azathioprine, mercaptopurine, or theophylline could increase plasma concentrations of these drugs resulting in severe toxicity. (7)-----------------------USE IN SPECIFIC POPULATIONS-----------------------• There is insufficient data in patients with severe renal impairment. No studies have been conducted in patients with severe hepaticimpairment. Caution should be exercised in these patients. (8.6, 8.7) • No studies have been conducted in patients with secondary hyperuricemia (including patients being treated for Lesch-Nyhansyndrome or malignant disease, or in organ transplant recipients);therefore, ULORIC is not recommended for use in these patients.(8.8)See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.Revised: February 2009FULL PRESCRIBING INFORMATION: CONTENTS* 1 INDICATIONSANDUSAGE2 DOSAGEANDADMINISTRATION2.1 Recommended Dose2.2 Special Populations2.3 Uric Acid Level2.4 Gout Flares3 DOSAGE FORMS AND STRENGTHS4 CONTRAINDICATIONS5 WARNINGSANDPRECAUTIONS5.1 Gout Flare5.2 Cardiovascular Events5.3 Liver Enzyme Elevations6 ADVERSEREACTIONS6.1 Clinical Trials Experience7 DRUGINTERACTIONS7.1 Xanthine Oxidase Substrate Drugs7.2 Cytotoxic Chemotherapy Drugs7.3 In Vivo Drug Interaction Studies8 USE IN SPECIFIC POPULATIONS8.1 Pregnancy8.3 Nursing Mothers8.4 Pediatric Use8.5 Geriatric Use8.6 Renal Impairment8.7 Hepatic Impairment8.8 Secondary Hyperuricemia10 OVERDOSAGE11 DESCRIPTION12 CLINICAL PHARMACOLOGY12.1 Mechanism of Action12.2 Pharmacodynamics12.3 Pharmacokinetics13 NONCLINICAL TOXICOLOGY13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility13.2 Animal Toxicology14 CLINICAL STUDIES14.1 Management of Hyperuricemia in Gout16 HOW SUPPLIED/STORAGE AND HANDLING17 PATIENT COUNSELING INFORMATION17.1 General Information17.2 FDA-Approved Patient Labeling*Sections or subsections omitted from the full prescribing information are not listedFULL PRESCRIBING INFORMATION1 INDICATIONS AND USAGEULORIC® is a xanthine oxidase (XO) inhibitor indicated for the chronic management of hyperuricemia in patients with gout.ULORIC is not recommended for the treatment of asymptomatic hyperuricemia.2 DOSAGE AND ADMINISTRATION2.1 Recommended DoseFor treatment of hyperuricemia in patients with gout, ULORIC is recommended at 40 mg or 80 mg once daily.The recommended starting dose of ULORIC is 40 mg once daily. For patients who do not achieve a serum uric acid (sUA) less than 6 mg per dL after 2 weeks with 40 mg, ULORIC 80 mg is recommended.ULORIC can be taken without regard to food or antacid use [see Clinical Pharmacology (12.3)].2.2 Special PopulationsNo dose adjustment is necessary when administering ULORIC in patients with mild to moderate renal impairment [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)]. The recommended starting dose of ULORIC is 40 mg once daily. For patients who do not achieve a sUA less than 6 mg per dL after 2 weeks with 40 mg, ULORIC 80 mg is recommended.No dose adjustment is necessary in patients with mild to moderate hepatic impairment [see Use in Specific Populations (8.7) and Clinical Pharmacology (12.3)].2.3 Uric Acid LevelTesting for the target serum uric acid level of less than 6 mg per dL may be performed as early as 2 weeks after initiating ULORIC therapy.2.4 Gout FlaresGout flares may occur after initiation of ULORIC due to changing serum uric acid levels resulting in mobilization of urate from tissue deposits. Flare prophylaxis with a non-steroidal anti-inflammatory drug (NSAID) or colchicine is recommended upon initiation of ULORIC. Prophylactic therapy may be beneficial for up to six months [see Clinical Studies (14.1)].If a gout flare occurs during ULORIC treatment, ULORIC need not be discontinued. The gout flare should be managed concurrently, as appropriate for the individual patient [see Warnings and Precautions (5.1)].3 DOSAGE FORMS AND STRENGTHS• 40 mg tablets, light green to green, round shaped, debossed with “TAP” and “40”• 80 mg tablets, light green to green, teardrop shaped, debossed with “TAP” and “80”4 CONTRAINDICATIONSULORIC is contraindicated in patients being treated with azathioprine, mercaptopurine, or theophylline [see Drug Interactions (7)].5 WARNINGS AND PRECAUTIONS5.1 Gout FlareAfter initiation of ULORIC, an increase in gout flares is frequently observed. This increase is due to reduction in serum uric acid levels resulting in mobilization of urate from tissue deposits.In order to prevent gout flares when ULORIC is initiated, concurrent prophylactic treatment with an NSAID or colchicine is recommended [see Dosage and Administration (2.4)].5.2 Cardiovascular EventsIn the randomized controlled studies, there was a higher rate of cardiovascular thromboembolic events (cardiovascular deaths, non-fatal myocardial infarctions, and non-fatal strokes) in patients treated with ULORIC [0.74 per 100 P-Y (95% CI 0.36-1.37)] than allopurinol [0.60 per 100 P-Y (95% CI 0.16-1.53)] [see Adverse Reactions (6.1)]. A causal relationship with ULORIC has not been established. Monitor for signs and symptoms of myocardial infarction (MI) and stroke.5.3 Liver Enzyme ElevationsDuring randomized controlled studies, transaminase elevations greater than 3 times the upper limit of normal (ULN) were observed (AST: 2%, 2%, and ALT: 3%, 2% in ULORIC and allopurinol-treated patients, respectively). No dose-effect relationship for these transaminase elevations was noted. Laboratory assessment of liver function is recommended at, for example, 2 and 4 months following initiation of ULORIC and periodically thereafter.6 ADVERSE REACTIONS6.1 Clinical Trials ExperienceBecause clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.A total of 2757 subjects with hyperuricemia and gout were treated with ULORIC 40 mg or 80 mg daily in clinical studies. For ULORIC 40 mg, 559 patients were treated for ≥ 6 months. For ULORIC 80 mg, 1377 subjects were treated for ≥ 6 months, 674 patients were treated for ≥ 1 year and 515 patients were treated for ≥ 2 years.Most Common Adverse ReactionsIn three randomized, controlled clinical studies (Studies 1, 2 and 3), which were 6 to 12 months in duration, the following adverse reactions were reported by the treating physician as related to study drug. Table 1 summarizes adverse reactions reported at a rate of at least 1% in ULORIC treatment groups and at least 0.5% greater than placebo.Table 1: Adverse Reactions Occurring in ≥ 1% of ULORIC-Treated Patients and at Least 0.5% Greater than Seen in Patients Receiving Placebo in Controlled StudiesAdverse Reactions Placebo ULORIC allopurinol* (N=134)40 mg daily(N=757)80 mg daily(N=1279) (N=1277)Liver Function AbnormalitiesNauseaArthralgiaRash 0.7%0.7%0%0.7%6.6%1.1%1.1%0.5%4.6%1.3%0.7%1.6%4.2%0.8%0.7%1.6%* Of the subjects who received allopurinol, 10 received 100 mg, 145 received 200 mg, and 1122 received 300 mg, based on level of renal impairment. The most common adverse reaction leading to discontinuation from therapy was liver function abnormalities in 1.8% of ULORIC 40 mg, 1.2% of ULORIC 80 mg, and in 0.9% of allopurinol-treated subjects.In addition to the adverse reactions presented in Table 1, dizziness was reported in more than 1% of ULORIC-treated subjects although not at a rate more than 0.5% greater than placebo.Less Common Adverse ReactionsIn phase 2 and 3 clinical studies the following adverse reactions occurred in less than 1% of subjects and in more than one subject treated with doses ranging from 40 mg to 240 mg of ULORIC. This list also includes adverse reactions (less than 1% of subjects) associated with organ systems from Warnings and Precautions.Blood and Lymphatic System Disorders: anemia, idiopathic thrombocytopenic purpura, leukocytosis/leukopenia, neutropenia, pancytopenia, splenomegaly, thrombocytopenia.Cardiac Disorders: angina pectoris, atrial fibrillation/flutter, cardiac murmur, ECG abnormal, palpitations, sinus bradycardia, tachycardia.Ear and Labyrinth Disorders: deafness, tinnitus, vertigo.Eye Disorders: vision blurred.Gastrointestinal Disorders: abdominal distention, abdominal pain, constipation, dry mouth, dyspepsia, flatulence, frequent stools, gastritis, gastroesophageal reflux disease, gastrointestinal discomfort, gingival pain, haematemesis, hyperchlorhydria, hematochezia, mouth ulceration, pancreatitis, peptic ulcer, vomiting.General Disorders and Administration Site Conditions: asthenia, chest pain/discomfort, edema, fatigue, feeling abnormal, gait disturbance, influenza-like symptoms, mass, pain, thirst. Hepatobiliary Disorders: cholelithiasis/cholecystitis, hepatic steatosis, hepatitis, hepatomegaly. Immune System Disorder: hypersensitivity.Infections and Infestations: herpes zoster.Procedural Complications: contusion.Metabolism and Nutrition Disorders: anorexia, appetite decreased/increased, dehydration, diabetes mellitus, hypercholesterolemia, hyperglycemia, hyperlipidemia, hypertriglyceridemia, hypokalemia, weight decreased/increased.Musculoskeletal and Connective Tissue Disorders: arthritis, joint stiffness, joint swelling, muscle spasms/twitching/tightness/weakness, musculoskeletal pain/stiffness, myalgia.Nervous System Disorders: altered taste, balance disorder, cerebrovascular accident, Guillain-Barré syndrome, headache, hemiparesis, hypoesthesia, hyposmia, lacunar infarction, lethargy, mental impairment, migraine, paresthesia, somnolence, transient ischemic attack, tremor.Psychiatric Disorders: agitation, anxiety, depression, insomnia, irritability, libido decreased, nervousness, panic attack, personality change.Renal and Urinary Disorders: hematuria, nephrolithiasis, pollakiuria, proteinuria, renal failure, renal insufficiency, urgency, incontinence.Reproductive System and Breast Changes: breast pain, erectile dysfunction, gynecomastia. Respiratory, Thoracic and Mediastinal Disorders: bronchitis, cough, dyspnea, epistaxis, nasal dryness, paranasal sinus hypersecretion, pharyngeal edema, respiratory tract congestion, sneezing, throat irritation, upper respiratory tract infection.Skin and Subcutaneous Tissue Disorders: alopecia, angio edema, dermatitis, dermographism, ecchymosis, eczema, hair color changes, hair growth abnormal, hyperhidrosis, peeling skin, petechiae, photosensitivity, pruritus, purpura, skin discoloration/altered pigmentation, skin lesion, skin odor abnormal, urticaria.Vascular Disorders: flushing, hot flush, hypertension, hypotension.Laboratory Parameters: activated partial thromboplastin time prolonged, creatine increased, bicarbonate decreased, sodium increased, EEG abnormal, glucose increased, cholesterol increased, triglycerides increased, amylase increased, potassium increased, TSH increased, platelet count decreased, hematocrit decreased, hemoglobin decreased, MCV increased, RBC decreased, creatinine increased, blood urea increased, BUN/creatinine ratio increased, creatine phosphokinase (CPK) increased, alkaline phosphatase increased, LDH increased, PSA increased, urine output increased/decreased, lymphocyte count decreased, neutrophil count decreased, WBC increased/decreased, coagulation test abnormal, low density lipoprotein (LDL) increased, prothrombin time prolonged, urinary casts, urine positive for white blood cells and protein. Cardiovascular SafetyCardiovascular events and deaths were adjudicated to one of the pre-defined endpoints from the Anti-Platelet Trialists’ Collaborations (APTC) (cardiovascular death, non-fatal myocardial infarction, and non-fatal stroke) in the randomized controlled and long-term extension studies. In the Phase 3 randomized controlled studies, the incidences of adjudicated APTC events per 100 patient-years of exposure were: Placebo 0 (95% CI 0.00-6.16), ULORIC 40 mg 0 (95% CI 0.00-1.08), ULORIC 80 mg 1.09 (95% CI 0.44-2.24), and allopurinol 0.60 (95% CI 0.16-1.53).In the long-term extension studies, the incidences of adjudicated APTC events were: ULORIC 80 mg 0.97 (95% CI 0.57-1.56), and allopurinol 0.58 (95% CI 0.02-3.24).Overall, a higher rate of APTC events was observed in ULORIC than in allopurinol-treated patients.A causal relationship with ULORIC has not been established. Monitor for signs and symptoms of MI and stroke.7 DRUG INTERACTIONS7.1 Xanthine Oxidase Substrate DrugsULORIC is an XO inhibitor. Drug interaction studies of ULORIC with drugs that are metabolized by XO (e.g., theophylline, mercaptopurine, azathioprine) have not been conducted. Inhibition of XO by ULORIC may cause increased plasma concentrations of these drugs leading to toxicity [see Clinical Pharmacology (12.3)]. ULORIC is contraindicated in patients being treated with azathioprine, mercaptopurine, or theophylline [see Contraindications (4)].7.2 Cytotoxic Chemotherapy DrugsDrug interaction studies of ULORIC with cytotoxic chemotherapy have not been conducted. No data are available regarding the safety of ULORIC during cytotoxic chemotherapy.7.3 In Vivo Drug Interaction StudiesBased on drug interaction studies in healthy subjects, ULORIC does not have clinically significant interactions with colchicine, naproxen, indomethacin, hydrochlorothiazide, warfarin or desipramine [see Clinical Pharmacology (12.3)]. Therefore, ULORIC may be used concomitantly with these medications.8 USE IN SPECIFIC POPULATIONS8.1 PregnancyPregnancy Category C: There are no adequate and well-controlled studies in pregnant women. ULORIC should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.Febuxostat was not teratogenic in rats and rabbits at oral doses up to 48 mg per kg (40 and 51 times the human plasma exposure at 80 mg per day for equal body surface area, respectively) during organogenesis. However, increased neonatal mortality and a reduction in the neonatal body weight gain were observed when pregnant rats were treated with oral doses up to 48 mg per kg (40 times the human plasma exposure at 80 mg per day) during organogenesis and through lactation period.8.3 Nursing MothersFebuxostat is excreted in the milk of rats. It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when ULORIC is administered to a nursing woman.8.4 Pediatric UseSafety and effectiveness in pediatric patients under 18 years of age have not been established.8.5 Geriatric UseNo dose adjustment is necessary in elderly patients. Of the total number of subjects in clinical studies of ULORIC, 16 percent were 65 and over, while 4 percent were 75 and over. Comparing subjects in different age groups, no clinically significant differences in safety or effectiveness were observed but greater sensitivity of some older individuals cannot be ruled out. The C max and AUC24 of febuxostat following multiple oral doses of ULORIC in geriatric subjects (≥ 65 years) were similar to those in younger subjects (18-40 years) [see Clinical Pharmacology (12.3)].8.6 Renal ImpairmentNo dose adjustment is necessary in patients with mild or moderate renal impairment (Cl cr 30-89 mL per min). The recommended starting dose of ULORIC is 40 mg once daily. For patients who do not achieve a sUA less than 6 mg per dL after 2 weeks with 40 mg, ULORIC 80 mg is recommended.There are insufficient data in patients with severe renal impairment (Cl cr less than 30 mL per min); therefore, caution should be exercised in these patients [see Clinical Pharmacology (12.3)].8.7 Hepatic Impairment No dose adjustment is necessary in patients with mild or moderate hepatic impairment (Child-Pugh Class A or B). No studies have been conducted in patients with severe hepatic impairment (Child-Pugh Class C); therefore, caution should be exercised in these patients [see Clinical Pharmacology (12.3)].8.8 Secondary Hyperuricemia No studies have been conducted in patients with secondary hyperuricemia (including organtransplant recipients); ULORIC is not recommended for use in patients whom the rate of urateformation is greatly increased (e.g., malignant disease and its treatment, Lesch-Nyhan syndrome). The concentration of xanthine in urine could, in rare cases, rise sufficiently to allow deposition in the urinary tract.10 OVERDOSAGEULORIC was studied in healthy subjects in doses up to 300 mg daily for seven days withoutevidence of dose-limiting toxicities. No overdose of ULORIC was reported in clinical studies.Patients should be managed by symptomatic and supportive care should there be an overdose. 11 DESCRIPTIONULORIC (febuxostat) is a xanthine oxidase inhibitor. The active ingredient in ULORIC is 2-[3-cyano4-(2-methylpropoxy) phenyl]-4-methylthiazole-5-carboxylic acid, with a molecular weight of 316.38. The empirical formula is C 16H 16N 2O 3S.The chemical structure is: ONC H 3C CH 3S NCH 32H Febuxostat is a non-hygroscopic, white crystalline powder that is freely soluble indimethylformamide; soluble in dimethylsulfoxide; sparingly soluble in ethanol; slightly soluble inmethanol and acetonitrile; and practically insoluble in water. The melting range is 205°C to 208°C. ULORIC tablets for oral use contain the active ingredient, febuxostat, and are available in two dosage strengths, 40 mg and 80 mg. Inactive ingredients include lactose monohydrate,microcrystalline cellulose, hydroxypropyl cellulose, sodium croscarmellose, silicon dioxide and magnesium stearate. ULORIC tablets are coated with Opadry II, green.12 CLINICAL PHARMACOLOGY12.1 Mechanism of ActionULORIC, a xanthine oxidase inhibitor, achieves its therapeutic effect by decreasing serum uric acid. ULORIC is not expected to inhibit other enzymes involved in purine and pyrimidine synthesis and metabolism at therapeutic concentrations.12.2 PharmacodynamicsEffect on Uric Acid and Xanthine Concentrations: In healthy subjects, ULORIC resulted in a dose dependent decrease in 24-hour mean serum uric acid concentrations, and an increase in 24-hour mean serum xanthine concentrations. In addition, there was a decrease in the total daily urinary uric acid excretion. Also, there was an increase in total daily urinary xanthine excretion. Percent reduction in 24-hour mean serum uric acid concentrations was between 40% to 55% at the exposure levels of 40 mg and 80 mg daily doses.Effect on Cardiac Repolarization: The effect of ULORIC on cardiac repolarization as assessed by the QTc interval was evaluated in normal healthy subjects and in patients with gout. ULORIC in doses up to 300 mg daily, at steady state, did not demonstrate an effect on the QTc interval.12.3 PharmacokineticsIn healthy subjects, maximum plasma concentrations (C max) and AUC of febuxostat increased in a dose proportional manner following single and multiple doses of 10 mg to 120 mg. There is no accumulation when therapeutic doses are administered every 24 hours. Febuxostat has an apparent mean terminal elimination half-life (t1/2) of approximately 5 to 8 hours. Febuxostat pharmacokinetic parameters for patients with hyperuricemia and gout estimated by population pharmacokinetic analyses were similar to those estimated in healthy subjects.Absorption: The absorption of radiolabeled febuxostat following oral dose administration was estimated to be at least 49% (based on total radioactivity recovered in urine). Maximum plasma concentrations of febuxostat occurred between 1 to 1.5 hours post-dose. After multiple oral 40 mg and 80 mg once daily doses, C max is approximately 1.6 ± 0.6 mcg per mL (N=30), and 2.6 ± 1.7 mcg per mL (N=227), respectively. Absolute bioavailability of the febuxostat tablet has not been studied.Following multiple 80 mg once daily doses with a high fat meal, there was a 49% decrease in C max and an 18% decrease in AUC, respectively. However, no clinically significant change in the percent decrease in serum uric acid concentration was observed (58% fed vs. 51% fasting). Thus, ULORIC may be taken without regard to food.Concomitant ingestion of an antacid containing magnesium hydroxide and aluminum hydroxide with an 80 mg single dose of ULORIC has been shown to delay absorption of febuxostat (approximately1 hour) and to cause a 31% decrease in C max and a 15% decrease in AUC∞. As AUC rather thanC max was related to drug effect, change observed in AUC was not considered clinically significant. Therefore, ULORIC may be taken without regard to antacid use.Distribution: The mean apparent steady state volume of distribution (V ss/F) of febuxostat was approximately 50 L (CV ~40%). The plasma protein binding of febuxostat is approximately 99.2%, (primarily to albumin), and is constant over the concentration range achieved with 40 mg and 80 mg doses.Metabolism: Febuxostat is extensively metabolized by both conjugation via uridine diphosphate glucuronosyltransferase (UGT) enzymes including UGT1A1, UGT1A3, UGT1A9, and UGT2B7 and oxidation via cytochrome P450 (CYP) enzymes including CYP1A2, 2C8 and 2C9 and non-P450 enzymes. The relative contribution of each enzyme isoform in the metabolism of febuxostat is not clear. The oxidation of the isobutyl side chain leads to the formation of four pharmacologically active hydroxy metabolites, all of which occur in plasma of humans at a much lower extent than febuxostat.In urine and feces, acyl glucuronide metabolites of febuxostat (~35% of the dose), and oxidative metabolites, 67M-1 (~10% of the dose), 67M-2 (~11% of the dose), and 67M-4, a secondary metabolite from 67M-1, (~14% of the dose) appeared to be the major metabolites of febuxostat in vivo.Elimination: Febuxostat is eliminated by both hepatic and renal pathways. Following an 80 mg oral dose of 14C-labeled febuxostat, approximately 49% of the dose was recovered in the urine as unchanged febuxostat (3%), the acyl glucuronide of the drug (30%), its known oxidative metabolites and their conjugates (13%), and other unknown metabolites (3%). In addition to the urinary excretion, approximately 45% of the dose was recovered in the feces as the unchanged febuxostat (12%), the acyl glucuronide of the drug (1%), its known oxidative metabolites and their conjugates (25%), and other unknown metabolites (7%).The apparent mean terminal elimination half-life (t1/2) of febuxostat was approximately 5 to 8 hours. Special PopulationsPediatric Use: The pharmacokinetics of ULORIC in patients under the age of 18 years have not been studied.Geriatric Use: The C max and AUC of febuxostat and its metabolites following multiple oral doses of ULORIC in geriatric subjects (≥ 65 years) were similar to those in younger subjects (18-40 years). In addition, the percent decrease in serum uric acid concentration was similar between elderly and younger subjects. No dose adjustment is necessary in geriatric patients [see Use in Specific Populations (8.5)].Renal Impairment: Following multiple 80 mg doses of ULORIC in healthy subjects with mild (Cl cr50-80 mL per min), moderate (Cl cr 30-49 mL per min) or severe renal impairment (Cl cr 10-29 mL per min), the C max of febuxostat did not change relative to subjects with normal renal function (Cl cr greater than 80 mL per min). AUC and half-life of febuxostat increased in subjects with renal impairment in comparison to subjects with normal renal function, but values were similar among three renal impairment groups. Mean febuxostat AUC values were up to 1.8 times higher in subjects with renal impairment compared to those with normal renal function. Mean C max and AUC values for 3 active metabolites increased up to 2- and 4-fold, respectively. However, the percent decrease in serum uric acid concentration for subjects with renal impairment was comparable to those with normal renal function (58% in normal renal function group and 55% in the severe renal function group).No dose adjustment is necessary in patients with mild to moderate renal impairment [see Dosage and Administration (2) and Use in Specific Populations (8.6)]. The recommended starting dose of ULORIC is 40 mg once daily. For patients who do not achieve a sUA less than 6 mg per dL after 2 weeks with 40 mg, ULORIC 80 mg is recommended. There is insufficient data in patients with severe renal impairment; caution should be exercised in those patients [see Use in Specific Populations (8.6)].ULORIC has not been studied in end stage renal impairment patients who are on dialysis.Hepatic Impairment: Following multiple 80 mg doses of ULORIC in patients with mild (Child-Pugh Class A) or moderate (Child-Pugh Class B) hepatic impairment, an average of 20-30% increase was observed for both C max and AUC24 (total and unbound) in hepatic impairment groups compared to subjects with normal hepatic function. In addition, the percent decrease in serum uric acid concentration was comparable between different hepatic groups (62% in healthy group, 49% in mild hepatic impairment group, and 48% in moderate hepatic impairment group). No dose adjustment is necessary in patients with mild or moderate hepatic impairment. No studies have been conducted in subjects with severe hepatic impairment (Child-Pugh Class C); caution should be exercised in those patients [see Use in Specific Populations (8.7)].。
非布司他说明书

非布司他说明书非布司他主要成份为非布佐司他,其化学名为2-[(3-氰基-4-异丁氧基)苯基]-4-甲基-5-噻唑羧酸。
为黄嘌呤氧化酶(XO)抑制剂,适用于具有痛风症状的高尿酸血症的长期治疗。
基本信息中文名称:非布司他别名:非布佐司他、非布索坦(通用名称)汉语拼音FeibuzuositaPian 外文名称:Febuxostat Tablets药品名称:非布司他成份本品主要成份为非布佐司他,其化学名为2-[(3-氰基-4-异丁氧基)苯基]-4-甲基-5-噻唑羧酸。
结构式:分子式:C16H16N2O3S分子量:316.37性状本品为白色粉末适应症适用于具有痛风症状的高尿酸血症的长期治疗。
不推荐本品用于治疗无症状性高尿酸血(症)。
剂型胶囊剂、片剂规格40mg/粒或片;80mg/粒或片用法用量推荐剂量用于治疗有痛风症状的高尿酸血症患者时,推荐本品剂量为40mg或80mg,每日一次。
推荐本品的起始剂量为40mg,给药本品时无需考虑食物或抗酸剂的影响。
特殊人群轻或中度肾功能损伤患者服用本品时不必调整剂量。
推荐本品的起始剂量为40mg,每日一次。
给药剂量40mg,持续两周后,对血清尿酸水平(sUA)仍高于6 mg/dl的患者,推荐给药剂量80mg。
轻中度肝功能损伤患者服用本品无需剂量调整。
对严重肝功能损伤患者使用本品尚无研究,因此给药本品应谨慎。
尿酸水平使用本品治疗2周后即可进行血清尿酸的再检验。
治疗目标是降低和维持血清尿酸水平使其低于6 mg/dl。
预防痛风急性发作推荐至少用药6个月(见【注意事项】)。
痛风发作变化的血清尿酸水平会导致沉积的尿酸盐活动,因此开始给药本品后会导致痛风发作。
推荐使用本品时,同时给药非甾体抗炎药(NSAID)或秋水仙碱,以预防痛风发作。
预防痛风急性发作推荐至少用药6个月。
如果在给药治疗期间发生痛风,不需要停药。
对个别患者的痛风应相应给予治疗。
续发性高尿酸血症对继发性高尿酸血症患者(包括器官移植患者)给药本品尚无研究;尿酸盐生成率增加的患者不推荐使用本品(例如恶性病及其治疗、莱-萘二氏综合征)。
氟苯丙胺说明书

氟苯丙胺说明书
【别名】安德力减肥丸;芬美啉片;盐酸氟苯丙胺 ,盐酸芬氟拉明
【外文名】Fenfluramine
【适应症】除用于单纯性肥胖症外,可用于患有高血压、糖尿病、冠心病及焦虑的肥胖病人服药后病人均能适应饮食控制,有明显减少脂肪积聚的效果,般服药12周后平均腹围可减少10cm,平均体重减轻6kg。
【用量用法】口服:第1周每日40mg,早、晚餐0.5~1小时服用;第2、3周每日60mg,早、中、晚餐前0.5~1小时服;以后根据疗效与耐受程度可继续维持原量,或逐渐增至每日80~100mg(每日100mg限用于较重的肥胖者),8~12周1疗程。
【注意事项】
1.治疗期间不要间歇性服药,疗程最后4或6周内逐渐减量而至停药,不宜突然停服。
连续服药时间不应超过6月,否则易发生耐药性及依赖心理。
2.不良反应主要有非腹泻性便次增、轻度腹痛、头晕、乏力、口干等,但一般均能耐受,持续用药可逐渐消除。
尚可有嗜睡,抑郁及夜尿增多。
不能耐受者应减量。
3.精神抑郁症、癫痫病人及孕妇忌用。
严重心律失常、高空作业者及驾驶员慎用。
4.对肥胖伴有高血压和糖尿病病人,如合并使用降压药或降糖药,可产生协同作用。
【规格】片剂:每片20mg。
索菲布韦详细说明书2013中文版

SOVALDITM (sofosbuvir)片,为口服使用美国初次批准:2013适应证和用途SOVALDI是一种丙型肝炎病毒(HCV)核苷酸类似物NS5B聚合酶抑制剂适用为慢性丙型肝炎(CHC)感染的治疗作为组合抗病毒治疗方案的一个组分。
(1)(1)SOVALDI疗效已在有HCV基因型1,2,3或4感染受试者中被确定,包括有肝细胞癌符合米兰[Milan]标准(等待肝移植)和有HCV/HIV-1共-感染受试者。
(1)剂量和给药方法(1)一片400 mg片每天1次有或无食物服用。
(2.1)(2)应与利巴韦林[ribavirin]联用或与聚乙二醇化干扰素[pegylated干扰素]和利巴韦林联用为CHC的治疗。
建议联合治疗:(2.1)(3)SOVALDI与利巴韦林联用共24周干扰素不合格可被考虑为被基因型1感染CHC 患者。
(2.1)(4)在有肝细胞癌等待肝移植直至48周或直至肝移植患者应被与联用利巴韦林为CHC 的治疗,以先发生为准。
(2.1)(5)对有严重肾受损或肾病终末期患者不能建议剂量。
(2.4,8.6)剂型和规格片:400 mg。
(3)禁忌证(1)当与聚乙二醇干扰素α/利巴韦林或单独利巴韦林联用时,对聚乙二醇干扰素α和/或利巴韦林的所有禁忌证也都应用于SOVALDI联合治疗。
(4)(2)因为利巴韦林可能引起出生缺陷和胎儿死亡,在妊娠妇女和男性其女性伴侣妊娠时禁忌SOVALDI与聚乙二醇干扰素α/利巴韦林或利巴韦林联用。
(4)警告和注意事项妊娠:利巴韦林可能致出生缺陷和胎儿死亡和动物研究已证明干扰素有流产效应;女性患者和男性患者的女性伴侣避免妊娠。
治疗开始前患者必须有一个阴性妊娠测试,使用至少2种有效非激素避孕方法和每月妊娠测试。
(5.1)不良反应SOVALDI与利巴韦林联用观察到最常见不良事件(发生率大于或等于20%,所有级别)是疲乏和头痛。
SOVALDI与聚乙二醇干扰素α和利巴韦林联用观察到最常见不良事件是疲乏,头痛,恶心,失眠和贫血。
非布司他片(菲布力)中文说明书

非布司他片(菲布力)中文说明书非布司他片菲布力 FebuxostatTab1.ets FeibusitaPian【成份】本品主要成份为非布司他。
辅料包括:乳糖水合物、部分预胶化淀粉、羟丙基纤维素、交联羟甲纤维素钠、硬脂酸镁、羟丙甲纤维素、聚乙二醇6000o 化学名称:2-[3-制基-4-(2-甲基丙氯基)苯基]-4-甲基噬哗-5-较酸 化学结构式:分子式:Ci 6Hi 6N 2O 3S 分子量:316.37【性状】本品为薄膜衣片,除去包衣后显白色。
【适应症】适用于痛风患者高尿酸血症的长期治疗。
不推荐用于无临床症状的高尿酸血症。
【规格】20mg>40mg o【用法用量】在降尿酸药物治疗初期可能导致血尿酸值急速降低诱发痛风性关节炎(痛风发作),故推荐本品初始剂量为20mg 每日1次,且可在给药开始4周后根据血尿酸值逐渐增加用量,每次增量20mg 。
每日最大剂量为80mg 。
血尿酸值达标(<6mg∕d 1.或<360UmO1./1.)后,维持最低有效剂量。
给药时,无需考虑食物和抗酸剂的影响。
特殊人群肝损害患者:轻、中度肝损害(ChiId-PughA.B 级)的患者无需调整剂量。
尚未进行重度肝损害患者(Chi1.d-PUghC 级)使用非布司他的疗效及安全性研究,因此此类患者应慎用非布司他。
肾损害患者:轻、中度肾损害(CICr30-89m1.∕min )的患者无需调整剂量。
尚无严重肾损害(CIer<30m1.∕min )患者的充足研究数据,因此此类患者应慎用非布司他。
痛风发作由于本品为降尿酸药物,在痛风性关节炎(痛风发作)发作时使用本品可使血尿酸值降低,加重痛风性关节炎(痛风发作),故在使用本品前有痛风性关节炎的患者,在症状稳定前,不可使用本品。
另外,在使用本品过程中发现有痛风性关节炎(痛风发作)时,可不改变本品用量继续用药,亦可根据具体症状合用秋水仙碱、非类固醇抗炎药、肾上腺皮质激素等药物。
Fluo-4, AM 产品说明书

Fluo-4, AMStorage upon receipt: -20°C, protected from lightIntroductionFluo-4, AM is an analog of Fluo-3, AM with the two chlorine substituents replaced by fluorines, which results in increased fluorescence excitation at 488 nm and consequently higher fluorescence signal levels. Fluo-4, AM is a membrane permeant probe and can be passively loaded in cells by simple incubation. Once inside the cell, Fluo-4 becomes fluorescent in the presence of free Ca2+. Fluo-4, AM is widely used for determining GPCR function via cell-based high-throughput screening.This indicator is useful for fluorescence and confocal microscopy, flow cytometry, and microplate screening applications.Fluo-4, AM is supplied as 0.5 mg solid (C219), special packing 10×50 µg (C220), and 1 mM solution in DMSO (C221).SpecificationsApplicationsCell Loading GuidelineNote: The following protocol is provided as an introductory guide only. The detailed procedures can be found from literatures.1,21. Prepare a Fluo-4 AM stock solution in anhydrous DMSO at 1-5 mM.2. Dilute an aliquot of Fluo-4 AM stock solution (1-5 mM) to a final concentration of 1-5 µM in the bufferedphysiological medium of choice. Addition of the non-ionic detergent Pluronic R F-127 can assist inPage 1dispersion of the nonpolar Fluo-4 AM ester in aqueous media. This can be conveniently accomplished by mixing the aliquot of Fluo-4 AM ester stock solution in DMSO with an equal volume of 20% (w/v) Pluronic in DMSO (Cat No. C021) before dilution into the loading medium, making the final Pluronic concentration about 0.02%.3. The organic anion-transport inhibitors probenecid (1-2.5 mM) may be added to the cell medium toreduce leakage of the de-esterified indicator.4. Cells are normally incubated with the Fluo-4 AM ester for 15–60minutes at 20–37°C. Exact loadingconcentration, time, and temperature will need to be determined empirically; in general it is desirable to use the minimum dye concentration required to yield fluorescence signals with adequate signal to noise.Subcellular compartmentalization, an inherent problem with the AM ester loading technique, is usually lessened by lowering the incubation temperature.5. Before fluorescence measurements are commenced, cells should be washed in indicator-free medium(containing an anion transport inhibitor, if applicable) to remove any dye that is nonspecificallyassociated with the cell surface, and then incubated for a further 30 minutes to allow completede-esterification of intracellular Fluo-4 AM ester.High-Throughput ScreeningIntracellular Ca2+ measurements in 96-well and 384-well microplates are an essential tool for high- throughput pharmacological screening. Cell samples in microplate wells are loaded with the AM ester form of the indicator using protocol basically similar to those described in Cell Loading Guideline. References:1. Methods Cell Biol 40, 155 (1994);2. Cell Biology: A Laboratory Handbook, 2nd Edition, J.E. Celis, Ed., Volume 3, pp 363–374,Academic Press (1998);Page 2。
比索洛尔说明书

比索洛尔说明书【药品名称】通用名称:比索洛尔商品名称:_____英文名称:Bisoprolol【成份】本品主要成份为比索洛尔。
【性状】本品为_____(具体剂型和外观描述)。
【适应症】用于高血压、冠心病(心绞痛)。
伴有心室收缩功能减退(射血分数≤35%,根据超声心动图确定)的中度至重度慢性稳定性心力衰竭。
在使用本品前,需要接受 ACE 抑制剂、利尿剂和选择性使用强心甙类药物治疗。
【规格】_____(具体规格)【用法用量】对于高血压和心绞痛患者:通常每日一次,每次5mg。
轻度高血压患者可以从25mg 开始治疗。
如果效果不明显,剂量可增至每日一次,每次 10mg。
对于慢性稳定性心力衰竭患者:本品必须在使用 ACE 抑制剂、利尿剂和选择性使用强心甙类药物治疗的基础上使用。
推荐的起始剂量为125mg,每日一次。
如能耐受,可在 1-2 周后增加至 25mg,每日一次;再过 1-2 周后增加至 375mg,每日一次;再过 1-2 周后增加至 5mg,每日一次;再过 1-2 周后增加至75mg,每日一次;再过 1-2 周后增加至 10mg,每日一次。
剂量应根据患者的个体情况进行调整,最大推荐剂量为 10mg,每日一次。
本品应在早晨并可以在进餐时服用。
用水整片送服,不应咀嚼。
【不良反应】1、神经系统异常:头晕、头痛等。
2、心血管系统异常:心动过缓、低血压等。
3、胃肠道异常:恶心、呕吐、腹泻、便秘等。
4、呼吸系统异常:支气管痉挛、呼吸困难等。
5、皮肤和皮下组织异常:瘙痒、皮疹等。
需要注意的是,不良反应的发生频率和严重程度可能因个体差异而有所不同。
如果出现严重的不良反应,应立即停药并就医。
【禁忌】1、急性心力衰竭或处于心力衰竭失代偿期需用静注正性肌力药物治疗的患者。
2、心源性休克者。
3、二度或三度房室传导阻滞者(无心脏起搏器)。
4、病窦综合征患者。
5、窦房阻滞者。
6、心动过缓者,治疗开始时心率少于 60 次/分钟。
卡菲全能版说明书

卡菲全能版说明书2019-03-20目录第 1 章: 产品介绍 (1)包装清单 (1)卡菲概览 (1)第 2 章: 开始使用 (3)充电 (3)快速安装 (3)如何更换电池? (5)固件升级 (5)第 3 章: iOS APP使用 (6)遥控拍摄 (7)浏览照片模式 (13)设置 (15)第 4 章: 使用索尼相机联机拍摄 (17)连接 (17)PC远程访问 (18)手动对焦,自动对焦和景深合成(适用于索尼a7 III 和 a7r III) (19)MTP 模式 (19)第 5 章: 第三方联机拍摄服务 (20)宾得相机和Lightroom无线联机拍摄 (21)第 6 章: 注意事项与常见问题解答 (24)注意事项 (24)常见问题解答 (25)第 1 章: 产品介绍包装清单卡菲概览1. 卡菲专业版2. 3200 毫安时锂电池3. 相机连接线4. USB 充电线5. 快速入门指南6.电池盖翘片USB 接口电源开关充电接口重置按键5G Wi-FiUSB 灯电源灯电池灯热靴插座电池盖电源灯亮起表示系统开始运行电池灯红色表示电量低;橙色表示电池正在充电;绿色表示电量已充满5G Wi-Fi 灯亮起表示5G Wi-Fi启动正常USB 灯亮起或闪烁表示USB正在连接充电使用 Micro USB 线连接手机充电器或者连接电脑USB 口来给CamFi Pro设备充电。
连接CamFi Pro连接电源第 2 章: 开始使用快速安装1. 连接CamFi Pro 至单反相机使用Mini USB 线连接CamFi Pro 至单反相机,并分别打开CamFi Pro和单反相机的电源开关。
获取CamFi 软件你需要在你的智能手机或电脑上安装CamFi软件。
CamFi软件支持iPhone, Android,Windows和Mac系统。
请访问:/cn/download.html。
获得支持的设备完整列表。
1. 在你的智能手机或电脑浏览器上访问:/cn/download.html2. 按照说明在你的智能手机或电脑上安装CamFi软件。
Feiba(抗抵抗血小板抗凝因子复合物)(阴性)说明书

Hemophilia Products – Anti-Inhibitor Coagulant Complex: Feiba(Intravenous)Document Number: IC-0337 Last Review Date: 06/01/2023Date of Origin: 12/16/2014Dates Reviewed: 12/2014, 4/2015, 5/2015, 09/2015, 12/2015, 03/2016, 06/2016, 12/2016, 06/2017, 09/2017, 11/2017, 11/2018, 03/2019, 02/2020, 06/2021, 06/2022, 06/2023I.Length of AuthorizationCoverage is provided for 3 months and may be renewed thereafter, unless otherwise specified* Note: The cumulative amount of medication the patient has on-hand will be taken into account for authorizations.*Initial and renewal authorization periods may vary by specific covered indicationII.Dosing LimitsA.Quantity Limit (max daily dose) [NDC unit]:−Feiba 500 IU (Orange) single-dose vial: 293 vials per 30-day supply−Feiba 1000 IU (Green) single-dose vial: 147 vials per 30-day supply−Feiba 2500 IU (Purple) single-dose vial: 59 vials per 30-day supplyB.Max Units (per dose and over time) [HCPCS Unit]:−146,625 billable units per 30 day supplyIII.Initial Approval Criteria 1-3,7-10Coverage is provided in the following conditions:Hemophilia A (congenital factor VIII deficiency) † Ф•Diagnosis of congenital factor VIII deficiency has been confirmed by blood coagulation testing; AND•Confirmation the patient has inhibitors to Factor VIII; AND•Used as treatment in at least one of the following:o Control and prevention of acute bleeding episodes (episodic treatment of acute hemorrhage); OR(*Authorizations valid for 1 month); ORo Routine prophylaxis to prevent or reduce the frequency of bleeding episodes; AND ▪Patient has at least two documented episodes of spontaneous bleeding intojoints; ORo Patient has a documented trial and failure of Immune Tolerance Induction (ITI);AND▪Patient has a documented trial and failure or contraindication toemicizumab-kxwh therapy.Hemophilia B (congenital factor IX deficiency aka Christmas disease) † Ф•Diagnosis of congenital factor IX deficiency has been confirmed by blood coagulation testing; AND•Confirmation the patient has inhibitors to Factor IX; AND•Used as treatment in at least one of the following:o Control and prevention of acute bleeding episodes (episodic treatment of acute hemorrhage); ORo Perioperative management(*Authorizations valid for 1 month); ORo Routine prophylaxis to prevent or reduce the frequency of bleeding episodes; AND ▪Patient has at least two documented episodes of spontaneous bleeding intojoints; ORo Patient has documented trial and failure of Immune Tolerance Induction (ITI) † FDA Approved Indication(s); ‡ Compendia Recommended Indication(s); Ф Orphan Drug IV.Dispensing Requirements for Rendering Providers (Hemophilia Management Program)−Prescriptions cannot be filled without an expressed need from the patient, caregiver or prescribing practitioner. Auto-filling is not allowed.−Monthly, rendering provider must submit for authorization of dispensing quantity before delivering factor product. Information submitted must include:▪Original prescription information, requested amount to be dispensed, vial sizes available to be ordered from the manufacturer, and patient clinical history (includingpatient product inventory and bleed history)▪Factor dose should not exceed +1% of the prescribed dose and a maximum of three vials may be dispensed per dose. If unable to provide factor dosing within therequired threshold, below the required threshold, the lowest possible dose able to beachieved above +1% should be dispensed.Prescribed dose should not be increased tomeet assay management requirements.−The cumulative amount of medication(s) the patient has on-hand should be taken into account when dispensing factor product. Patients should not have more than 5 extra doseson-hand for the treatment of acute bleeding episodes.−Dispensing requirements for renderings providers are a part of the hemophilia management program. This information is not meant to replace clinical decision makingwhen initiating or modifying medication therapy and should only be used as a guide.V.Renewal Criteria 1-3,7Coverage can be renewed based upon the following criteria:•Patient continues to meet indication-specific relevant criteria identified in section III; AND •Absence of unacceptable toxicity from the drug. Examples of unacceptable toxicity include: symptoms of allergic-anaphylactic reactions (anaphylaxis, dyspnea, rash, etc.),thromboembolic events (venous thrombosis, pulmonary embolism, myocardial infarction,stroke, etc.), development of neutralizing antibodies (inhibitors), etc.; AND •Any increases in dose must be supported by an acceptable clinical rationale (i.e. weight gain, half-life study results, increase in breakthrough bleeding when patient is fully adherent totherapy, etc.); AND•The cumulative amount of medication(s) the patient has on-hand will be taken into account when authorizing. The authorization will allow up to 5 doses on-hand for the treatment ofacute bleeding episodes as needed for the duration of the authorization; AND Control and prevention of acute bleeding episodes•Renewals will be approved for a 6 month authorization periodPerioperative management of surgical bleeding•Coverage may NOT be renewedRoutine prophylaxis to prevent or reduce the frequency of bleeding episodes•Renewals will be approved for a 12 month authorization period; AND•Patient has demonstrated a beneficial response to therapy (i.e., the frequency of bleeding episodes has decreased from pre-treatment baseline)Dosage/Administration1-3Control and prevention of bleeding Congenital Hemophilia A / Hemophilia B with inhibitors Joint hemorrhageAdminister 50—100 units/kg IV every 12 hours until pain and acute disabilities are improvedMucous Membrane BleedingAdminister 50—100 units/kg IV every 6 hours for at least 1 day or until bleeding is resolvedSoft tissue hemorrhageAdminister 100 units/kg IV every 12 hours until resolution of bleedOther severe hemorrhageAdminister 100 units/kg IV every 6—12 hours until resolution of bleed Routine ProphylaxisCongenitalHemophilia A/Hemophilia B withinhibitorsAdminister 85 units/kg IV every other dayPerioperative management Congenital Hemophilia A / Hemophilia B with inhibitors PreoperativeAdminister 50—100 units/kg IV administered as a 1 time dose immediately prior to surgeryPostoperativeAdminister 50 – 100 units/kg IV administered every 6 – 12 hours postoperatively until resolution of bleed and healing is achievedVI.Billing Code/Availability Information HCPCS Code & NDC:Feiba Baxalta US Inc J7198 1 IU 500 units 64193-0426-xx 1000 units 64193-0424- xx 2500 units 64193-0425- xxVII.References1.Feiba [package insert]. Lexington, MA; Baxalta US Inc. March 2023. Accessed May 2023.2.MASAC Recommendations Concerning Products Licensed for the Treatment of Hemophiliaand Selected Disorders of the Coagulation System. Revised May 2023 National HemophiliaFoundation. MASAC Document #276; May 2023. Available at: .Accessed May 2023.3.Guidelines for the Management of Hemophilia. 3rd Edition. World Federation ofHemophilia. 2020. Available at: https:///publications/files/pdf-1863.pdfAccessed May 2023.4.Graham A1, Jaworski K. Pharmacokinetic analysis of anti-hemophilic factor in the obesepatient. Haemophilia. 2014 Mar;20(2):226-9.5.Croteau SE1, Neufeld EJ. Transition considerations for extended half-life factor products.Haemophilia. 2015 May;21(3):285-8.6.Mingot-Castellano, et al. Application of Pharmacokinetics Programs in Optimization ofHaemostatic Treatment in Severe Hemophilia a Patients: Changes in Consumption,Clinical Outcomes and Quality of Life. Blood. 2014 December; 124 (21).7.MASAC Recommendation Concerning Prophylaxis for Hemophilia A and B with andwithout Inhibitors. 2022 National Hemophilia Foundation. MASAC Document #267; April 2022. Available at: . Accessed May 2023.8.Sjamsoedin LJ, Heijnen L, Mauser-Bunschoten EP, et al. The effect of activatedprothrombin-complex concentrate (FEIBA) on joint and muscle bleeding in patients withhemophilia A and antibodies to factor VIII. A double-blind clinical trial. N Engl J Med. 1981 Sep 24;305(13):717-21.9.Hilgartner M, Knatterud G, and the FEIBA Study Group. The Use of Factor Eight InhibitorBy-Passing Activity (FEIBA Immuno) Product for Treatment of Bleeding Episodes inHemophiliacs With Inhibitors. Blood, Vol 6. No. 1 (January). 1983.10.Stasyshyn S, Antunes S, Mamonov V, et al. Prophylaxis with anti‐inhibitor coagulantcomplex improves health‐related quality of life in haemophilia patients with inhibitors:results from FEIBA NF Prophylaxis Study. Haemophilia, 03 March 2014.https:///10.1111.hae.12390.11.First Coast Service Options, Inc. Local Coverage Article: Billing and Coding: HemophiliaClotting Factors (A56482). Centers for Medicare & Medicaid Services Inc. Updated on10/28/2022 with effective date 10/01/2022. Accessed May 2023.12.Palmetto GBA. Local Coverage Article: Billing and Coding: Guidance for Anti-InhibitorCoagulant Complex (AICC) National Coverage Determination (NCD) 110.3 (A56065).Centers for Medicare & Medicaid Services Inc. Updated on 11/14/2022 with effective date11/24/2022. Accessed May 2023.13.Novitas Solutions, Inc. Local Coverage Article: Billing and Coding: Hemophilia FactorProducts (A56433). Centers for Medicare & Medicaid Services Inc. Updated on 10/14/2022 with effective date 10/01/2022. Accessed May 2023.Appendix 1 – Covered Diagnosis CodesD66Hereditary factor VIII deficiencyD67Hereditary factor IX deficiencyAppendix 2 – Centers for Medicare and Medicaid Services (CMS)Medicare coverage for outpatient (Part B) drugs is outlined in the Medicare Benefit Policy Manual (Pub. 100-2), Chapter 15, §50 Drugs and Biologicals. In addition, National Coverage Determination (NCD), Local Coverage Determinations (LCDs) and Local Coverage Articles (LCAs) may exist and compliance with these policies is required where applicable. They can be found at: https:///medicare-coverage-database/search.aspx. A dditional indications may be covered at the discretion of the health plan.Medicare Part B Covered Diagnosis Codes (applicable to existing NCD/LCD/LCA):Jurisdiction(s): J,M NCD/LCD Document (s): A56065https:///medicare-coverage-database/new-search/search-results.aspx?keyword=a56065&areaId=all&docType=NCA%2CCAL%2CNCD%2CMEDCAC%2CTA%2CMCD%2 C6%2C3%2C5%2C1%2CF%2CPJurisdiction(s): H,L NCD/LCD Document (s): A56433https:///medicare-coverage-database/new-search/search-results.aspx?keyword=a56433&areaId=all&docType=NCA%2CCAL%2CNCD%2CMEDCAC%2CTA%2CMCD%2 C6%2C3%2C5%2C1%2CF%2CPJurisdiction Applicable State/US Territory ContractorE (1) CA, HI, NV, AS, GU, CNMI Noridian Healthcare Solutions, LLCF (2 & 3) AK, WA, OR, ID, ND, SD, MT, WY, UT, AZ Noridian Healthcare Solutions, LLC5 KS, NE, IA, MO Wisconsin Physicians Service Insurance Corp (WPS)6 MN, WI, IL National Government Services, Inc. (NGS)H (4 & 7) LA, AR, MS, TX, OK, CO, NM Novitas Solutions, Inc.8 MI, IN Wisconsin Physicians Service Insurance Corp (WPS) N (9) FL, PR, VI First Coast Service Options, Inc.J (10) TN, GA, AL Palmetto GBA, LLCM (11) NC, SC, WV, VA (excluding below) Palmetto GBA, LLCL (12) DE, MD, PA, NJ, DC (includes Arlington &Novitas Solutions, Inc.Fairfax counties and the city of Alexandria in VA)K (13 & 14) NY, CT, MA, RI, VT, ME, NH National Government Services, Inc. (NGS)15 KY, OH CGS Administrators, LLC。
菲布力说明书

1.成份:本品主要成份为非布佐司他,其化学名为2-[(3-氰基-4-异丁氧基)苯基]-4-甲基-5-噻唑羧酸。
结构式:分子式:C16H16N2O3S分子量:316.372.性状:本品为白色粉末3.适应症:本品为黄嘌呤氧化酶(XO)抑制剂,适用于具有痛风症状的高尿酸血症的长期治疗。
不推荐本品用于治疗无症状性高尿酸血(症)。
4.剂型:胶囊剂、片剂5.规格:40mg/粒或片;80mg/粒或片6.用法用量:推荐剂量用于治疗有痛风症状的高尿酸血症患者时,推荐本品剂量为40mg或80mg,每日一次。
推荐本品的起始剂量为40mg,给药本品时无需考虑食物或抗酸剂的影响。
特殊人群轻或中度肾功能损伤患者服用本品时不必调整剂量。
推荐本品的起始剂量为40mg,每日一次。
给药剂量40mg,持续两周后,对血清尿酸水平(sUA)仍高于6 mg/dl的患者,推荐给药剂量80mg。
轻中度肝功能损伤患者服用本品无需剂量调整。
对严重肝功能损伤患者使用本品尚无研究,因此给药本品应谨慎。
尿酸水平 使用本品治疗2周后即可进行血清尿酸的再检验。
治疗目标是降低和维持血清尿酸水平使其低于6 mg/dl 。
预防痛风急性发作推荐至少用药6个月(见【注意事项】)。
痛风发作 变化的血清尿酸水平会导致沉积的尿酸盐活动,因此开始给药本品后会导致痛风发作。
推荐使用本品时,同时给药非甾体抗炎药(NSAID )或秋水仙碱,以预防痛风发作。
预防痛风急性发作推荐至少用药6个月。
如果在给药治疗期间发生痛风,不需要停药。
对个别患者的痛风应相应给予治疗。
续发性高尿酸血症对继发性高尿酸血症患者(包括器官移植患者)给药本品尚无研究;尿酸盐生成率增加的患者不推荐使用本品(例如恶性病及其治疗、莱-萘二氏综合征)。
稀有病例的尿中黄嘌呤的浓度会显着增加,在尿路中沉积。
7.不良反应:临床试验中,共对2757例患高尿酸血症和痛风患者给药本品,每天40mg 或80mg 。
对559例患者给药本品40mg ,持续≥ 6个月。
抗痛风新药菲布力
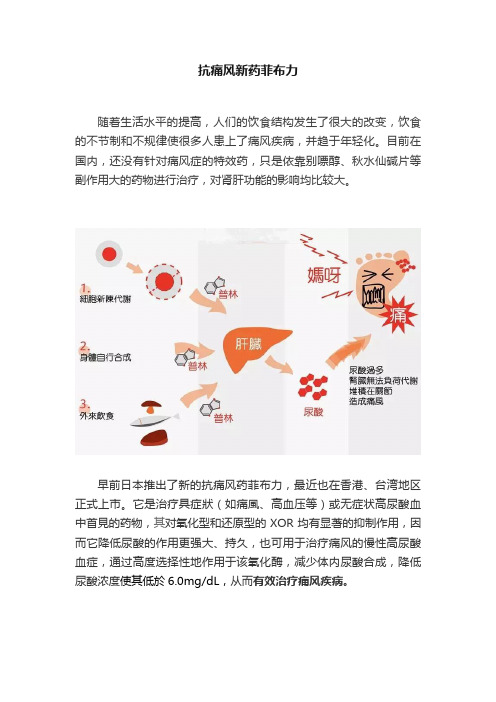
抗痛风新药菲布力随着生活水平的提高,人们的饮食结构发生了很大的改变,饮食的不节制和不规律使很多人患上了痛风疾病,并趋于年轻化。
目前在国内,还没有针对痛风症的特效药,只是依靠别嘌醇、秋水仙碱片等副作用大的药物进行治疗,对肾肝功能的影响均比较大。
早前日本推出了新的抗痛风药菲布力,最近也在香港、台湾地区正式上市。
它是治疗具症狀(如痛風、高血压等)或无症状高尿酸血中首見的药物,其对氧化型和还原型的XOR均有显著的抑制作用,因而它降低尿酸的作用更强大、持久,也可用于治疗痛风的慢性高尿酸血症,通过高度选择性地作用于该氧化酶,减少体内尿酸合成,降低尿酸浓度使其低於6.0mg/dL,从而有效治疗痛风疾病。
与别嘌醇相比,菲布力具有明显的优势:(1)别嘌醇只对还原型的XOR有抑制作用,而菲布力对氧化型和还原型的XOR均有显著的抑制作用,因而其降低尿酸的作用更强大、持久;(2)由于别嘌醇为嘌呤类似物,不可避免的造成涉及嘌呤及吡啶代谢其他酶活性的影响。
因此别嘌醇治疗中,需要重复大剂量给药来维持较高的药物水平。
由此也带来由于药物蓄积所致的严重甚至致命的不良反应。
而菲布力为非嘌呤类XOR抑制剂,所以具有更好的安全性。
一项多中心、双盲、随机Ⅱ期临床研究评价了菲布力的安全性和对痛风的疗效。
总共有136名男性和17名女性痛风病人随机接受安慰剂或本品(40、80或120mg/d),4周后检测发现,本品各剂量组病人血清尿酸浓度较治疗前均显著降低,按剂量由低至高各组分别平均降低37%、44%和59%,而安慰剂组病人仅降低了2%;绝大多数病人坚持完成了试验,本品和安慰剂组不良反应发生率相近,并且这些不良反应大多轻微,具有自限性,常见的有腹泻、疼痛、背痛、头痛和关节痛。
维安医生提醒:服用治疗痛风的新药,必须在医生的指导下进行,根据体质和病情状况给予合理的处方和使用方法,定期复诊,合理调整剂量,避免造成不必要的伤害,以到达最佳的治疗效果。
蛋白琥珀酸铁口服溶液(菲普利)的说明书

蛋白琥珀酸铁口服溶液(菲普利)的说明书合理饮食,正常的作息时间,这是保持健康身体的首要条件。
如今的生活质量提高了,许多人在饮食方面喜欢偏食,这样是不利于身体健康的。
因此,我们需要用药物来不充体内的微量元素。
今天我们就为您介绍一种叫做蛋白琥珀酸铁口服溶液(菲普利)的药物,它可以有效不充各种人体所需的微量元素。
【药品名称】通用名称:蛋白琥珀酸铁口服溶液商品名称:蛋白琥珀酸铁口服溶液(菲普利)英文名称:Iron Proteinsuccinylate Oral Solution拼音全码:DanBaiHuPoSuanTieKouFuRongYe(FeiPuLi)【主要成份】本品主要成份为蛋白琥珀酸铁800毫克(相当于三价铁40毫克)。
【性状】本品为深红色液体;气芳香,味甜。
【适应症/功能主治】绝对和相对缺铁性贫血的治疗,由于铁摄入量不足或吸收障碍、急性或慢性失血以及各种年龄患者的感染所引起的隐性或显性缺铁性贫血的治疗,妊娠与哺乳期贫血的治疗。
【规格型号】15ml*10瓶【用法用量】本品均由口服。
成人:每天1-2瓶(相当于三价铁40-80毫克),遵医嘱分两次在饭前口服。
儿童:每天按体重1.5毫升/公斤(相当于每天三价铁4毫克/公斤体重),应遵医嘱分两次于饭前口服。
【不良反应】偶有发生。
尤其用药过量时易发生胃肠功能紊乱(如腹泻、结肠痉挛、恶心、呕吐、上腹部疼痛),在减量或停药后可消失。
【禁忌】对本药品过敏者以及含铁血黄素沉着、血色素沉着、再生障碍性贫血、溶血性贫血、铁利用障碍性贫血、慢性胰腺炎合肝硬化患者禁用。
【注意事项】在开始治疗前,应先找出产生贫血的原因。
本品尤其适用于妊娠与哺乳期妇女贫血的治疗。
本品不会影响病人的反应(驾驶及机器的操作)。
本品不会引起成瘾性,除了持续性出血、月经过多及怀孕外,不应服用本品超过六个月。
【儿童用药】儿童每天按体重1.5毫升/公斤(相当于每天三价铁4毫克/公斤体重),应遵医嘱分两次于饭前口服。
夏桑菊颗粒(菲德力)的说明书

夏桑菊颗粒(菲德力)的说明书清内火解内毒才能让人体保持一个非常健康愉悦的状态,才能以正常的心态去面对日益竞争激烈的社会。
如今大多数人比较浮躁,多半是跟身体没有清热解毒有关系。
清热解毒药物夏桑菊颗粒(菲德力)的出现能很好的帮您解决这一问题,它对于人体没有任何副作用,可以放心使用,下面来看看对于夏桑菊颗粒(菲德力)的介绍吧。
【药品名称】通用名称:夏桑菊颗粒商品名称:夏桑菊颗粒(菲德力)【适应症/功能主治】清肝明目,疏风散热,除湿痹,解疮毒。
用于风热感冒,目赤头痛,头晕耳鸣,咽喉肿痛,疔疮肿毒等症,并可作清凉饮料。
【规格型号】10g*20袋【用法用量】口服,一次10-20克,一日3次。
【注意事项】1.忌烟、酒及辛辣、生冷、油腻食物。
2.不宜在服药期间同时服用滋补性中成药。
3.风寒感冒者不适用,其表现为恶寒重,发热轻,无汗,头痛,鼻塞,流清涕,喉痒咳嗽。
4.高血压、心脏病、肝病、糖尿病、肾病等慢性病严重者应在医师指导下服用。
5.服药三天后症状无改善,或症状加重,或出现新的严重症状如胸闷、心悸等应立即停药,并去医院就诊。
6.小儿、年老体弱者、孕妇应在医师指导下服用。
7.脾胃虚寒,症见腹痛、喜暖、泄泻者慎用。
8.对本品过敏者禁用,过敏体质者慎用。
9.本品性状发生改变时禁止使用。
【有效期】0 月【批准文号】国药准字Z51020889【生产企业】四川菲德力制药有限公司【药品类别】本品为内科感冒类非处方药药品。
【性状】本品为棕褐色的颗粒;味甜。
上述内容充分为您介绍了关于夏桑菊颗粒(菲德力)的各种功效,希望能给您带来帮助。
清热解毒属于常见治疗方式,绝大多数人体内或多或少都患有温毒和体热的症状,只要您能正确认识这种疾病,并且及时服药,就能很快治愈。
琥珀酸亚铁片说明书

琥珀酸亚铁片说明书通用名:琥珀酸亚铁片生产厂家: 金陵药业股份有限公司南京金陵制药厂批准文号:国药准字H10930005药品规格:100mg*20片药品价格:¥30元【通用名称】琥珀酸亚铁片【商品名称】琥珀酸亚铁片速力菲【拼音全码】HuPoSuanYaTiePianSuLiFei【主要成份】主要活性成分为琥珀酸亚铁。
【性状】琥珀酸亚铁片速力菲为薄膜衣片,除去薄膜衣后显暗黄色。
【适应症/功能主治】缺铁性贫血的预防及治疗用。
【规格型号】100mg*20s【用法用量】口服。
用于预防:成人一日1片,孕妇一日2片,儿童一日0.5片。
用于治疗:成人一日2~4片,儿童一日1~3片,分次服用。
【不良反应】1、可见胃肠道不良反应,如恶心、呕吐、上腹疼痛、便秘。
2、琥珀酸亚铁片速力菲可减少肠蠕动,引起便秘,并排黑便。
【禁忌】1.肝肾功能严重损害,尤其是伴有未经治疗的尿路感染禁用;2.铁负荷过高、血色病或含铁血黄素沉着症患者禁用;3.非缺铁性贫血如地中海贫血患者禁用。
4.对琥珀酸亚铁片速力菲过敏者禁用。
【注意事项】1.用于日常补铁时,应采用预防量。
2.治疗剂量不得长期使用,应在医师确诊为缺铁性贫血后使用,且治疗期间应定期检查血象和血清铁水平。
3.下列情况慎用:酒精中毒、肝炎、急性感染、肠道炎症、胰腺炎、胃与十二指肠溃疡、溃疡性肠炎。
4.琥珀酸亚铁片速力菲不应与浓茶同服。
5.琥珀酸亚铁片速力菲宜在饭后或饭时服用,以减轻胃部刺激。
6.如服用过量或出现严重不良反应,应立即就医。
7.对琥珀酸亚铁片速力菲过敏者禁用,过敏体质者慎用。
8.琥珀酸亚铁片速力菲性状发生改变时禁止使用。
9.请将琥珀酸亚铁片速力菲放在儿童不能接触的地方。
10.儿童必须在成人监护下使用。
【儿童用药】尚不明确。
【老年患者用药】尚不明确。
【孕妇及哺乳期妇女用药】尚不明确。
【药物相互作用】1.维生素C与琥珀酸亚铁片速力菲同服,有利于琥珀酸亚铁片速力菲吸收。
2.琥珀酸亚铁片速力菲与磷酸盐类、四环素类及鞣酸等同服,可妨碍铁的吸收。
多糖铁复合物胶囊琥珀酸亚铁缓释片,

多糖铁复合物胶囊琥珀酸亚铁缓释片,益血生胶囊:阿胶、龟甲胶、鹿角胶、鹿血、牛髓、紫河车、鹿茸、茯苓、黄芪(蜜制)、白芍、当归、党参、熟地黄、白术(麸炒)、制何首乌、大枣、炒山楂、炒麦芽、炒鸡内金、知母(盐制)、大黄(酒制)、花生衣健脾补肾,生血填精。
用于脾肾两虚,精血不足所致的面色无华、眩晕气短、体倦乏力、腰膝痠软;缺铁性贫血、慢性再生障碍性贫血见上述证候者。
力蜚能说明书•通用名称:•多糖铁复合物胶囊•功能主治:•用于治疗单纯性缺铁性贫血。
•用法用量:•成人每日一次,每次口服1-2粒;儿童需在医生的指导下使用。
•剂型:•胶囊剂•不良反应:•极少出现胃肠刺激或便秘。
速力菲琥珀酸亚铁缓释片,本品主要成份为琥珀酸亚铁。
抗贫血药,用于治疗缺铁性贫血。
饭后口服。
成人预防量:每次1片,隔日服一次(即隔日0.2g)。
成人治疗量:每次1~2片,每日一次,(即每日0.2g~0.4g)。
或遵医嘱。
血红蛋白正常后仍需继续服用1~2月。
可能出现食欲减退、恶心、呕吐、腹泻等。
可适当减少服用量或停药。
1.血色病或含铁血黄素沉着症及不伴缺铁的其他贫血(如地中海贫血)禁用。
2.肝、肾功能严重损害者禁用。
1.酒精中毒、肝炎、肝肾功能不良、急性感染、肠道炎症、胰腺炎、消化性溃疡慎用。
2.服时忌茶,以免被鞣质沉淀而无效。
3.用药期间应定期检查血红蛋白、网织红细胞、血清铁蛋白及血清铁。
4.与制酸药如碳酸氢盐同用、易产生沉淀影响吸收。
5.与四环素类药物同用可形成络合物而妨碍吸收。
6.与维生素C同服可增加本品吸收,而易致胃肠道反应。
7.应用本品可使大便隐血试验阳性而干扰上消化道出血的诊断。
8.本品应整片吞服。
9.治疗同时需寻找缺铁的原因。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
1.成份:本品主要成份为非布佐司他,其化学名为2-[(3-氰基-4-异丁氧基)苯基]-4-甲基-5-噻唑羧酸。
结构式:分子式:C16H16N2O3S分子量:316.372.性状:本品为白色粉末3.适应症:本品为黄嘌呤氧化酶(XO)抑制剂,适用于具有痛风症状的高尿酸血症的长期治疗。
不推荐本品用于治疗无症状性高尿酸血(症)。
4.剂型:胶囊剂、片剂5.规格:40mg/粒或片;80mg/粒或片6.用法用量:推荐剂量用于治疗有痛风症状的高尿酸血症患者时,推荐本品剂量为40mg或80mg,每日一次。
推荐本品的起始剂量为40mg,给药本品时无需考虑食物或抗酸剂的影响。
特殊人群轻或中度肾功能损伤患者服用本品时不必调整剂量。
推荐本品的起始剂量为40mg,每日一次。
给药剂量40mg,持续两周后,对血清尿酸水平(sUA)仍高于6 mg/dl的患者,推荐给药剂量80mg。
轻中度肝功能损伤患者服用本品无需剂量调整。
对严重肝功能损伤患者使用本品尚无研究,因此给药本品应谨慎。
尿酸水平使用本品治疗2周后即可进行血清尿酸的再检验。
治疗目标是降低和维持血清尿酸水平使其低于6 mg/dl。
预防痛风急性发作推荐至少用药6个月(见【注意事项】)。
痛风发作变化的血清尿酸水平会导致沉积的尿酸盐活动,因此开始给药本品后会导致痛风发作。
推荐使用本品时,同时给药非甾体抗炎药(NSAID)或秋水仙碱,以预防痛风发作。
预防痛风急性发作推荐至少用药6个月。
如果在给药治疗期间发生痛风,不需要停药。
对个别患者的痛风应相应给予治疗。
续发性高尿酸血症对继发性高尿酸血症患者(包括器官移植患者)给药本品尚无研究;尿酸盐生成率增加的患者不推荐使用本品(例如恶性病及其治疗、莱-萘二氏综合征)。
稀有病例的尿中黄嘌呤的浓度会显著增加,在尿路中沉积。
7.不良反应:临床试验中,共对2757例患高尿酸血症和痛风患者给药本品,每天40mg或80mg。
对559例患者给药本品40mg,持续≥ 6个月。
给药本品80mg的患者中,1377例患者持续≥ 6个月,674例患者治疗≥1年,515例患者治疗≥2年。
常见不良反应 三项随机、对照临床研究(研究1、2、3),持续6-12个月,报道了由于药物引起的不良反应。
表1 概述了本品给药组至少1%发生率的不良反应和比空白对照组发生率至少高0.5%的不良反应 本品给药组不良反应发生率≥ 1%和比空白对照组至少高0.5%的不良反应不良反应空白对照组非布佐司他组 别嘌呤醇组* (N=134)每日40mg (N=757)每日80mg (N=1279) (N=1277) 肝功能异常恶心关节痛疹 0.7% 0.7% 0% 0.7% 6.6% 1.1% 1.1% 0.5% 4.6% 1.3% 0.7% 1.6% 4.2% 0.8% 0.7% 1.6% *根据肾功能损伤,给药别嘌呤醇患者组中,10例给药100mg ,145例给药200mg ,1122例给药300mg 。
导致治疗过程中停药的最常见不良反应为肝功能异常,本品40mg 给药组1.8%,80mg 组1.2%,别嘌呤醇给药组0.9%。
除了表1列出的不良反应,本品给药组中报道的头晕不良反应也大于1%,但与空白对照组相比低于0.5%。
较少发生的不良反应2期和3期临床研究中,以下不良反应发生率低于1%,而本品给药范围为40mg-240mg 时更高些。
下列包括注意事项中器官系统的不良反应(低于1%)。
血液和淋巴系统:贫血、特发性血小板减少性紫癜、白细胞增多/减少嗜中性白血球减少症、全血细胞减少症、脾大、血小板减少症。
心脏疾患:心绞痛、心房纤维性颤动、心杂音、ECG 异常、心悸、窦性心动过缓、心动过速。
耳朵和内耳方面的异常:耳聋、耳鸣、眩晕。
眼部疾病:视觉模糊。
胃肠道病症:腹胀、腹痛、便秘、口干燥、消化不良、肠胃气胀、频便、胃炎、胃食管返流疾病、胃肠不适、龈痛、呕血、胃酸过多、便血、口腔溃疡形成、胰腺炎、消化性溃疡、呕吐。
一般病症和给药情况:虚弱、胸痛/不适、水肿、疲劳、情绪异常、步态障碍、流行性感冒症状、痞气、疼痛、口渴。
肝胆管病症:胆石病/胆囊炎、肝脂肪变性、肝炎、肝肿大。
免疫系统疾病:超敏反应。
传染及感染:带状疱疹。
并发症:挫伤。
新陈代谢病症:厌食症、食欲减退/增强、脱水、糖尿病、高胆固醇血症、高血糖、高脂血症、高甘油三酸酯血症、低钾血、体重减少/增加。
肌骨骼和结缔组织病症:关节炎、关节僵直、关节肿胀、肌肉痉挛/肌肉颤搐/紧缩/无力、肌骨骼疼痛/四肢强直、肌痛。
神经系统病症:味觉改变、平衡紊乱、中风综合征、Guillain-Barré综合症、头痛、轻偏瘫、感觉迟钝、嗅觉减退、腔隙梗塞、瞌睡、精神损害、偏头痛、感觉异常、瞌睡、短暂性缺血发作、震颤。
精神病症:激动、焦虑、抑郁、失眠、易怒、性欲降低、神经过敏、恐慌发作、人格变化。
肾脏、泌尿系统病症:血尿、肾石病、尿频、蛋白尿、肾衰竭、肾机能不全、尿急、失禁。
生殖系统和乳腺变化:乳房痛、勃起机能障碍、男子女性型乳房。
呼吸、胸部及纵膈病症:支气管炎、咳嗽、呼吸困难、鼻出血、鼻发干、鼻窦分泌物过多、咽水肿、呼吸道充血、喷嚏、咽喉刺激、上呼吸道感染。
皮肤和皮下组织病症:脱发、血管性水肿、皮炎、皮肤划痕现象、淤斑、湿疹、头发变色、毛发生长异常、多汗、皮肤脱皮、瘀点、光过敏、瘙痒、紫癜、皮肤变色、皮肤病损、皮肤气味异常、荨麻疹。
血管病症:面红、热潮红、高血压、低血压。
实验室参数:活化部分凝血激酶时间延长、肌酸增加、重碳酸盐减少、钠增加、EEG异常、葡萄糖增加、胆固醇增加、甘油三酯增加、淀粉酶增加、钾增加、TSH增加、血小板计数计数减少、血细胞比容减少、血红素减少、MCV增加、RBC减少、肌酸酐增加、血尿素增加、BUN/肌酸酐比率增加增加、肌酸磷酸激酶(CPK)增加、碱性磷酸(酯)酶增加、LDH 增加、PSA增加、尿排出量增加/减少、淋巴细胞计数减少、中性白细胞计数减少、WBC增加/减少、检尿蛋白质异常、低密度脂蛋白(LDL)增加、凝血酶原时间延长、尿管型、尿液白细胞及蛋白质阳性。
心血管安全随机对照长期扩展研究中,心血管事件及死亡属于APTC事件(心血管死亡、非致命性心肌梗塞、非致命性中风)一项预定义终末点指标。
3期随机对照研究中,报道的100例患者APTC事件发病率:空白对照组0(95% CI 0.00-6.16)、本品40mg组0(95% CI 0.00-1.08)、本品80mg组1.09(95% CI 0.44-2.24)、别嘌呤醇0.60(95% CI 0.16-1.53)。
一项长期扩展研究中,报道的APTC事件发病率为:本品80mg组0.97 (95% CI0.57-1.56)、别嘌呤醇0.58(95% CI 0.02-3.24)。
研究发现,本品APTC事件发病率高于别嘌呤醇给药组患者。
相关机制尚不确定。
应监测MI和中风的体征和症状。
8.注意事项:痛风发作开始应用本品治疗后,可观察到痛风发作增加。
这是由于变化的血清尿酸水平减少导致沉积的尿酸盐活动引起的。
为预防给药本品时发生痛风发作,推荐同时给药非甾体抗炎药或秋水仙碱(见【用法用量】)。
心血管病症随机对照研究中,使用本品[0.74 per 100 P-Y (95% CI 0.36-1.37)]的患者比给药别嘌醇[0.60 per 100 P-Y (95% CI 0.16-1.53)]患者更易发生心血管血栓事件(心血管死亡、非致命性心肌梗塞、非致命性中风)(见【不良反应】)。
相关原因尚未明确。
应对心肌梗塞(MI)及中风的体征和症状进行监测。
肝脏酶增加随机对照研究中,观察到转氨酶水平比正常上限的3倍还高(给药本品及别嘌呤醇患者分别提高,AST:2%,2%、ALT:3%,2%)。
未发现转氨酶的提高具有量效关系。
肝功能实验室分析推荐,应用本品治疗2月和4月,此后周期性治疗。
9.禁忌:正在服用硫唑嘌呤、巯嘌呤或胆茶碱的患者禁用本品。
10.特殊用药:孕妇及哺乳期妇女用药1.孕妇C类药物:对孕妇尚无足够充分的对照研究。
只有在对婴儿的潜在益处与风险相当的情况下,孕妇才能服用本品。
在器官生成期对大鼠和兔子口服给药非布佐司他,剂量达到48mg/kg(按照体表面积计算分别为人给药80mg/天的40和50倍)时,无致畸性。
在器官生成期及泌乳期,对怀孕期大鼠口服给药本品,剂量达到48mg/kg(为人给药80mg/天的40倍),发现新生儿死亡率增加,新生儿体重增加减少。
2.哺乳期妇女非布佐司他经大鼠乳汁分泌。
非布佐司他是否经人体乳汁分泌尚未知。
由于很多药物通过人乳汁分泌,故对哺乳期妇女给药本品时应谨慎。
儿童用药18以下儿童患者使用本品的安全性及有效性尚未确定。
老年用药老年患者使用本品无需剂量调整。
与其他年龄组相比,在安全性及有效性方面无临床显著差异,但不排除有些老年患者对本品较敏感。
老年患者(≥65岁)多剂量口服给药非布佐司他后的Cmax及AUC24与年轻患者(18-40岁)相似。
11.药物相互作用:巯嘌呤/硫唑嘌呤等非布佐司他是黄嘌呤氧化酶(XO)抑制剂。
虽然本品与通过XO代谢药物(例如胆茶碱、巯嘌呤、硫唑嘌呤)的相互作用尚无研究,但本品对XO的抑制作用会这些药物在血浆中浓度的增加从而产生毒性。
正在服用硫唑嘌呤、巯嘌呤或胆茶碱患者禁止使用本品。
细胞毒素化疗药物本品与细胞毒素化疗药物的相互作用尚未进行研究。
本品在细胞毒素化疗期间的安全性尚无可靠数据。
体内药物相互作用研究根据健康受试者的药物相互作用研究,本品与秋水仙碱、萘普生、吲哚美辛、氢氯噻嗪、华法林、地昔帕明无显著临床意义的相互作用。
因此,本品可以与这些药物联用。
12.药物过量:对健康受试者给药本品剂量达到每天300mg,持续7天,无剂量限制性毒性。
没有药物过量的病例报道。
药物过量患者应进行对症和支持疗法。
13.药理毒理:作用机制非布佐司他是黄嘌呤氧化酶抑制剂,通过减少血清尿酸达到疗效。
本品在治疗浓度下不会抑制嘌呤和嘧啶的合成及代谢过程中的其他酶。
毒理研究以比格犬进行的为期12个月的毒性研究发现,15mg/kg剂量时(约为人给药80mg/天的4倍)在肾脏中发现黄嘌呤沉积物结晶和结石。
对大鼠给药48mg/kg(约为人给药80mg/天的35倍),为期6个月的研究中,由于黄嘌呤结晶沉积也发现了类似的结石形成。
致癌性:以F344大鼠和B6C3F1小鼠进行为期两年的致癌性研究。
对雄性大鼠和雌性小鼠分别给药24 mg/kg (为人推荐最大给药剂量80mg/天的25倍)、18.75 mg/kg (为人给药剂量80mg/天的12.5倍),分别发现移行细胞乳头状瘤及膀胱癌增加。
膀胱肿瘤是肾脏和膀胱结石形成的。
致突变性:本品在有或无代谢活化剂时,中国仓鼠肺成纤维细胞体外染色体畸变试验结果为阳性。