1,3-二羰基化合物的氧二氟甲基化反应
各类官能团的反应

CH3 H
CH3 D
H
OH
(1) (C H 3C O O )2H g / H 2O (2) N aB H 4, N aO H
OH D CH3 H
.
Cope 消除
β-碳上有氢的氧化胺加热到150~200 oC时发生热分解, 生成羟胺和烯烃。由亚瑟·科普(Arthur C. Cope)发现
.
Chugaev反应
.
脂环烃
1. 环烷烃的自由基卤代反应、催化加氢反应; 2. 环丙烷衍生物加H2反应与加X2、HX反应的区别; 3. 三元环与烯烃的鉴别(KMnO4)。
.
芳烃
1. 苯环的五大类亲电取代反应(-X, -NO2, -SO3H, -R, COR)及常用的亲电反应试剂;
2. 知道与苯环相连时哪些是吸电子基团、供电子基团; 3. 知道单取代苯亲电反应的定位规则并且会用共振论来
(如何使羟基转变成一个好的离去基团) • 知道试剂的碱性和亲核性的大致关系; • 卤代烃制备格式试剂,格氏试剂的用途; • 知道苄卤和烯丙基卤的特殊活泼性。
.
溶剂对亲核取代反应的影响
溶剂的分类:质子溶剂、偶极、非极性溶剂
溶剂对反应影响的规律
1 质子溶剂对SN1反应有利,对SN2的影响要作具体分析 。
加少量碘即可促进反应。
R- Cl I-
Nu-
R-Nu
R-I Nu-
利用碘负离 子是一个好 的离去基团
.
两位负离子
定义:一个负离子有两个位置可以发生反应,则称其具 有双位反应性能,具有双位反应性能的负离子称为两位 负离子。
*1 RCH2Br + NaNO2
SN2 乙醚
SN1 *2 R2CHBr + AgNO2 乙醚
α–二羰基化合物

α–二羰基化合物
α–二羰基化合物是一类具有两个羰基基团的化合物,通常具有较高的化学稳定性和反应性,广泛应用于有机合成、材料科学以及医药等领域。
其分子结构中,两个羰基基团位于相邻碳原子上,因此也被称为1,2-二羰基化合物。
α–二羰基化合物的合成方法包括基于酮或醛的反应、酰基化反应以及羧酸衍生物的还原反应等。
此外,α–二羰基化合物还可以通过不同的化学反应进行进一步的修饰和转化,例如通过羟甲基化反应形成α-羟甲基-β-二羰基化合物,或者通过胺基化反应形成α-胺基-β-二羰基化合物等。
这些方法为α–二羰基化合物的合成和应用提供了广泛的可能性和应用前景。
- 1 -。
有机化学反应的总结

有机化学一、烯烃1、卤化氢加成 (1)CHCH 2RHXCH CH 3RX【马氏规则】在不对称烯烃加成中,氢总是加在含碳较多的碳上。
【机理】CH 2CH 3CH +CH 3CH 3X +CH 3CH 3+H +CH 2+C3X +CH 3X主次【本质】不对称烯烃的亲电加成总是生成较稳定的碳正离子中间体。
【注】碳正离子的重排 (2)CHCH 2RCH 2CH 2R BrHBrROOR【特点】反马氏规则 【机理】 自由基机理(略)【注】过氧化物效应仅限于HBr 、对HCl 、HI 无效。
【本质】不对称烯烃加成时生成稳定的自由基中间体。
【例】CH 2CH3BrCH CH 2BrC H 3CH +CH 3C H 3HBrBrCH 3CH 2CH 2BrCH CH 3C H 32、硼氢化—氧化CHCH 2R CH 2CH 2R OH1)B 2H 62)H 2O 2/OH-【特点】不对称烯烃经硼氢化—氧化得一反马氏加成的醇,加成是顺式的,并且不重排。
【机理】2CH33H323H32CH CH2CH32CH CH=CH(CH3CH2CH2)3-H3CH2CH2C22CH3CH2OCH2CH2CH33CH2CH2C2CH2CH3+O H-OHB-OCH2CH2CH3CH2CH2CH3H3CH2CH2B OCH2CH2CH3CH2CH2CH32CH2CH3HOO-B(OCH2CH2CH3)3B(OCH2CH2CH3)3+3NaOH3NaOH3HOCH2CH2CH33+Na3BO32【例】CH31)BH32)H2O2/OH-CH3HHOH3、X2加成C CBr/CClC CBrBr【机理】CCC CBrBrCBr+C CBrOH2+-H+C CBrOH【注】通过机理可以看出,反应先形成三元环的溴鎓正离子,然后亲和试剂进攻从背面进攻,不难看出是反式加成。
不对称的烯烃,亲核试剂进攻主要取决于空间效应。
【特点】反式加成 4、烯烃的氧化1)稀冷高锰酸钾氧化成邻二醇。
精细有机合成—构成环状化合物的反应

由Knorr合成法得到的产物可水解脱羧,生成取代吡咯。许多其 他吡咯,尤其是用于卟啉合成中的吡咯,也是利用这一方法制备, 不过改变不同的取代基而已。为了方便地脱去烷氧羰基,在Knorr合 成中,用苯基和叔丁基酯取代了乙酯基。如原料中的氨苯上有烃基, 则可得到N-烃基吡咯。
α,β-不饱和羰基化合物是极活泼的亲二烯体系,并且代表了该合成方 法中最有价值的组分,其典型的例子有丙烯醛、丙烯酸及其酯、顺丁烯 二酸及其酸酐和丁炔二酸:
+ +
+
+
(2) Robinson增环反应 活泼亚甲基化合物与α,β-不饱和酮、酯、腈等起Michael反应,然后起
醇醛缩合反应称之为Robinson增环反应,常用于合成环状化合物。在合 成六元环烃,特别在甾体化合物的合成上具有重要作用。这种方法分两个 阶段进行。先起Michael加成反应,接着起分子内的羟醛缩合反应,增环 生成环己酮。一般采用催化量的碱,主要得到1,4-加成产物,采用当量碱 则主要得到环合产物.这样可以利用两步合一的反应方便地合成六元环。
二卤环丙烷用AgNO3处理,可转化为烯丙基化合物,这是用卡 宾增长碳链的另一种方法。
卡宾与杂环体系的烯键加成,形成扩环产物,这在合成上十分有用:
3.1.2 四元环衍生物 用1,3-二卤代烷对活性亚甲基化合物进行分子内烷基化,例如
在强碱存在下,丙二酸酯与1,3二溴丙烷成环,生成环丁烷衍生物。
四元环除由丙二酸酯法合成外,还可以由[2+2]环加成反应合成。[2+2] 环加成是由两个烯分子组成四元环的反应。简单的烯烃在加热时不能生成 环丁烷衍生物,丙烯腈容易二聚成顺-和反-1,2—二氰基环丁烷:
有机化学反应机理+范例+原理

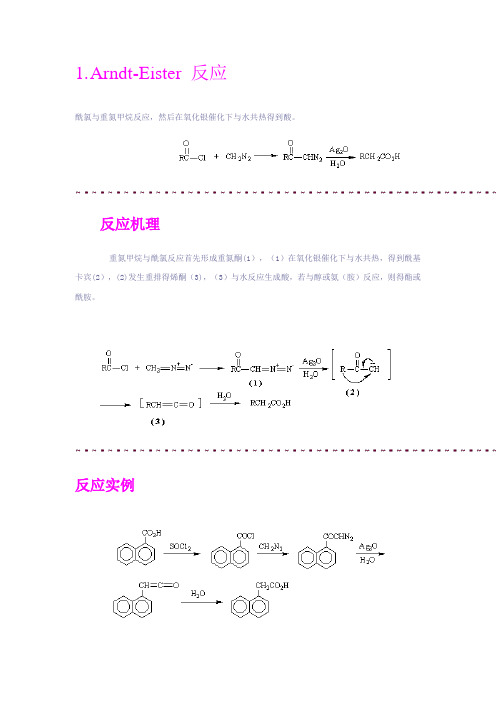
1.A rndt-Eister 反应酰氯与重氮甲烷反应,然后在氧化银催化下与水共热得到酸。
反应机理重氮甲烷与酰氯反应首先形成重氮酮(1),(1)在氧化银催化下与水共热,得到酰基卡宾(2),(2)发生重排得烯酮(3),(3)与水反应生成酸,若与醇或氨(胺)反应,则得酯或酰胺。
反应实例2.Baeyer----Villiger 反应反应机理过酸先与羰基进行亲核加成,然后酮羰基上的一个烃基带着一对电子迁移到-O-O-基团中与羰基碳原子直接相连的氧原子上,同时发生O-O键异裂。
因此,这是一个重排反应具有光学活性的3---苯基丁酮和过酸反应,重排产物手性碳原子的枸型保持不变,说明反应属于分子内重排:不对称的酮氧化时,在重排步骤中,两个基团均可迁移,但是还是有一定的选择性,按迁移能力其顺序为:醛氧化的机理与此相似,但迁移的是氢负离子,得到羧酸。
反应实例酮类化合物用过酸如过氧乙酸、过氧苯甲酸、间氯过氧苯甲酸或三氟过氧乙酸等氧化,可在羰基旁边插入一个氧原子生成相应的酯,其中三氟过氧乙酸是最好的氧化剂。
这类氧化剂的特点是反应速率快,反应温度一般在10~40℃之间,产率高。
3.Beckmann 重排肟在酸如硫酸、多聚磷酸以及能产生强酸的五氯化磷、三氯化磷、苯磺酰氯、亚硫酰氯等作用下发生重排,生成相应的取代酰胺,如环己酮肟在硫酸作用下重排生成己内酰胺:反应机理在酸作用下,肟首先发生质子化,然后脱去一分子水,同时与羟基处于反位的基团迁移到缺电子的氮原子上,所形成的碳正离子与水反应得到酰胺。
迁移基团如果是手性碳原子,则在迁移前后其构型不变,例如:反应实例4.Birch还原芳香化合物用碱金属(钠、钾或锂)在液氨与醇(乙醇、异丙醇或仲丁醇)的混合液中还原,苯环可被还原成非共轭的1,4-环己二烯化合物。
反应机理首先是钠和液氨作用生成溶剂化点子,然后苯得到一个电子生成自由基负离子(Ⅰ),这是苯环的л电子体系中有7个电子,加到苯环上那个电子处在苯环分子轨道的反键轨道上,自由基负离子仍是个环状共轭体系,(Ⅰ)表示的是部分共振式。
(完整版)羰基的亲核加成及相关反应

羰基的亲核加成及相关反应羰基化合物包括醛、酮、羧酸及衍生物和CO 2。
5.1 羰基的结构CO δ+δ-亲电中心羰基碳的活性较大,易被亲核试剂进攻而发生亲核加成反应和亲核取代反应。
5.2 亲核加成反应的历程及影响因素 5.2.1 HCN 的加成 反应为碱催化。
]CN ][CO [k v ->=OH -+HCNCN -+ H 2O快-COδ+δ-CO -CNCOHCN+OH - 反应的平衡位置受电子效应和空间效应的影响。
酮正向反应的趋势较小(空阻大)。
二、亲核加成反应的一般特点 1.反应可以被酸或碱催化酸催化可提高羰基的亲电活性。
CO +H ++OH碱催化提高亲核试剂的亲核性。
NuH +OH --+H 2ONu H ->2.多数醛酮的亲核加成为可逆反应,用于分离与提纯。
5.2.2 影响羰基亲核加成反应活性的因素 一、羰基化合物的结构 1.电子效应羰基碳的正电性越大,亲核加成速度越大,反应活性越大。
羰基碳所连的吸电基(-I ,-C )使其亲核加成反应的活性增加,而供电基(+I ,+C )则使其活性降低。
活泼顺序:ClCHO > HCHO > RCHO > CH 3COR > RCOOR' > RCONR'2 > RCOO --I > +C(+C)(+C,空阻)( +C > -I)(+C)CO RR'活性极低(1)π-π共轭效应(增加其稳定性);(2)+C 效应(降低羰基碳的正电性);(3)加成产物失去共轭能,反应活化能高;(4)产物的张力大幅增加。
2.立体效应CO -sp 2活性:O CHH OC CH 3H OC CH 3CH 3O OC CH 3CH 2CH 2CH 3OC Ph Ph>>>>>二、试剂的亲核性对同一羰基化合物,试剂的亲核性越大,平衡常数越大,亲核加成越容易。
1.带负电荷的亲核试剂比起共轭酸(中性分子)的亲核性强。
6 第六章 羰基化合物的反应

CH3CH2CH2CHO + CH2CHO -OH CH2CH3
OH
I2
CH3CH2CH2CHCHCHO
CH2CH3
CH3CH2CH2CH=CCHO CH2CH3
醛的自身缩合
酮的自身缩合
~ ~
~
O Soxhlex 提取器 Ba(OH)2
2CH3CCH3 O
-H2O I2
(CH3)2C=CH C CH=C(CH3)2
C2H5 H C
C6H5
O C
CH3
LiAlH4 乙醚
C2H5
OH
H
H
CC
+
C6H5
CH3
75%
H2O
C2H5 H CC
C6H5 25%
H OH
CH3
当羰基和一个手性中心连接时,反应符合 克莱姆规则一。
6.5 碳负离子
O R C CH3 -OH
}O
RC CH2
烯醇负离子
[O RC CH2
碳负离子
O (CH3)2C=CH C CH3
O CH3CCH3
H+
H
O H
CH3
CH2-H 1,4加成 HO
CH3 CH3
插烯系规则
CH3 互变异构 O
CH3 CH3
分子内缩合
CH3 CH3
CH3
*2 交叉羟醛缩合反应 两种不同的醛、酮之间发生的羟醛缩 合反应称为交叉的羟醛缩合反应。
有两种情况 (1)一种醛或酮有-H,另一种醛或酮无-H。 (2)两种醛酮都有-H。(在定向羟醛缩 合反应中讨论。)
A 甲醛的羟甲基化反应
-OH
CH2O + H-CH2CHO
基础有机化学反应总结

基础有机化学反应总结一、烯烃1、卤化氢加成(1)【马氏规则】在不对称烯烃加成中,氢总是加在含碳较多的碳上。
【机理】【本质】不对称烯烃的亲电加成总是生成较稳定的碳正离子中间体。
【注】碳正离子的重排(2)【特点】反马氏规则【机理】自由基机理(略)【注】过氧化物效应仅限于HBr、对HCl、HI无效。
【本质】不对称烯烃加成时生成稳定的自由基中间体。
【例】2、硼氢化—氧化【特点】不对称烯烃经硼氢化—氧化得一反马氏加成的醇,加成是顺式的,并且不重排。
【机理】【例】3、X2加成【机理】【注】通过机理可以看出,反应先形成三元环的溴鎓正离子,然后亲和试剂进攻从背面进攻,不难看出是反式加成。
不对称的烯烃,亲核试剂进攻主要取决于空间效应。
【特点】反式加成4、烯烃的氧化1)稀冷高锰酸钾氧化成邻二醇。
3H 33H3稀冷KMnO 433M nO OOO H 2O 3H 33H 3 2)热浓酸性高锰酸钾氧化3)臭氧氧化4)过氧酸氧化5、烯烃的复分解反应【例】6、共轭二烯烃1)卤化氢加成2)狄尔斯-阿德尔(Diels-Alder )反应【描述】共轭二烯烃和烯烃在加热的条件下很容易生成环状的1,4加成产物。
【例】二、脂环烃1、环丙烷的化学反应【描述】三元环由于张力而不稳定,易发生加成反应开环,类似碳碳双键。
【特点】环烷烃都有抗氧化性,可用于区分不饱和化合物。
【注】遵循马氏规则【例】2、环烷烃制备1)武兹(Wurtz)反应【描述】通过碱金属脱去卤素,制备环烷烃。
【例】2)卡宾①卡宾的生成A、多卤代物的α消除B、由某些双键化合物的分解②卡宾及烯烃的加成反应【特点】顺式加成,构型保持【例】③类卡宾【描述】类卡宾是一类在反应中能起到卡宾作用的非卡宾类化合物,最常用的类卡宾是ICH2ZnI。
【特点】顺式加成,构型保持【例】三、炔烃1、还原成烯烃1)、顺式加成2)、反式加成2、亲电加成1)、加X 2【机理】中间体Br+R 2R 1【特点】反式加成 2)、加HXR R HBr RR Br H (一摩尔的卤化氢主要为反式加成)3)、加H 2O【机理】【特点】炔烃水合符合马式规则。
精细化学有机合成必备——缩合反应

HO CH2 O HO CH2 C HO CH2
O
(Cannizzaro反应)
HO CH2 CH2 OH + HCOONa HO CH2
季戊四醇
C H + H C H + NaOH
HO CH2 C
6
醛的歧化
(Cannizzaro反应)
无α-氢的醛,如甲醛、2,2-二甲基丙醛、苯甲醛和呋喃醛 等,在碱的催化下,可以发生歧化反应,生成等摩尔的羧酸
Cannizzaro反应也可发生在两个不同的无α-氢的醛分子间,当
其中之一为甲醛时,总是甲醛被氧化,而另一醛被还原成醇。
7
例如:
CHO COOH CH2OH
+ NaOH(浓)
+
O
O
(CH3)2CH-C-H + H-C-H(过量) CH3
OH- (浓) , HCHO
OH- (浓)
CH3 O HOCH2-C-C-H CH3
该类反应需在缩合剂或催化剂酸、碱、盐、金属、醇钠等 存在下才能顺利进行,这里我们选择一些重要的缩合反应进行 讨论。
12.1 醛酮缩合反应
12.1.1 羟醛缩合( Aldol缩合、醇醛缩合) 含有活泼氢的醛或酮在碱或酸的催化下生成β-羟基酮的反应 称为Aldol缩合反应,即醛醇缩合反应,包括醛-醛缩合、酮-酮 缩合、醛-酮交叉缩合三大类。
H+转移
R R'
N-CH2-OH 2
-H2O
R N=CH2 R'
活化的C=N
O CH3C-CH 2 H
O H CH3-C=CH 2
R CH2=N R'
O CH3-C-CH 2-CH 2-N
[理学]第六章 羰基化合物的反应
![[理学]第六章 羰基化合物的反应](https://img.taocdn.com/s3/m/f28edb13f111f18583d05a59.png)
sp3杂化 四面体
产物中基团拥挤程度增大。
R 越大,妨碍Nu:从背后进攻C原子。
6.2 羰基加成反应及产物
a. 与水加成
O
H+或OH-
OH R' + H2O R C OH R'
R
C
除甲醛、多卤代醛外,其它醛的水合反应平衡偏向 左边。
(2)与ROH的加成
H+
OCH2CH3 CH3CH OH
CH3CH=O + CH3CH2OH
H
O
C2H5
PhH
Ph H H C2H5 O
R H H Ph C2H5 OH
+
H
O
C2H5
1 RMgX 2 H2O
PhH
OH H H Ph C2H5 R
35oC R CH3 C6H5 (CH3)2CH (CH3)3C -70oC R (CH3)3C CH3
主 2.5 > 4 5 49
: : : :
H O O
L
R
S
Nu-
C2H5 H C6H5
O C C CH3
C 2 H5 H C6H5
LiAlH4 乙醚
OH H CH3
H2 O
C
C
+
C2H5 H C6H5
C
C
H OH CH3
75%
25%
当羰基和一个手性中心连接时,反应符合 克莱姆规则一。
6.5 碳负离子
O R C CH3
-
OH
O R C
CH2
第六章 羰基化合物的反应
6.1 羰基化合物的反应机理 6.2 羰基加成反应及产物 6.3 加成-消除反应 6.4 羰基化合物的反应活性和加成的立体选择性 6.5 碳负离子 6.6 各种重要的缩合反应 6.7 羰基与叶子立德的反应 6.8 羧酸衍生物的亲核加成 6.9 亲核性碳 6.10 分子内催化作用
羰基的亲核加成及相关反应

亲核试剂从空阻小的一边进攻羰基
❖ 换用仲丁基硼氢化锂,内侧进攻产物99.6%, 外侧产物0.4%
醛酮的简单亲核加成
强酸强碱
❖ 仅限醛、脂肪甲基酮及<8个碳原子的环酮
可极化性及亲核性:
O CH2-CH=CH2
66%
曼尼希(Mannich)反应
具有α-氢的醛酮或其它含活泼氢的化合物与 甲醛及胺(或氨)反应生成含胺甲基的化合 物
COCH3 + CH2O + NH3
COCH2 CH2NH2
酸组分
醛组分 碱组分
曼尼希碱
❖ 活泼氢被胺甲基化后,加热失去氨,生成
αβ-不饱和羰基化合物(PhCOCH=CH2)
醛、酮等)在强碱下可形成α-碳负离子(强亲核
试剂),然后与羰基进行亲核加成反应生成β-羟
基醛(酮)(也称醇醛)
❖ β-羟基醛(酮)脱水生成α,β-不饱和醛酮
羟醛缩合反应
❖ pKa酸式离解常数:
丙烯≈38、乙炔≈25、丙酮≈20
❖ CH3→CH=CH2σ-π超共轭效应及+I效应利于亲电加 成
❖ CH3→CH=O σ-π超共轭效应及+I效应不利于亲核 加成
(C6H5)3P + CH3CH2Br C6H6
(C6H5)3P+CH2CH3Br季 鏻盐
(C6H5)3P+ CH2CH3Br- + C6H5Li
(C6H5)3P=CHCH3 + C6H6+ LiBr Wittig试剂
烃代亚甲基三苯基膦
❖ 季鏻盐酸性较弱,要强碱(C6H6Li)才能夺取其中的 质子
羰基化合物的反应

(79%)
醛、酮的混合缩合(又称交叉缩合),即克莱森-施密特缩合,要得到单一的产物,
其中一个组分应无 α-H。
C6H5CHO + CH3CHO 羰基组分 α-H组分
NaOH 20oC
OH
C6H5CH CH2CHO ∆
C6H5CH=CHCHO
由于大共轭体系的形成,使乙醛的自缩合反应成为次要的
O C6H5CHO +
R1CH H
OH
C NR2 R2
-H2O
R1CH C NR2 R2
R1CH C NR2 β R2
烯胺是重要的有机合成中间体,其 β-C 有很强的亲核性,具有碳负离子的性
质。
O
+ N H
N β
RX
NX R
H3O
O R
+ N
HH
RX 可以是 α-卤代酮,α-卤代酯,卤代苄,酰氯等。
烯胺的烃基化产物水解后即脱去仲胺,恢复原来的羰基。
CN O + H2C
NH4OAc
COOH
CN
C
+ H2O
COOH
CH3NO2 NaOH
CH=CHNO2 + H2O
CHO
CH3COCH2COOEt Et3N
COCH3 CH=C
COOEt
+ H2O
CHO + CH2(COOH)2 吡啶
CH=CHCOOH + CO2 + H2O
O2N
O2N
4.柏金反应,芳香醛与酸酐在相应的羧酸盐(钾盐或钠盐)催化下缩合生成 α,
H
O H3C
H
B
(过量 )
3.克诺文诺盖尔反应,醛酮与其它含活泼氢化合物的缩合。反应历程和羟醛缩
第六章 羰基化合物的反应

H+或OH-
OH R' + H2O R C OH R'
R
C
除甲醛、多卤代醛外, 除甲醛、多卤代醛外,其它醛的水合反应平衡偏向 左边。 左边。
(2)与ROH的加成 ) 的加成
H+
OCH2CH3 CH3CH OH
CH3CH=O + CH3CH2OH
半缩醛
CH3CH2OH, H+
CH3CH
OCH2CH3 OCH2CH3
6.5 碳负离子
O R C CH3
-
OH
O R C
CH2
} [
OH
动力学产物
O C CH2
O R C CH2
R
]
烯醇负离子 H+
碳负离子
烯醇负离子
O
热力学产物
R C CH3 + R C CH2
O
Br2
R C CH2Br
烯醇负离子是一个两位负离子,氧碱性强,碳亲核性强。 烯醇负离子是一个两位负离子,氧碱性强,碳亲核性强。
A C B + H Nu OH
H A
-A
A C OH
B
A C OH
Nu
A
B
Nu H A C B OH
A C B + HA OH
羰基质子化,可以提高羰基的反应活性, 羰基质子化,可以提高羰基的反应活性, 羰基质子化后,氧上带有负电荷,很不稳定, 羰基质子化后,氧上带有负电荷,很不稳定, π电子发生转移,使碳原子带有正电荷。 电子发生转移, 电子发生转移 使碳原子带有正电荷。 决定反应速率的一步, 决定反应速率的一步,是Nu -进攻中心碳原子的 一步。 一步。
有机化学人名反应B

有机人名反应 B需予科研家园资料整理Hofmann烷基化卤代烷与氨或胺发生烷基化反应,生成脂肪族胺类:由于生成的伯胺亲核性通常比氨强,能继续与卤代烃反应,因此本反应不可避免地产生仲胺、叔胺和季铵盐,最后得到的往往是多种产物的混合物。
用大过量的氨可避免多取代反应的发生,从而可得到良好产率的伯胺。
反应机理反应为典型的亲核取代反应(S N1或S N2)反应实例Hofmann消除反应季铵碱在加热条件下(100--200°C)发生热分解,当季铵碱的四个烃基都是甲基时,热分解得到甲醇和三甲胺:如果季铵碱的四个烃基不同,则热分解时总是得到含取代基最少的烯烃和叔胺:反应实例Hofmann重排(降解)酰胺用溴(或氯)在碱性条件下处理转变为少一个碳原子的伯胺:反应机理反应实例参见: Curtius 反应Lossen 反应Schmidt 反应Houben-Hoesch反应酚或酚醚在氯化氢和氯化锌等Lewis酸的存在下,与腈作用,随后进行水解,得到酰基酚或酰基酚醚:反应机理反应机理较复杂,目前尚未完全阐明反应实例Hunsdiecker反应干燥的羧酸银盐在四氯化碳中与卤素一起加热放出二氧化碳,生成比原羧酸少一个碳原子的卤代烃:X = Br , Cl , I反应机理反应实例Kiliani氰化增碳法糖在少量氨的存在下与氢氰酸加成得到 羟基腈,经水解得到相应的糖酸,此糖酸极易转变为内酯,将此内酯在含水的乙醚或水溶液中用钠汞齐还原,得到比原来的糖多一个碳原子的醛糖。
反应实例Knoevenagel反应含活泼亚甲基的化合物与醛或酮在弱碱性催化剂(氨、伯胺、仲胺、吡啶等有机碱)存在下缩合得到 不饱和化合物。
反应机理反应实例Knorr反应氨基酮与有 亚甲基的酮进行缩合反应,得到取代吡咯:反应实例Koble反应脂肪酸钠盐或钾盐的浓溶液电解时发生脱羧,同时两个烃基相互偶联生成烃类:如果使用两种不同脂肪酸的盐进行电解,则得到混合物:反应机理反应实例Koble-Schmitt反应酚钠和二氧化碳在加压下于125-150 ºC反应,生成邻羟基苯甲酸,同时有少量对羟基苯甲酸生成:反应产物与酚盐的种类及反应温度有关,一般来讲,使用钠盐及在较低的温度下反应主要得到邻位产物,而用钾盐及在较高温度下反应则主要得对位产物:邻位异构体在钾盐及较高温度下加热也能转变为对位异构体:反应机理反应机理目前还不太清楚。
有机化学反应机理(整理版)
1.A rndt-Eister 反应酰氯与重氮甲烷反应,然后在氧化银催化下与水共热得到酸。
反应机理重氮甲烷与酰氯反应首先形成重氮酮(1),(1)在氧化银催化下与水共热,得到酰基卡宾(2),(2)发生重排得烯酮(3),(3)与水反应生成酸,若与醇或氨(胺)反应,则得酯或酰胺。
反应实例2.Baeyer——--Villiger 反应反应机理过酸先与羰基进行亲核加成,然后酮羰基上的一个烃基带着一对电子迁移到—O—O—基团中与羰基碳原子直接相连的氧原子上,同时发生O—O键异裂。
因此,这是一个重排反应具有光学活性的3-——苯基丁酮和过酸反应,重排产物手性碳原子的枸型保持不变,说明反应属于分子内重排:不对称的酮氧化时,在重排步骤中,两个基团均可迁移,但是还是有一定的选择性,按迁移能力其顺序为:醛氧化的机理与此相似,但迁移的是氢负离子,得到羧酸。
反应实例酮类化合物用过酸如过氧乙酸、过氧苯甲酸、间氯过氧苯甲酸或三氟过氧乙酸等氧化,可在羰基旁边插入一个氧原子生成相应的酯,其中三氟过氧乙酸是最好的氧化剂。
这类氧化剂的特点是反应速率快,反应温度一般在10~40℃之间,产率高。
3.Beckmann 重排肟在酸如硫酸、多聚磷酸以及能产生强酸的五氯化磷、三氯化磷、苯磺酰氯、亚硫酰氯等作用下发生重排,生成相应的取代酰胺,如环己酮肟在硫酸作用下重排生成己内酰胺:反应机理在酸作用下,肟首先发生质子化,然后脱去一分子水,同时与羟基处于反位的基团迁移到缺电子的氮原子上,所形成的碳正离子与水反应得到酰胺。
迁移基团如果是手性碳原子,则在迁移前后其构型不变,例如:反应实例4.Birch还原芳香化合物用碱金属(钠、钾或锂)在液氨与醇(乙醇、异丙醇或仲丁醇)的混合液中还原,苯环可被还原成非共轭的1,4-环己二烯化合物。
反应机理首先是钠和液氨作用生成溶剂化点子,然后苯得到一个电子生成自由基负离子(Ⅰ),这是苯环的л电子体系中有7个电子,加到苯环上那个电子处在苯环分子轨道的反键轨道上,自由基负离子仍是个环状共轭体系,(Ⅰ)表示的是部分共振式.(Ⅰ)不稳定而被质子化,随即从乙醇中夺取一个质子生成环己二烯自由基(Ⅱ).(Ⅱ)在取得一个溶剂化电子转变成环己二烯负离子(Ⅲ),(Ⅲ)是一个强碱,迅速再从乙醇中夺取一个电子生成1,4—环己二烯。
有机化学(下)课件10.3α-氢原子的活泼性

(B) C=O [H] CH2
Zn-Hg/HCl (Clemmensen还原法,酸性介质) NH2NH2,NaOH,三乙二醇。醚 (黄鸣龙还原法,碱性介质)
(b.p约200 C)
(1)催化加氢
在金属催化剂Pt、Ni、Pd、Cu等存在下,醛或酮与 氢气作用,发生加成反应,分别生成伯醇或仲醇。
CH3CH2CH2CHO
酮-烯醇互变
这种能够互相转变而又同时存在的异构体叫互变异构体。 酮和烯醇的这种互变异构体叫酮-烯醇互变异构.
酮
烯醇
在微量的酸和碱的存在下,酮和烯醇互相转变,很快就能达到动态平衡
含有一个羰基的结构较简单的醛、酮的 烯醇式在互变异构的混合物中比例很少。
β-二羰基化合物
由于共轭效应的影响,烯醇式的能量降低,增加其稳 定性,所以在互变异构的混合物中含量要高的多。
二丙酮醇
含有α-氢原子的酮也能发生类似的羟醛缩合反应, 最后生成α,β-不饱和酮。
O 2 CH3 C CH3
Ba(OH)2 慢
bp: 56oC
OH
O
CH3 C CH2 C CH3
CH3
二丙酮醇
bp: 164oC
I2
CH3
O
C CH C CH3
H3C
异丙叉丙酮
Ba(OH)2
丙酮
Soxhlet 提取器
O
10% NaOH
CH3 C H + CH2 C H
α
5℃
CH3
CH
β
CαH2C
H
β-羟基醛
一分子醛的α-氢原子加到另一分子醛的羰基氧原子上; 其余部分通过α-羰加到羰基的碳原子,生成β-羟基醛。
I)反应机理
羰基化合物的反应

K=38
ECt H3CHO + H2O
ECt H3CHO CO
+ HCN
NaHSO3
CCNH3CH(OH)2
CH3CHOH
K≈ 1 K≈ 104
Et2CCONH K=4× 10-4
Et
SO3Na
第14页/共104页
7.2 羰基加成反应
与氢氰酸的加成 与亚硫酸氢钠的加成 与金属有机物的加成
与水的加成 与醇的加成 与胺及氨的衍生物的加成 与格氏试剂的加成
CH3
S
C6H13 H
C
CH3
COOH
R
在较高温度时, 转化 很快达到平衡, 外消 旋化
第38页/共104页
2.碳负离子的生成 C-H键的异裂
C H+ B
共轭酸
碳原子酸
C + HB
共轭碱
第39页/共104页
HC CH
NaNH3 液 NH3
HC CNa
NH3
Ph3C H
NaNH3 液 NH3
Ph3CNa
能与羰基氧H形成羟基的基团时
Ph Ph
空OH间RL亲OC阻H核3S碍试CY小HO剂3L的i从N一uP含Ph边h氢H对键O羰环基LOCi的-HC加3HRL3成OS
YH
HO Nu
Ph
OH Ph
CH3 CH3
第30页/共104页
Cornforth规则
当酮的α-手性碳原子上连卤素原子 由于卤原O子与羰基的偶极作用O
9
11
10
第47页/共104页
杂化作用
C-H键中碳的s成分越多 H越易以质子形式解离
H C CH
H2C CH2
羰基化合物的反应

O
O
O
O > RCCH3 > RCR' > ArCR
羰基具有平面结构,亲核剂可从平面的两边进攻羰基碳原子。如果羰基平面
两边的空间条件不同,进攻试剂将主要从空间阻碍较小的一边进攻。
樟脑分子中的桥环不能翻转,有碳桥的一边位阻很大。因此,负氢从位阻小
的碳桥对面进攻羰基碳。
O
1.NaBH4 2.H3O
H
OH +
OH H
(主)
若 Nu 体积很小时,3,5-位碳上直立氢的干扰趋于缓解,但加成过程中新生成的
C-O 单键与 2,6-位平伏 C-H 健之间产生的张力则转化为主要的影响因素。二者的
距离越接近,产生的张力越大。
b
HO
t-Bu
+ HBH3Na H
a
H3O a t-Bu
H3O t-Bu b
OH H
H (次) H
曼尼赫反应在合成上的重要性是由于曼尼赫碱受热立即分解生成 α、β-不饱和羰
NaOH
O CH
+ H2O
HCH=O + CH3CHO (过量 )
无 a-H 羰基活性大
OH
HCHO
(HOCH2)3C-CHO
C(CH2OH)4 + HCOOH
歧化反应
三次羟甲基化
季戊四醇
按碰撞理论,容易理解分子内的羟醛缩合进行得更快。特别重要的是鲁宾逊扩环 反应,它包括麦克尔加成和分子内的羟醛缩合两步:
C O + ROH
OH C
OR
ROH / H3O
半缩醛(酮)
缩羰在有机合成中是重要的保护基。例如(1):
OR C
OR
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
1,3-二羰基化合物的氧二氟甲基化反应
目前通过二氟卡宾试剂对O-H键的插入构建OCF<sub>2</sub>H基团的方法应用最广,因此许多优秀的二氟宾试剂相应地发展起来。
这些试剂产生二氟卡宾中间体的方式各有不同,多数需要加强碱才能引发反应,有的需要高温或低温条件下方可顺利产生二氟甲基中间体,还有些反应条件苛刻,甚至用到高毒,低沸点一些环境不友好试剂。
不仅如此,这些二氟卡宾试剂实现O-H键插入反应时,底物大多局限于酚类和醇类,只有几例实现了对于1,3-二羰基化合物的氧二氟甲基化。
相比之下,我们课题组发展的二氟亚甲基鏻内盐
(Ph<sub>3</sub>P<sup>+</sup>CF<sub>2</sub>CO<sub>2</sub><sup>-</sup> ,PDFA),作为二氟卡宾试剂,在温和的条件下,不需要加入额外的添加剂,就可以高效地产生干净的二氟卡宾中间体,并实现一系列二氟卡宾相关的反应。
因此,利用这个干净温和的二氟卡宾源,我们发现1,3-二酮化合物与二氟亚甲基鏻內
盐PDFA配比1:2,对二甲苯2 mL作溶剂,氮气环境下,反应温度60 <sup>o</sup>C,反应时间3 h,反应效果最佳。
通过对一系列的1,3-二酮的底物普适性拓展,13个常见的1,3-环己二酮、1,3-环戊二酮底物,在内盐的作用下都可以以良好到优秀的产率得到1,3二酮的氧二氟甲基化的产物。
但是该反应还存在一些不足的地方,对于链状的1,3-二羰基底物效果不好,在二氟亚甲基鏻内盐的作用下,只有较少的产物。
总的来说该反应操作简单,非常高效,条件温和,官能团容忍性高,产率中等到优秀,有望用于在合成的后期,向具有环状1,3-二酮类结构的复杂分子中引入二氟甲基。