RNA IP流程
RNAi的实验设计及使用步骤

RNAi 的实验设计及使用步骤RNA 干扰(RNA interfenng , RNAi )现象是由与靶基因序列同源的双链 RNA( double-stranded RNA , dsRNA )引发的广泛存在于生物体内的序列特异性 基因转录后的沉默过程.细胞中的核糖核酸酶III 家族成员之一的,dsRNA 特异 性的核酸酶Dicer 将dsRNA 裂解成由21・25个核甘酸组成的小干扰RNA(small interfenng RNA , siRNA),随后siRNA 作为介寻子引起特异性地降解相同序列的 niRNA ,从而阻断相应基因表达的转录后基因沉默机制. B. RNAi 实验流程 siRNA 的转染 (siRNA Transfection) siRNA 设计A.哺乳动物siRNA 设计基因抑制的有效性很大程度取决于目的基 因序列的选择.目的序列可以随机选择也可以通过在目的基因的不同区域上测试 不同的序列以决定何种序列是最有效的.哺乳动物siRNA 设计需要注意的几个 方面1 .从转录本(mRNA )的AUG 起始密码开始,寻找“AA 〞二连序列,并记 下其31端的19个碱基序列,作为潜在的siRNA 靶位点.正义链和反义链都采 用这19个碱基(不包括AA 重复)来设计.2 .防止在起始密码子或无义区域附 近选择目的序列.3 . siRNA 序列的GC 含量应为30%-60%左右.4 .在设计 siRNA 时不要针对5和3端的非编码区(untranslatedregions , UTRs ),原因是 这些地方有丰富的调控蛋白结合区域,靶基因确状 (TargetGene Identification) siRNA 设计 CsiRMA Design) siRNA 的规备 (siRNA Generation)功能研究(Function Research) RNAI 结果检测 1/ f (Measure Knockdown)匕 H而这些UTR结合蛋白或者译起始复合物可能会影响siRNA核酸内切酶复合物结合mRNA从而影响siRNA的效果. 5.将挑选的序列在公共数据库中进行比拟以保证目的序列与其它基因没有同源性.将潜在的序列和相应的基因组数据库〔人,或者小鼠,大鼠等等〕进行比拟, 排除那些和其他编码序列/EST同源的序列.例如使用BLAST〔 /BLAST/ 〕o 7 .选出适宜的目标序列进行合成.通常一个基因需要设计多个靶序列的siRNA,以找到最有效的siRNA序列.8 .阴性对照: 一个完整的siRNA实验应该有阴性对照,作为阴性对照的siRNA应该和选中的siRNA序列有相同的组成,但是和mRNA没有明显的同源性.通常的做法是将选中的siRNA序列打乱,同样需要检查它和其他基因是否具有同源性.9 .有结果显示,UU结尾和dTdT结尾的siRNA在效果上没有区别,由于这个突出端无需和靶序列互补.合成siRNA时可直接提供以AA打头的21个碱基序列.siRNA转染的方法哺乳动物转染的常见方法有:磷酸钙共沉淀、电穿孔法、DEAE葡聚糖和polybrene.机械法〔例如,显微注射和基因枪\阳离子脂质体试剂,其中阳离子脂质体试剂转染法是目前最常用的转染方法〔具体转染方法可参考In\'itrogen Lipofectaniine 2000,.贴壁细胞转染程序以24孔板为例,假设要检测基因沉默或者过表达效果,推荐最低RNA oligo终浓度为50nM 〔不同细胞可以上下优化2-4倍浓度〕;遵循以下操作方法可以高效地将RNA oligo转染贴壁和悬浮培养的多种真核细胞.但是对某些特殊的细胞系和培养条件,或特殊应用等,也需要单独特别优化.1 .细胞铺板转染时细胞密度:一般来说,当细胞密度到达60・80%时进行转染可以取得较高的转染效率.然而,不同细胞的最适转染空度都不尽相同.因此在初次转染某种细胞时,可以通过预实验先确认该细胞最正确的状态及转染密度.2.复合物的制备及转染1〕使用前将GP・transfect・Mate转染试剂放置于室温中,使用前轻轻混匀;2〕在1.5 nil无菌离心管中参加50 口无血清培养基或OPTI-MEM ,并添加适量的转染试剂,用移液器轻轻混匀,室温静置5 min ;3〕同时在另一1.5 ml无菌离心管中参加50可无血清培养基或OPTLMEM,并添加适量的RNAoligo/DNA 〔参见表2〕,用移液器轻轻混匀,室温静置5 min.4〕将GP・tl•ansfect・Mate・培养基混合物滴加至RNA oligo/DNA.培养基混合物中,用移液器轻轻混匀,室温静置15-20 min后,立即转染.注:复合物尽量在60 niin内使用,并且GP-tiansfect- Mate.培养基混合物和RNAoligo/DNA・培养基混合物的混合次序非常重要,切勿颠倒.3.转染过程1〕趁静置时,给24孔板换液,每孔换上400 m预热的新鲜培养基;2〕将100卜11转染混合物参加孔中,终体系为500可.加完后将板轻轻晃动以使复合物均匀分布;3〕37℃静置培养细胞,4 -6h换成完全培养基.2 4-72 h检测mRNA表达,4 8-96 h检测蛋白表达.13操作流程〔24孔板〕以24孔板为例,假设要检测基因沉默的效果,推荐最低siRNA终浓度1011M :假设要在显微镜下从荧光强度看转染效率,因荧光信号被检测到需要一定的敏感度,推荐siRNA终浓度50iiM o遵循以下操作方法可以高效地将siRNA转染贴壁和悬浮培养的多种真核细胞.A.细胞铺板贴壁细胞数量:在转染实验之前的18・24个小时,在每个孔的500m 生长培养基中参加1.535x104个细胞〔保证转染时细胞密度在30-50% \ 悬浮细胞数量:转染实验的当天,在500卜d生长培养基中参加1-2x105个细胞. 注意:转染时应保证细胞没有过度生长或处于休止期.B.转染复合物的制备1.将siRNA-Mate转染试剂放置于室温中,使用前轻轻混匀;2.将100可OPTI-MEM 或无血清培养基参加无菌EP管中;3.在上述含有无血清培养基的EP管中参加lOpmol (140ng)待转染的siRNA ,并充分混匀.随后在管子中参加2可siRNA・Mate转染试剂,快速涡旋10s使其完全混匀;4.室温静置10分钟,使siRNA 和转染试剂形成转染复合物.静置时间不要超过30min°C.转染过程1.趁静置时, 给24孔板换液,每孑眼上0.5ml预热的新鲜培养基;2.将静置完毕的100P l转染复合物参加孔中,终体系为600M , siRNA 终浓度为16.7iiM o加完后将细胞培养板轻晃摇匀.3.37.(3静置培养细胞,检测基因沉默水平可在24-72h内收样检测mRNA ( RT-PCR实验等)或48-9611内检测蛋白(Western Blot实验等工常见问题L针对人体基因设计的siRNA对其他物种是否也有效?一般siRNA都具有物种特异性,很少与其他物种有相同的靶位点,所以针对人体基因设计的siRNA不会沉默其他物种的同源序列.然而,也有研究说明siRNA 经过特异性设计后能对两个或两个以上的物种有效,这需要仔细进行siRNA设计和生物信息学分析.2.你们提供的siRNA是怎样装运的?如果在常温下放置了一个星期,还有效吗?siRNA是冻干粉包装的,在常温下运输.这些冻干的样品在室温下能稳定保存2-4个星期,所以放置一个星期不会影响其沉默效果.但我们建议您收到样品后最好保存于-2(rc或-7(TC有霜冷冻箱中.3.在体外实验中,需要多少量的siRNA ?我们建议您用于实验的siRNA的浓度为lOOnM.1 nmol siRNA的量对于一个24孔板或是96孑版的实验已经足够了.4.用100nM的siRNA转染时只得到50%沉默效率,我可以将siRNA 的浓度增加到200nM甚至400nM吗?增加siRNA的浓度一般不能改良沉默效率.高浓度的siRNA将可能导致去靶作用和对细胞的毒性.siRNA的高基因沉默效率来自于合理的设计,在100nM甚至更低的浓度都可能有75%的沉默效率.另外,低的转染效率会导致低的沉默效率,建议您更进一步优化siRNA的导入条件.5.24定量RNA的公式是什么?研究者可以用Beer法那么定量RNA :吸光度(260nm)=(摩尔消光系数)*(浓度)*(路径氏度,cm).为了便于理解,等式变为:浓度二(吸光度,260nm)/[(摩尔消光系数)*(路径氏度,cm)].当使用一个标准的10mm比色皿时,在公式中路径氏度这个变量等于1.6m浓度为10uM的siRNA溶解液中含有多少卜唱的siRNA ?首先计算含有多少nmol 的siRNA : a.等式:?nmol=(6|il)(10uniol/L) ; b. 单位换算:?nmol= (6|il)( 10|imol/L)( 1 L/l ,000,000pl)(1,000nmol/jimol) ; c.答案:?nmol=0.06nmol.然后利用siRNA 的平均分子量13,300g/mol)*nmol 变为pg : a. 等式:? Hg=(0.06miiol)( 13,3 OOg/mol) ; b. 单位换算:?Ng=(0.06nmol)(13,300g/mol)(lmol/L000,000,00 Omnol)(l,OOO,OOO|.ig/g) ; c.答案:?卜唱=0.798约0.8卜唱.所以6m浓度为10gM 的siRNA溶解液中含有0.8卜唱的siRNA.我需要浓度为20uM的样品,如何计算重悬siRNA缓冲液的量?样品浓度的计算如下:(siRNA的量,nmol)/(重悬体积,卜il)=样品浓度…mol/L.在解答前应先统一单位,保证单位可以抵消.例如:您购置了20nmol的siRNA , 想溶解为5 OpM的样品.可按以下方法计算重悬缓冲液体积:a.等式:(20 mnol)/?|il=50umol/L ;b.解答未知量:?卜il=(20nmol)(lL/50umol) ;c.单位换算:?m=(20nmol)(lL/50 卜imol)(lNmol/l,000nniol)(l,000,000Nl/lL) ; d.答案:?j.d=400j.d o因此,应该使用400|.d缓冲液去重悬20nmol的siRNA ,溶解后为50pM的样品.一定需要阴性对照吗?是,阴性对照是RNA干扰实验中不可缺少的.由于在超过200nM的浓度下,siRNA有可能会导致非特异性的压力反响,在实验体系中必需设置阴忸寸照.它能够帮助我们确认基因表达水平的降低是否是序列特异性的RNAi的结果.由于siRNA的合成方法和工艺以及转染试剂等因素可能导致广泛的基因沉默现象.如果没有阴性对照,研究人员很可能错误地将广泛的、非特异性基因沉默当作由RNAi引起的基因特异性沉默.常用的阴性对照有哪些类型?常用的阴性对照大体分为两种,一种是使用通用阴性对照序列,该序列已经被上千篇文章使用;另一类是使用和靶基因siRNA打乱序列的siRNA作为阴性对照,这样的阴性对照和通用序列相比拟,一方面合成是根据定制产品价格合成的,另一方面,由于打乱序列是没有经过验证的序列,有可能会产生.ff target 现象.因此,除非有非常特殊的要求,最好使用通用序列作为阴性对照.在阴性对照体系中和实验体系中观察到同样的实验结果,这是什么原因?这个结果充分说明了设置阴性对照的必要性.该结果说明您所观察到的表型不是序列特异性knockdown产生的表型,您需要降低siRNA的工作浓度.为什么说阳性对照在RNA干扰实验中很重要?阳性对照作为一个实验系统检查是很重要的.也就是说,当您看到siRNA 阳性对照的预期实验结果时,您能保证在您的实验方法中您的转染、RNA提取物和检测方法是可靠的.通常最好的阳性对照是内在的对照.你们能提供预先合成的siRNA对照么?可以.提供许多预先合成的用于RNA干扰实验的对照双链.用于对照的siRNA的最正确浓度是多少?阳性对照和阴性对照的siRNA的浓度都应该与基因特异性的siRNA的浓度相同.在siRNA上进行标记的最正确位点在哪里?反义链的51端标记会影响siRNA的沉默活性,所以不推荐标记这个位点.在其他三个末端进行标记对沉默活性几乎没有影响.有研究说明正义链的51端标记是最有效的化学合成位点.荧光标记的siRNA是不是可以作为良好的对照?如何检测荧光标记的siRNA ?已经有为数不少的实验室研究过荧光标记的siRNA的荧光检测结果和knockdown效率之间的正相关关系.荧光标记双链是最常用的优化转染条件的方法.可以用流式细胞仪或者荧光共聚隹显微镜检测标记的siRNA.FAM的最大吸收率和I双射率各在哪个波段?FAM在495nm处有最大吸收率,在520nm处有最大发射率.如何筛选转染试剂?好的转染试剂有以下特点:1.对siRNA有较高的转染率;2.对细胞的毒性小.在mRNA水平检测siRNA导入的效果比用看家基因的siRNA阳性对照检测更为准确.转染过程中发现大量细胞死亡,我应该如何处理?如果出现细胞大量死亡,意味着您的转染条件仍然需要优化:转染条件的优化一般包括以下几个方面:1 .调整转染试剂的浓度;2.在转染后适当的时间内更换无转染试剂的培养液;3.调整细胞的生长状态:一般处于良好生长状态的细胞对转染试剂就有更好的耐受性;4.调整转染试剂和siRNA的比例:如果同一管转染试剂在不同实验中对细胞的毒性有差异,一般说来应该是实验过程本身带来的差异:如果已经做了以上几项工作,转染效率仍然得不到提升,建议您更换一种转染试剂.如何将siRNA导入到细胞内?使用什么样的转染方法,很大程度上取决于您使用的细胞系:1.贴壁的、易转染的细胞,我们推荐使用Lipofectamin 2000或siRNA Mate转染试剂.2.悬浮的或原代的细胞,我们推荐使用电击转化方法;3.电击转化效率仍然很低的细胞, 需要选择载体系统.吉玛基因除提供化学合成的siRNA以外,还提供完整的载体系统.我的课题主要是研究细胞凋亡相关基因,我手上有一些新基因需要使用RNA干扰进行深入研究,我该如何选择对照系统?用RNAi进行pathway研究, 是RNAi主要应用之一.在这样的课题中,对照系统的选择尤其重要.已经发表的和已经验证的siRNA为RNA干扰研究提供了系统性对照.用RNAi进行细胞凋亡相关基因的研究,以往已经有众多的文献.我刚刚开始接触RNA干扰技术,从文献上看,不同细胞应该选用不同的转染试剂,不同的研究目的,应该使用不同的siRNA ,我的经费有限,如何有效确定我的研究体系?在开始RNA干扰研究之初,需要确定以下几个方面:使用化学合成的siRNA 还是使用载体构建shRNA ?我们推荐您使用化学合成的方法来筛选有效的目的片段,然后把您筛选出来的目的片段插入到载体,进行抗性筛选,得到稳定表达的细胞株,再做长期研究.反义核酸和RNAi有何差异?反义核酸和RNAi从作用原理到使用的范围都有很大差异.这里只能做一个简单的介绍:从原理上说,反义核酸是一段与靶基因配对的单链DNA或类似DNA的片段与靶基因结合,结果阻止靶基因的转录或是译,以往的研究说明在反义核酸的研究中,序列的有效性和有效序列的非特异性往往有比拟高的正相关性,这就在一定程度上限制了反义核酸的应用.RNAi的作用机理是双链的RNA进入细胞内,导致靶基因的切割和降解.siRNA的序列可以选择在高度特异针对靶基因的位置,也就为RNAi作为药物研究提供了高效、低副作用的空间.自从RNAi技术问世以来,国外很多专业从事反义核酸研究的公司都纷纷转向RNAi研究,ISIS是一个非常就有代表性的例子.他们认为,如果使用反义核酸可以进行的研究,以及反义核酸可以涉及的研究领域,RNAi均可以毫不示弱的开展,并且应该会得到更加令人满意的结果.我使用合成的双链DNA克隆载体,但是测序结果不正确,是DNA合成的问题,还是其他问题?首先,质量良好的、并且是退火状态良好的dsDNA是克隆成功的关键因素.一般情况下, 需要拟2不止一个克隆测序〔通常是2个以上〕,如果两个克隆的序列都是错误的,并且错误的碱基是相同的,那根本可以确定是DNA oligo合成的问题.如果两个克隆的不同碱基发生错误,有可能是合成片段本身的问题,也有可能是克隆过程造成的问题,可以再多挑1-2个克隆测序确证.大多数情况下,两个阳性克隆中,一般至少会有一个克隆是正确的,这种个别碱基在个别分子中的错误是合成和克隆过程共同造成的,但一般发生的几率较低.我方案使用RT-PCR检测基因knockdown结果,之前应该注意哪些问题?我们推荐RT-PCR的引物应该设计在target位置的两侧,而不是同侧.目前已经知道的siRNA介导的基因knockdown机制是RSIC先介导靶mRNA的切割,切割导致靶mRNA的降解,由于切割和降解可能具有不同的时间点,因此, 设计于同侧的PCR 引物可能导致假阴性结果或knockdown效率的低估.另外, RT-PCR结果会由于靶基因mRNA降解时间不同、细胞内靶基因mRNA丰度不同而导致假阴性结果,特别对于丰度较高的基因,推荐使用的检测方式包括量PCR、Northern检测、Western 检测等.这些方法会更加真实的反映RNAi的结果.开始体内实验前需要注意什么问题?体内实验设计包含动物模型的选择、给药途径、剂量和给药次数等等.siRNA使用的数量及浓度主要取决于给药靶点的性质,诸如肿瘤的类型,组织的类型,靶基因表达水平,动物模型个体大小等等,针对不同的研究模型,最好先查阅相关资料.在体内研究中,最好的给药途径是什么?给药频率是多少?寡核昔酸可以通过大剂量给药或使用ALZET微小泵持续给药.大剂量给药时要慎重,由于己有研究说明:一些毒性与寡核昔酸反义链的浓度有关,快速给药时可能会导致动物下肢瘫痪或致死,所以给药时要注意观察.很多己发表的论文实验是通过尾静脉给药的.任何给药方式都需要优化,以保证最正确的导入方式和动物的健康.一般拿静脉注射来说,每天注射一到两次,连续注射一到两周.siRNA是否可以用于动物水平的实验?siRNA经过严格HPLC纯化.已经被用户通过局部注射用于小鼠的体内实验.并且得到满意的实验结果.小鼠体内实验需要多少siRNA ?迄今为上还没有明确的siRNA体内实验使用量的计算方式,在实验前最好先从文献中查询是否已经有相关的文章发表.对于常规实验,一般使用100可的注射体积,浓度为10 to 500卜iM.原先用antisense的研究说明,当剂量大于20 mg/kg/day(416mM)时.会观察到明显的毒性.最好针对您的实验绘制剂量反响由线.常规情况下,给药剂量可以以〜5mg/kg/day[〜7.7nmol/day or 100mMper day]作为优化起点.需要注意的是,这个剂量只是一个预实验的起点,最后的给药剂量取决于动物模型、靶基因、靶组织和给药方式等因素.沉默效果不理想,应该如何处理?最常见的影响沉默效果的两个原因是:转染效率低?口siRNA序列设计的效果不理想.如果您初次使用siRNA或采用了新的细胞系,并发现沉默效果不佳, 我们建议您对转染效率进行检测,并选择优化转染条件.如果您已经对实验转染条件进行优化但是问题依然存在,我们建议您换用另一种转染试剂或是采用其他技术,这也许能提升转染效率.如果已经提升了转染效率但是沉默效果仍然未达到要求,可能是由于siRNA序列设计的效果不理想.。
RNAi的实验原理和操作实用技术Word版

RNAi的实验原理和操作实用技术几十年来生物学上最重要的进展,也许是关于RNA分子能调节基因表达的发现。
RNA干涉(RNAi)是指双链RNA分子使基因表达沉寂的现象,是在线虫中发现的,在 1998年的一篇Nature论文中被公诸于众。
此后,科学家们明白,RNAi还有其他形式,它既是一种了解基因功能的强大工具,又是很多生物的基因组所采用的一种在演化上来讲很古老的防卫方法。
RN Ai肯定有很多新用途,RNAi还可能具有一些仍然有待去发现的天然功能。
RNAi实验原理与方法近年来的研究表明,将与mRNA对应的正义RNA和反义RNA组成的双链R NA(dsRNA)导入细胞,可以使mRNA发生特异性的降解,导致其相应的基因沉默。
这种转录后基因沉默机制(post-transcriptional gene silencing, PTGS)被称为RNA干扰(RNAi)。
一、RNAi的分子机制通过生化和遗传学研究表明,RNA干扰包括起始阶段和效应阶段(inititation and effector steps)。
在起始阶段,加入的小分子RNA被切割为21-23核苷酸长的小分子干扰RNA片段(small interfering RNAs, siRNAs)。
证据表明;一个称为Dicer的酶,是RNase III家族中特异识别双链RNA的一员,它能以一种ATP依赖的方式逐步切割由外源导入或者由转基因,病毒感染等各种方式引入的双链RNA,切割将RNA降解为19-21bp的双链RNAs(siRNAs),每个片段的3’端都有2个碱基突出。
在RNAi效应阶段,siRNA双链结合一个核酶复合物从而形成所谓RNA诱导沉默复合物(RNA-induced silencing complex, RISC)。
激活RISC需要一个ATP依赖的将小分子RNA解双链的过程。
激活的RISC通过碱基配对定位到同源mRNA转录本上,并在距离siRNA3’端12个碱基的位置切割mRNA。
rna-ip条件 -回复

rna-ip条件-回复RNAi(RNA干扰)条件:从原理到应用一、简介和背景RNA干扰(RNA interference,简称RNAi)是一种在分子水平上抑制基因表达的机制,通过介导RNA分子的降解或转录后基因沉默,对基因功能进行干扰。
它是由安德鲁·费尔南德斯和克雷格·梅洛共同发现,并在这一发现之后逐渐引起了广泛的关注和研究。
RNAi被广泛地应用于基因功能研究、基因治疗和农业生物技术等领域,为科学家们提供了一种强有力的工具。
二、RNAi的原理RNAi的原理基于双链RNA分子的存在。
双链RNA分子可由外源性RNA(例如合成siRNA)或内源性RNA(例如miRNA或siRNA)产生。
这些双链RNA分子能够与靶基因的mRNA结合,并通过多种途径抑制基因表达。
1. siRNA的生成和靶向siRNA具有两个链段,即导引链和被释放链。
导引链与靶基因的mRNA相互配对,从而诱导被释放链降解,抑制基因表达。
siRNA可通过人工合成或由内源性RNA产生。
2. miRNA的生成和调控miRNA是一类与内源性siRNA相关的小分子RNA,与靶基因的mRNA部分配对,使其降解或转录后抑制。
miRNA由pri-miRNA分子生成,并通过多步骤的加工过程形成成熟的miRNA。
三、RNAi在基因功能研究中的应用1. 基因沉默通过RNAi实现基因的沉默,使研究人员能够研究特定基因在生理和病理过程中的功能。
利用siRNA或miRNA技术,可以选择性地抑制目标基因,研究其对细胞活动和疾病发展的影响。
2. HTP筛选高通量筛选技术(HTP)结合RNAi,可在大规模样本中快速有效地检测多个基因的功能。
这种方法广泛应用于药物发现和靶向治疗的研究中。
3. 基因治疗RNAi还具有潜在的基因治疗应用价值。
通过靶向特定的疾病相关基因,如癌症基因或遗传性疾病基因,可实现定向治疗,并减少对健康细胞的伤害。
四、RNAi在农业生物技术中的应用1. 抗虫性和抗病性作物利用RNAi技术,可以通过抑制具有害影响的昆虫或病原体基因的表达,使植物获得更强的抗虫性和抗病性能力。
RNAiso Plus提取总RNA
-1-
●实验操作
1. RNAiso Plus的使用量情况如下。 样品量
10 cm2的贴壁培养细胞 107的悬浮培养细胞 100 µl 的白细胞 50~100 mg 的普通组织样品 50~100 mg 的特殊组织样品(肝、脾、骨及软骨等) 15~30 mg 的植物材料(多糖和多酚含量不高的)
RNAiso Plus 使用量(ml) 1
●制品组成
RNAiso Plus*
100 ml
* RNAiso Plus 中含有强变性剂,应避免与皮肤、衣物等接触。若 不小心接触到眼睛或皮肤时,请立即到医院进行处理。
【试剂盒之外所需准备试剂】
◆ 氯仿 ◆ 异丙醇 ◆ 75%乙醇(DEPC 处理水配制) ◆ RNase-free 水(制备方法:使用 RNase-free 的玻璃瓶,向超纯水中加入 DEPC 至终浓度 0. 1%(v/v),
过夜搅拌后,高温高压灭菌。)
●保存和运输
1. 可以在室温下保存,建议在 2-8℃下避光保存,效果更佳。 2. 可以在常温下运输。
●RNA 提取实验前的准备
RNA 制备的关键是要抑制细胞中的 RNA 分解酶和防止所用器具及试剂中的 RNA 分解酶的污染。因此,在 实验中必须采取以下措施:戴一次性干净手套;使用 RNA 操作专用实验台;在操作过程中避免讲话等等。 通过以上办法可以防止实验者的汗液、唾液中的 RNA 分解酶的污染。
-2-
4. RNA 沉淀的清洗。 小心弃去上清,缓慢地沿离心管壁加入 75%的乙醇 l ml(切勿触及沉淀),轻轻上下颠倒洗涤离心管 管壁,12,000 g 4℃离心 5 分钟后小心弃去乙醇(为了更好地控制 RNA 中的盐离子含量,应尽量除净 乙醇)。
5. RNA 的溶解。 室温干燥沉淀 2~5 分钟(不可以离心或加热干燥,否则 RNA 将会很难溶解,有关 RNA 溶解可以参考 Troubleshooting 中的相关说明),加入适量的 RNase-free 水溶解沉淀,必要时可用移液枪轻轻吹打 沉淀,待 RNA 沉淀完全溶解后于-80℃保存。
细胞RNA-IP protocol
2hr;
5.使用GROSS buffer洗涤,buffer中加入mg/ml yeast tRNA和1 U/ml SUPERase inhibitor, protease ainhibitor cocktail 1:100,梯度高盐洗涤,第一遍150mm的NaCl 4度rotate 5min,离心3000rpm, 3min;而后每洗一遍NACL浓度提高100mm,直至550mm,550mm洗两遍;
6.取1/10 beads用做western,剩余用于TRIZOL抽提RNA;
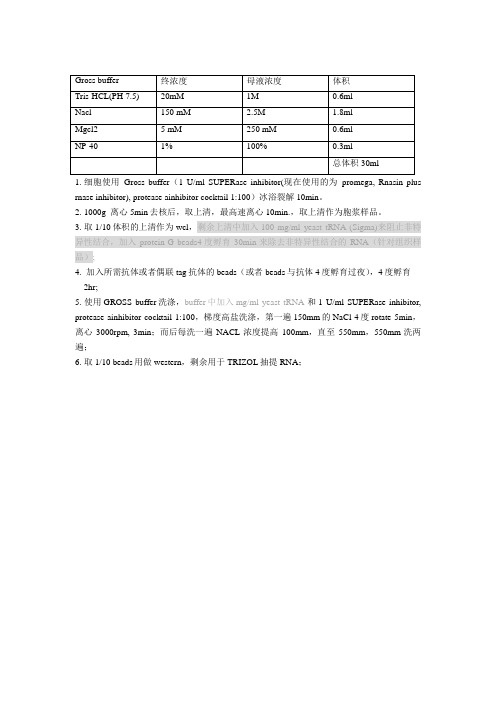
Gross buffer
终浓度
母液浓度
体积
Tris-HCL(PH 7.5)
20mM
1M
0.6ml
Nacl
150 mM
2.5M
1.8ml
Байду номын сангаасMgcl2
5 mM
250 mM
0.6ml
NP-40
1%
100%
0.3ml
总体积30ml
1.细胞使用Gross buffer(1 U/ml SUPERase inhibitor(现在使用的为promega, Rnasin plus rnase inhibitor), protease ainhibitor cocktail 1:100)冰浴裂解10min。
2.1000g离心5min去核后,取上清,最高速离心10min.,取上清作为胞浆样品。
3.取1/10体积的上清作为wcl,剩余上清中加入100 mg/ml yeast tRNA (Sigma)来阻止非特异性结合,加入protein-G beads4度孵育30min来除去非特异性结合的RNA(针对组织样品);
rna结合蛋白免疫沉淀原理

rna结合蛋白免疫沉淀原理免疫沉淀(Immunoprecipitation,IP)是一种常用的研究蛋白质相互作用的实验方法。
通过使用特定的抗体,将与目标蛋白结合的蛋白质沉淀下来,然后对沉淀的蛋白质进行检测和分析。
在Ma结合蛋白的免疫沉淀实验中,通常使用标记的Ma蛋白作为抗原,与细胞提取物中的蛋白质进行结合,然后通过免疫沉淀技术将结合的蛋白质沉淀下来,最后对沉淀的蛋白质进行检测和分析。
一、实验原理免疫沉淀实验的基本原理是利用抗体与抗原之间的特异性结合作用,将与目标蛋白结合的蛋白质沉淀下来。
抗体通常是一种能够识别并结合特定抗原的蛋白质,通过抗体的结合,将抗原-抗体复合物从溶液中沉淀下来,从而达到分离和纯化抗原的目的。
在Ma结合蛋白的免疫沉淀实验中,首先需要制备标记的Ma蛋白,并将其作为抗原添加到细胞提取物中。
细胞提取物通常包含大量的蛋白质,其中一些蛋白质可能与Ma蛋白结合。
通过抗原-抗体之间的特异性结合作用,将与Ma蛋白结合的蛋白质沉淀下来。
二、实验步骤1. 制备标记的Ma蛋白:首先需要将目标Ma蛋白进行标记,通常使用生物素、荧光染料等标记剂进行标记。
标记后的Ma蛋白具有更高的特异性和灵敏度,可以用于后续的免疫沉淀实验。
2. 细胞提取物的制备:从待研究的细胞中提取蛋白质,通常使用细胞裂解液将细胞溶解,然后通过离心、洗涤等步骤去除细胞碎片和其他杂质。
3. 抗原-抗体反应:将标记的Ma蛋白添加到细胞提取物中,与其中的蛋白质进行结合。
在适当的温度下保持一段时间,使抗原和抗体充分结合。
4. 免疫沉淀:通过加入抗Ma蛋白的抗体,将与Ma蛋白结合的蛋白质沉淀下来。
抗体通常与固相支持物(如微珠或凝胶)相结合,通过离心、洗涤等步骤将抗原-抗体复合物从溶液中分离出来。
5. 蛋白质检测和分析:对沉淀的蛋白质进行检测和分析,通常使用Western blot、质谱等方法对蛋白质进行定性和定量分析。
三、实验结果通过免疫沉淀实验,可以检测到与Ma蛋白结合的蛋白质,并对其进行鉴定和分析。
IP原理与步骤

免疫沉淀(Immunoprecipitation, IP)原理IP是利用抗原蛋白质和抗体的特异性结合以及细菌蛋白质的“protein A/G"特异性地结合到抗体(免疫球蛋白)的FC片段的现象开发出来的方法。
目前多用protein A/G预先结合在argarose beads上,使之与含有抗原的溶液及抗体反应后,beads上的prorein A/G就能达到吸附抗原的目的。
通过低速离心,可以从含有目的抗原的溶液中将目的抗原与其它抗原分离。
免疫沉淀实验的操作步骤比较多,同时由于在非变性条件下进行实验,所以要得到一个完美的实验结果,不仅需要高质量的抗体,同时对免疫沉淀体系也需要有严格的控制指标。
免疫沉淀实验从:蛋白样品处理;抗体-agarose beads孵育;抗体-agarose beads复合物洗涤到最后的鉴定,每步都非常关键,需要严格控制实验流程中每个关键步骤的质量,才能最终达到你的实验目的。
IP实验步骤基本实验步骤(1)收获细胞,加入适量细胞IP裂解缓冲液(含蛋白酶抑制剂),冰上或者4℃裂解30min, 12,000g离心30 min后取上清;(2)取少量裂解液以备Western blot分析,剩余裂解液将1μg相应的抗体和10-50 μl protein A/G-beads加入到细胞裂解液,4°C缓慢摇晃孵育过夜;(3)免疫沉淀反应后,在4°C 以3,000 g速度离心5 min,将protein A/G-beads离心至管底;将上清小心吸去,protein A/G-beads用1ml裂解缓冲液洗3-4次;最后加入15μl的2×SDS 加样缓冲液,沸水煮10分钟;(4)SDS-PAGE, Western blotting或进行质谱分析。
一、样品处理:免疫沉淀实验成功与否,第一步处理样品非常关键。
免疫沉淀实验本质上是处于天然构象状态的抗原和抗体之间的反应,而样品处理的质量决定了抗原抗体反应中的抗原的质量,浓度以及抗原是否处于天然构象状态。
免疫沉淀 Immunoprecipi tion IP 原理

免疫沉淀(Immunoprecipitation, IP)原理IP是利用抗原蛋白质和抗体的特异性结合以及细菌蛋白质的“protein A/G"特异性地结合到抗体(免疫球蛋白)的FC片段的现象开发出来的方法。
目前多用protein A/G预先结合在argarose beads上,使之与含有抗原的溶液及抗体反应后,beads上的prorein A/G就能达到吸附抗原的目的。
通过低速离心,可以从含有目的抗原的溶液中将目的抗原与其它抗原分离。
免疫沉淀实验的操作步骤比较多,同时由于在非变性条件下进行实验,所以要得到一个完美的实验结果,不仅需要高质量的抗体,同时对免疫沉淀体系也需要有严格的控制指标。
免疫沉淀实验从:蛋白样品处理;抗体-agarose beads孵育;抗体-agarose beads复基本实验步骤(1)收获细胞,加入适量细胞IP裂解缓冲液(含蛋白酶抑制剂),冰上或者4℃裂解30min, 12,000g离心30 min后取上清;(2) 取少量裂解液以备Western blot分析,剩余裂解液将1μg相应的抗体和10-50 μl protein A/G-beads加入到细胞裂解液,4°C缓慢摇晃孵育过夜;(3)免疫沉淀反应后,在4°C 以3,000 g速度离心5 min,将protein A/G-beads离心至管底;将上清小心吸去,protein A/G-beads用1ml裂解缓冲液洗3-4次;最后加入15μl的2×SDS 加样缓冲液,沸水煮10分钟;(4)SDS-PAGE, Western blotting或进行质谱分析。
一、样品处理:免疫沉淀实验成功与否,第一步处理样品非常关键。
免疫沉淀实验本质上是处于天然构象状态的抗原和抗体之间的反应,而样品处理的质量决定了抗原抗体反应中的抗原的质量,浓度以及抗原是否处于天然构象状态。
所以制备高质量的样品以用于后续的抗体-agarose beads孵育对免疫沉淀实验是否成功非常关键。
rna-ip条件

rna-ip条件1.引言【1.1 概述】概述部分主要介绍文章涉及的主题,即RNA-IP条件。
RNA-IP(RNA Immunoprecipitation)即RNA免疫共沉淀技术,是一种用于研究RNA 与特定蛋白质相互作用的实验方法。
通过该技术,可以帮助科研人员深入了解RNA的功能,揭示RNA在细胞生物学调控中的重要作用。
RNA-IP的基本原理是利用抗体选择性地富集靶向RNA及其相关蛋白质,从而分析特定RNA与特定蛋白质间的相互作用关系。
这种方法不仅能够帮助研究者鉴定RNA结合蛋白质,还能揭示RNA分子在转录后调控、翻译控制和RNA代谢等方面的调控机制。
通过RNA-IP的应用,我们可以解析RNA的功能和调控网络,为研究RNA介导的细胞过程提供重要的线索。
在本文的背景知识部分将介绍RNA-IP技术的起源及其发展历程,以及目前已有的不同的RNA-IP条件和方法。
随后,我们将详细讨论RNA-IP 的基本原理,包括抗体的选择、样品的制备和处理、共沉淀实验的步骤等。
通过阐述RNA-IP技术的基本原理,我们将向读者提供一个全面的认识,使其能够更好地理解和应用该技术。
本文的目的是系统地总结RNA-IP的应用前景,并对其在生命科学研究中的价值进行评估。
通过综合分析已有的研究结果和前沿进展,我们将提供一个综合性的视角,从细胞信号传导、转录后调控以及疾病研究等多个方面展望RNA-IP技术的未来发展方向。
结论部分将对本文的内容进行总结,并提出一些对未来研究有价值的建议。
通过深入研究RNA-IP条件与方法,我们可以更好地理解RNA分子的功能和调控,为解析细胞内复杂的基因调控机制提供重要的实验手段和理论依据。
本文将对RNA-IP技术在基础科学研究中的应用进行全面介绍,为相关领域的科学家提供参考和借鉴。
同时,我们也希望本文能够激发更多研究者对RNA-IP技术的兴趣,推动该领域的进一步研究和发展。
在接下来的章节中,我们将首先介绍RNA-IP技术的背景知识,为读者提供相关的前置知识。
RNA免疫沉淀(RIP)实验方案

此为临时链接,仅用于预览,将在短期内失效。
RNA 免疫沉淀 (RIP) 实验方案
abcam abcam 2018-08-23 请关注: ↑Abcam 助您更快实现研究使命
3 染色质剪切
3.1.将重悬后的细胞核分成两份,每份 500 mL(用于模拟实验和 IP)。 3.2.使用杜恩斯匀浆器捣碎 15 - 20 次对染色质进行机械剪切。 针对不同的细胞系可能需要优化剪切条件。 3.3.13,000 rpm 离心 10 分钟沉淀细胞核膜和碎片。 在液氮中冷冻一份裂解物用于对照 RNA 分离。
5 洗去未结合的物质
5.1. 2,500 rpm 离心 30 s 沉淀磁珠,移去上清液,在 500 mL RIP 缓冲液中重悬磁珠 对protein A/G 磁珠沉淀的严格清洗至关重要,并且可能需要优化。 5.2. RIP中重复清洗共三次,随后在 PBS 中清洗一次 第二次清洗后,将5%的磁珠进行冷冻,用于 SDS-PAGE 分析(例如,如果您的磁珠悬浮液总体积为 100 µL,则冷冻 5 µL 的磁珠悬浮液)。
查找您进行 RIP 实验所需要的一切,包括一份正确试剂的列表以及实验方案每个步骤的疑 难解答提示。
RIP 是一种基于抗体的技术,用于定位体内的 RNA-蛋白质相互作用。将所关注的 RNA 结合蛋白 (RBP) 与其结合的 RNA 一起进行免疫沉淀,鉴定结合转录RNA(mRNA、非编码 RNA 或病毒 RNA)。可以 通过实时 PCR、微阵列或测序检测。
◦ Hendrickson DG, Hogan DJ, McCullough HL, Myers JW, Herschlag D, Ferrell JE, and Brown
Magna(SIGMA) RIP RNA IP 说明书 中文

RIP流程规划RIP实验的裂解物要求●计算所需免疫沉淀的数量。
样本包括目的抗体(用户提供)和与目的抗体相同种属来源的阴性对照IgG。
抗SNRNP70(Cat.CS203216)和阴性对照正常兔IgG(Cat#PP64B)可以用作RIP程序的控件,这两个组件都包含在EZ Magna RIP套件(目录#17-701)。
●获得浓缩的细胞裂解液对于免疫沉淀的成功至关重要。
通常,一次RIP反应(使用一种抗体的一次免疫沉淀)需要100μL来自约2.0 x 107个细胞或一个15 cm平板的细胞裂解物。
RIP实验所需的裂解缓冲液体积的计算基于收获细胞的量。
该体积可能因使用的细胞类型而异。
请记住一旦你在某个细胞中用候选抗体证明RIP成功背景下,可以根据需要减少或进一步优化每个RIP的裂解物数量。
●必须优化每个RIP使用的细胞总数或蛋白质总量基于所研究的RNA结合蛋白的丰度以及RNA检测的计划方法。
一、裂解液制备完整RIP裂解缓冲液制备根据收获的细胞数量准备适量的完整的RIP裂解缓冲液。
(冰上操作)100μL RIP裂解缓冲液+ 0.5μL蛋白酶抑制剂鸡尾酒+ 0.25μL RNA酶抑制剂对于悬浮细胞:1.将细胞收获到15 mL锥形管中。
使用血细胞仪计数细胞。
2.在4°C下以1500 rpm的转速离心5分钟,收集细胞,并丢弃细胞上清液。
3、将细胞重新悬浮在10毫升冰冷的PBS中进行清洗。
通过离心收集细胞1500 rpm,在4°C下持续5分钟,并丢弃上清液。
4、重复步骤3进行一次额外清洗。
5、用等量的完整RIP裂解缓冲液重新悬浮细胞。
通过移液管混合直到混合物看起来均匀为止。
在冰上培养裂解物5分钟。
分配约200μL每种裂解液放入无核酸酶微量离心管中,并储存在-80°C下。
虽然每个抗体对应的裂解物分配量并不关键,但是每个RIP反应通常为100μL,每个RIP中通常使用阳性和阴性抗体。
实验表明,200μL细胞裂解液与单次使用的小份相关。
rna-ip条件 -回复

rna-ip条件-回复RNA IP (RNA Immunoprecipitation) 是一种分子生物学实验技术,旨在研究RNA分子与相关蛋白之间的相互作用。
通过使用RNA抗体,研究人员可以选择性地富集RNA结合蛋白,并进一步分析它们的相互作用及功能。
本文将一步一步地回答有关RNA IP的主题,包括技术原理、实验步骤、数据分析以及该技术的应用领域和挑战。
第一步:技术原理RNA IP的原理基于免疫沉淀技术,其中使用一种RNA特异性抗体与细胞/组织中的靶RNA结合。
为了实现这一目标,首先需要将细胞/组织中的蛋白质与RNA一起交联。
然后使用具有对RNA特异性的抗体,将抗体与交联后的RNA蛋白复合物结合。
免疫反应完成后,通过洗涤步骤去除非特异性结合的物质,最终可以纯化蛋白- RNA复合物。
第二步:实验步骤RNA IP的实验步骤如下:1.细胞/组织交联:使用适当的交联剂,如形醛或二硫化羟亚胺,将目标细胞/组织中的RNA与蛋白质交联。
2.细胞/组织裂解:使用细胞裂解缓冲液或组织断裂器将细胞/组织裂解,并释放RNA蛋白复合物。
3.免疫沉淀:将交联后的产物与特异性抗体结合,并通过抗体的亲和力选择性地富集RNA结合蛋白。
4.洗涤:通过多次洗涤步骤去除非特异性结合物,以提高样品纯度。
5.洗脱:将富集的RNA蛋白复合物从抗体上洗脱,以进一步分析。
第三步:数据分析分析RNA IP实验结果通常涉及以下步骤:1.蛋白质鉴定:通过质谱分析等技术鉴定与RNA结合的蛋白质,并确定其功能。
2.RNA测序:通过测序技术对富集的RNA进行分析,以确定与RNA 结合的蛋白质及其作用位点。
3.功能鉴定:利用基因表达谱分析等方法确定RNA结合蛋白对基因转录和翻译的调控作用。
第四步:应用领域RNA IP在许多生物学研究领域中发挥着重要作用,如:1.转录调控研究:通过分析转录因子与RNA的相互作用,可以揭示转录调控机制。
2.病理学研究:通过在疾病标本中应用RNA IP,可以鉴定与疾病进展和治疗相关的RNA结合蛋白。
rna免疫沉淀技术

rna免疫沉淀技术
RNA免疫沉淀技术(RNA immunoprecipitation, RIP)是一种用于富集细胞中靶RNA与其结合的蛋白质复合物的技术。
该技术已被广泛应用于探究RNA参与的生物学过程,如转录、剪切、稳定性、定位和翻译等。
下面,我们将分步骤阐述RNA免疫沉淀技术的操作过程。
一、样本准备
首先,需要制备含有感兴趣的RNA的样品。
可以从细胞或组织中提取RNA,也可以使用已知RNA序列的RNA质粒。
如果从细胞或组织中提取RNA,需要对RNA毒性和质量进行评估。
二、抗体选择
选择能够结合到目标RNA结合蛋白的抗体。
这可以通过文献研究和试验确定。
三、免疫沉淀
沉淀过程是将抗体与细胞中靶RNA与其结合的蛋白质复合物结合进行富集。
将样品加入含有抗体的磁珠或珠子中,通过简单的凝胶电泳或其他有效的清除步骤去除非特异性结合的蛋白质。
四、RNA提取
通过加入适当的反转录酵母(Reverse Transcriptase)和引物,将免疫沉淀的RNA反转录成cDNA。
然后可以使用PCR,RNA测序等技术来确定免疫沉淀的RNA顺序和数量。
在该技术中的关键步骤是抗体选择和副反应清除。
正确选择抗体可以提高RNA免疫沉淀的有效性,从而减少假阳性结果。
在沉淀过程中,非特异性抗体也会与非靶标蛋白质结合形成复合物,因此需要通过反应后实行有效清除。
总之,RNA免疫沉淀技术是一种重要的技术,可以用于富集细胞中靶RNA与其结合的蛋白质复合物,从而探究RNA参与的生物过程。
在实验过程中,需要正确选择抗体和优化副反应的清除步骤,以提高实验的效准确性。
RNA互作网络的构建和分析

RNA互作网络的构建和分析生命中的许多重要过程和疾病都涉及到RNA,当然,RNA分子之间的互作也很重要。
在过去的十年里,RNA互作网络得到了广泛关注。
本文将介绍RNA互作网络的构建和分析方法,并讨论它们在RNA功能研究和临床实践中的应用。
一、RNA互作网络的构建RNA互作网络的构建可以分为两个步骤:RNA-RNA交联实验和高通量测序。
1. RNA-RNA交联实验RNA-RNA交联实验(Cross-linking and immunoprecipitation,CLIP)是构建RNA互作网络的重要实验方法。
CLIP的基本原理是将含有RNA列表的细胞或组织与交联交联剂交联,然后利用抗体特异性地富集与目标蛋白RNA结合的复合物。
复合物经过切割和扩增后,就可以进行高通量测序来分析RNA-RNA交联位点和交联RNA列表。
2. 高通量测序在RNA-RNA交联实验后,RNA-RNA交联位点和RNA列表需要进行高通量测序。
这次测序可以利用新一代测序技术(NGS)进行。
NGS对DNA和RNA的高通量测序革命了基因组学和转录组学。
基于NGS的RNA互作网络分析可以鉴定出RNA交联位点,并找到RNA之间的相互作用关系。
二、RNA互作网络分析RNA互作网络中的节点可以是RNA分子、蛋白质以及其他含有结构域的蛋白质。
现在,已经有许多工具可以进行RNA互作网络的分析。
以下是常见的RNA互作网络分析方法:1. 差异表达基因的RNA互作网络通过分析差异表达基因的RNA互作网络,可以发现和深入探究已知的调控网络和新的RNA调控网络。
这个方法可以预测转录因子对RNA的调控、辅助因子之间的相互作用以及RNA在不同细胞中的功能作用。
2. GeneMANIAGeneMANIA是一款在线工具,可以进行RNA互作网络分析。
使用基于文献的演化网络和基于PPI(protein-protein interaction,蛋白质相互作用)的临近外推来预测基因功能以及基于RNA-RNA作用关系的预测。
RNA IP流程

RNA IP流程2篇RNA IP流程是指在申请RNA(注册实用新型)专利时所需进行的一系列步骤和程序。
本文将介绍RNA IP流程的基本原理和操作步骤,并简要探讨其意义和应用领域。
首先,RNA IP流程是指利用RNA技术申请专利的一套完整流程和程序。
RNA技术是一种生物技术,可以通过改变或修饰RNA分子的结构和功能,实现对基因和基因表达的调控。
在这个流程中,申请人需要提交RNA技术相关的专利申请,经过审查和评估后,方可获得专利权。
RNA IP流程的步骤和程序分为以下几个阶段:第一阶段:初始准备。
此阶段的关键是明确RNA技术的创新点和应用领域,并制定申请战略。
申请人需要对现有的技术和专利进行调研和分析,以确保自己的创新具有可实施性和独特性。
第二阶段:申请撰写。
在此阶段,申请人需要撰写专利申请文件,包括专利申请书、说明书和权利要求书。
这些文件需要清晰地描述RNA 技术的创新和应用,以及其与现有技术的区别和优势。
第三阶段:申请提交。
在此阶段,申请人需要将撰写好的专利申请文件提交给所属的知识产权机构,如专利局或国家知识产权局。
申请人需要支付相应的申请费用,并确保填写的信息准确无误。
第四阶段:审查和审查。
在此阶段,知识产权机构将对专利申请文件进行审查和评估。
这个过程可能需要一段时间,并且可能涉及对申请人的进一步交流和补充资料的要求。
审查和审查的目的是确保专利申请符合专利法律和标准。
第五阶段:专利授予。
在通过审查和审查后,如果专利申请被认为符合专利法律和标准,申请人将获得专利权。
专利权的授予意味着申请人可以在一定的时间内独占RNA技术的应用和利用。
RNA IP流程的意义和应用领域广泛。
首先,通过RNA技术申请专利,申请人可以获得对其创新的独有权益,从而有助于保护和推动自己的研究和开发工作。
其次,RNA技术具有广泛的应用前景,涵盖了农业、医药、生物工程等多个领域。
通过RNA IP流程,相关企业和机构可以更好地保护和应用这一技术,促进技术创新和行业发展。
IP步骤

IP(Immunoprecipitation)基本步骤1.细胞采用PBS洗涤(冰上预冷)3遍,每皿(直径=10cm)加入RIPA完全裂解液(冰上预冷)(含蛋白酶抑制剂及磷酸酶抑制剂)300~500微升,刮取细胞(刮刀预冷),转移入EP管(管子预冷)中,放入冰上,裂解2h,12000rpm×30min,收上清,弃沉淀。
此时可于上清中取数十微升,与等体积2×SDS细胞裂解液混合,作为WB上样的input组。
2.其余上清与适量IP的抗体混合(每10cm皿加入1~2ug抗体),4℃结合,过夜。
3.第二天,根据抗体类型不同,如下表,根据抗体片段中IgG的类型选择。
一个简单的方式是鼠抗采用protein A,兔抗采用protein G沉淀。
(有时可两个凝胶等比例混合用),用量参照说明书或每10cm皿20ul凝胶足够。
将protein G 或(和)protein A 采用RIPA不完全裂解液(不含抑制剂和甘油)洗涤3次,每次1min×2000rpm×4℃,后采用RIPA完全裂解液(预冷)重悬,均匀分配到过夜结合的样本中,4℃结合(最短3h,也可4℃过夜);4. 最后4℃13200rpm×30min,上清可留作,沉淀再次用RIPA半裂解液洗两次(5min×3000g×4℃),后加1×SDS煮15min,定量,保存备用,液体配方:RIPA裂解液:10ml10%去氧胆酸钠0.25mlEDTA(500mmol/L, ph 7.4) 20ul100* cocktail 相应体积100*PMSF (100mmol/L) 相应体积磷酸酶抑制剂相应体积Protein G calbiochem 货号539207Protein A invitrogen 货号404489RIPA基础液(温和型)可从大部分试剂公司购买到一下摘自网络:1.除了选择特异性好的抗体以及选择合适的阴性对照外,去除免疫沉淀实验非特异性的一个办法是对抗体-agarose beads复合物进行多次洗涤。
RNA互作网络的构建与分析

RNA互作网络的构建与分析随着基因组学、转录组学和蛋白质组学等技术的不断发展,我们对生物学的认识也在日益深入。
其中,RNA在许多生物学过程中扮演着非常重要的角色。
近年来,越来越多的研究表明,RNA差别表达和RNA相互作用网络是解析生命现象的有力工具。
本文旨在介绍RNA互作网络的构建与分析。
一、RNA互作网络的构建RNA互作网络是由各种RNA分子之间的相互作用所构成的网络。
这种相互作用可以包括RNA与蛋白质之间的相互作用,也可以包括RNA与RNA之间的相互作用。
构建RNA互作网络的过程需要多种方法,例如RNA pull-down、IP-MS、CLIP-seq和PAR-CLIP等。
1. RNA pull-downRNA pull-down技术是通过DNA或RNA探针将特定的RNA捕获并与其相关联的蛋白质或RNA共沉淀,从而获取RNA相互作用的信息。
该技术的优点在于其简单易行,不需要特殊仪器设备。
2. IP-MSIP-MS(免疫共沉淀-质谱法)技术是在免疫共沉淀的基础上结合质谱技术来鉴定RNA互作网络。
这种方法可以在蛋白质水平上获得RNA互作网络的信息,其优点在于其高通量和高灵敏度。
3. CLIP-seq和PAR-CLIPCLIP-seq和PAR-CLIP技术是一种高通量测序技术,可以鉴定RNA与RNA或RNA与蛋白质之间的相互作用。
它们都是通过交联的方式来捕获RNA与靶分子的相互作用情况,然后经过测序分析,得到RNA互作网络信息。
二、RNA互作网络的分析RNA互作网络的分析包括两个方面的内容,即RNA差别表达分析和RNA相互作用网络分析。
1. RNA差别表达分析RNA差别表达分析是比较不同条件下RNA表达的差异,并通过统计学方法预测功能上富集的基因。
这项分析常用的方法包括生物信息分析、功能注释和通路分析等。
生物信息分析主要是对RNA测序数据进行药物基因组、基因定位、基因识别、基因表达和序列比对等生物学分析。
new tech--RNA pull down protein(RNA IP方法)

lncRNA与蛋白互作技术:RNA-Protein Pull-Down/RIP(2013-07-05 11:29:49)lncRNA与蛋白互作技术:RNA-Protein Pull-Down/RIP1、Thermo fisher试剂盒近日,赛默飞世尔科技全新推出了一款Pierce Magnetic RNA-Protein Pull-Down Kit,让研究人员能够以末端标记的RNA作为诱饵,轻松富集蛋白质-RNA的相互作用。
与抗体捕获相比,这种方法的优势在于脱硫生物素化的目标RNA能够直接富集RBP(或复合物)。
蛋白质与RNA的相互作用是许多细胞功能的核心,如蛋白质合成、mRNA组装、病毒复制、细胞发育调控等。
了解它们之间相互作用的分子机制对理解这些生物学过程非常重要。
然而,之前的分析方法往往受限于使用放射性标记,或实验步骤过多,不仅耗时费力,也增加了实验结果的不稳定。
此试剂盒利用脱硫生物素末端标记的RNA和链霉亲和素磁珠标记的来高效富集RNA结合蛋白(RBP)。
与抗体捕获相比,这种方法的优势在于脱硫生物素化的目标RNA能够直接富集RBP(或复合物)。
此外,试剂盒还提供了经过验证的对照,适用于标记和pull-down分析,也与多个下游应用兼容,如Western blotting和质谱(MS)。
试剂盒中包含了Pierce RNA 3’-End Desthiobiotinylation Kit。
它利用T4 RNA连接酶将单个脱硫生物素化的胞苷二磷酸连接到单链RNA的3’端。
3’端的末端标记不干扰RNA结构,因此,比标记核苷酸的随机掺入更加理想。
每个标记反应适合50 pmol RNA;不过,如有必要的话,标记反应也可扩展(从1 pmol 到1 nmol)。
标记反应需要20倍过量的脱硫生物素化核苷酸。
对于不太复杂的RNA,孵育时间可为37°C 30分钟,若是更长或更复杂的RNA,时间也延长到4-16°C过夜。
rnapulldown实验原理

rnapulldown实验原理RNA拉down实验是一种用于检测RNA-蛋白相互作用的技术,可以用来测定RNA和特定蛋白之间的相互作用,从而探究RNA在细胞环境中的功能。
本文将介绍RNA拉down实验的背景、原理、流程以及其优势和缺点。
一、背景RNA拉down实验是一种发展自蛋白免疫沉淀实验(immunoprecipitation,IP)的新技术,简称“RNA-PD”。
它是基于蛋白质-RNA互作的一种检测方法,主要用于鉴定哪些RNA被某种蛋白结合,从而发现RNA的功能性相互作用,从而可以让我们更深入的了解RNA的功能。
二、原理RNA拉down实验将靶蛋白与可结合的核酸探针或核酸抗体结合,然后将全细胞中的RNA转换成具有抗体修饰的纳米级磁珠,最终通过磁分离,将蛋白质免疫沉淀珠从细胞中提取出来。
有了这一细胞抽取的纳米级磁珠,研究人员便可以检测出它们中结合蛋白的RNA序列,并将这些 RNA列及其表达水平与对照组、全细胞中的 RNA析进行比较,从而发现蛋白与RNA的相互作用。
三、流程RNA拉down实验的流程包括以下几个步骤:(1)细胞膜去除:首先,细胞膜应该去除,以便将蛋白质依赖性及质粒依赖性的RNA分离出来。
(2)细胞抽取:其次,细胞抽取,这一部分采用磁珠沉淀技术,通过使用带有抗体的纳米级磁珠,将细胞中特定的RNA聚集起来,其中的RNA聚集物可以进行更详细的分析。
(3)RNA水解:之后,RNA水解,利用水解酶将RNA水解,并加入 RNase inhibitor 以避免非特定性水解。
(4)检测:最后,利用实时定量PCR或者高通量测序技术检测细胞中RNA的表达水平,获得RNA的特定性绑定蛋白的结果。
四、优势RNA拉down实验是一种高灵敏度和特异性的技术,有很多优势: 1.以分离出具有特定RNA结合蛋白的细胞,以检测特定RNA-蛋白相互作用,进而揭示RNA的功能性。
2.能在低浓度的情况下,可以高效的从大量的细胞样本中检测出特定RNA-蛋白的相互作用,同时也避免了重复实验。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
RNA的免疫共沉淀分离及检测
非编码RNA的发现使得RNA领域再次成为了生命科学研究关注的焦点。
因为RNA是一种不稳定的生物大分子,绝大多数的RNA都需要与特定的RNA结合蛋白质结合形成RNA/
蛋白复合物才能稳定存在于细胞中;不仅如此,RNA与RNA结合蛋白之间的动态关联贯穿和伴随了RNA的转录合成、加工和修饰、胞内运输和定位、功能发挥及降解的整个生命循环。
鉴于此,利用RNA结合蛋白分离或发现鉴定功能性RNA分子是RNA研究领域中一个不可或缺的研究方法。
简单地说,就是利用RNA结合蛋白的抗体免疫沉淀RNA/蛋白复合物,再从沉淀的RNA/蛋白复合物中分离得到特定RNA结合蛋白的RNA;分离得到的RNA 可以通过末端标记和变性胶电泳对RNA分子的大小进行鉴定,也可以利用高通量RNA测序方法对RNA序列进行分析。
●全部所用的溶液试剂均经过DEPC处理或由DEPC水配制。
●RNA抽提过种中乙醇漂洗步骤可以省略。
●RNA样品溶解与保存:TE(pH8。
0)缓冲液。
80℃(如果下一步操作涉及酶反应)100%de-ionicFormamide(如果下一步是电泳检测)
●熟练的RNA操作非常重要。
RNase的污染可通过使用RNAZap处理操作环境得到降低。
1. 由组织或细胞裂解液免疫沉淀RNP复合物
(1)可预先UV照射组织和细胞固定RNA/结合蛋白复合物。
(2)使用预先冰浴冷却的PBS洗组织或细胞两次。
(3)将裂解液加入组织样品中进行匀浆(细胞样品匀浆步骤可省略)。
裂解液:
25 mmol/L Tris-HClpH7.5,150 mmol/L KCl,2 mmol/L EDTA,0.5%NP40,1 mmol/L NaF,1 mmol/L DTT,100 U/ml RNasin核糖核酸酶抑制剂,EDTA-free蛋白酶抑制剂。
(4)样品裂解液在4℃下14000 r/min(16000 g)离心10 min,上清液将用于免疫沉淀。
(5)将预平衡的ProteinA/GSepharosebeads与适量的抗体混匀,于4℃旋转混匀6 h左右。
(6)将第3步骤中准备的上清液样品加入beads-抗体混合液中于4℃旋转混匀6 h以上或过夜。
结合时间也取决抗体本身的质量和效率。
(7)将混匀液于4℃,1000 r/min离心10 s,弃上清液,用冰浴冷的低盐漂洗液洗beads 两次,每次5 min,冰上或4℃进行。
随后再用高盐漂洗液洗beads两次,每次5 min,冰
上或4℃进行。
低盐漂洗液:(高盐漂洗液使用300 mmol/L的NaCl)50 mmol/L
Tris-HClpH7.4,150 mmol/L NaCl(300 mmol/L),1 mmol/L MgCl2,0.05%NP40,2 mmol/L EDTA,1 mmol/L DTT,100 U/ml RNasin核糖核酸酶抑制剂。
2. 小分子RNA的抽提
(1)用ProteinaseK溶液重悬beads,55℃消化10 min。
(2)再用常规的TRizol法抽提RNA,用乙醇沉淀。
3. RNA5'端标记和电泳检测
(1)用CalfIntestinalPhosphatase处理抽提得到的RNA,以去掉5'端磷酸基团。
(2)经酚氯仿抽提和乙醇沉淀后,使用γ-32PATP标记RNA分子的5'末端。
(3)将标记后的RNA分子通过15%聚丙烯酰胺尿素变性胶进行电泳分离,随后经放射自显影进行检测。
4. Solexa高通量RNA测序。