美国对照临床试验研究进展
最新:单克隆抗体药物治疗视神经脊髓炎谱系疾病的临床试验研究进展

最新:单克隆抗体药物治疗视棉经脊髓炎谱系疾病的临床试验研究避展视神经脊髓炎谱系疾病(neuromyelitis optica spectrum disorders , NMOSD)是一种体液免疫介导的中枢禅经系统炎性脱髓鞠疾病,多累及视神经和脊髓,真高高复发、高致残的特点,主要的致病抗体为抗水通道蛋白4( A QP4)抗体。
约70%的NMOSD患者AQP4抗体阳性,女性较男性高发(9 : 1 ),平均发病年龄为40岁。
NMOSD急性期的治疗方法包括大剂量糖皮质激素、丙种球蛋白冲击治疗和血浆置换等。
经典的缓解期预防复发药物包括口服糖皮质激素、硫瞠瞟岭、他克莫司、环抱素等免疫抑制剂。
虽然这些药物仍是当前不可替代的治疗NMOSD的常用药物,但部分患者难以忍受真带来的广泛的免疫抑制、骨髓抑制等不良反应,因此开发安全高效的药物,对阜期治疗、预防复发以及提高NMOSD患者的生活质量尤为重要。
近年来,己再多种单克隆抗体药物用于治疗NMOSD 因真靶点相对明确,作用效应较集中,不良反应相对较少,为该疾病的治疗带来新希望。
为确定这些药物的中远期疗效、安全性和不良反应等,已开展了一系列临床t鹉金。
本文就单克隆抗体药物治疗NMOSD的作用机制和临床试验的最新进展进行综述。
01、NMOSD的发病机制AQP4抗体在NMOSD的发生发展中起重要作用。
AQP4是一种位于细胞膜的转运蛋白,可根据渗透压的变化控制水分子进出细胞,在整个中枢神经系统(包括脊髓、视神经和脑)的星形胶质细胞中表达。
AQP4抗体主要由外周的B细胞产生。
真在一定的条件下可通过血脑屏障(BBB)并与血管周围的星形胶质细胞足突表面的AQP4抗原结合,来诱导补体依赖性细胞毒性(CDC)和抗体依赖性细胞毒性(ADCC);前者指AQP4抗体和抗原结合后激活补体经典途径,形成膜攻击复合物,对星型胶质细胞发挥裂解效应;ADCC是指AQP4抗体和抗原结合后,抗体的Fe段与杀伤细胞(NK细胞、巨睦细胞等)表面的Fe受体结合,杀伤细胞通过程放穿孔素、颗粒酶等细胞毒物质直接杀伤星型胶质细胞。
什么是新药临床试验

什么是新药临床试验新药临床试验,也叫作新药临床研究,简单的说就是新药在上市前,在人体(病人或健康志愿者)进行药物的系统性研究,从临床试验中我们可以了解一种新的药物在人体上应用有没有疗效,有没有毒副作用,副作用有多大等等情况。
无论是在中国,还是在其它国家,所有的新药在上市前都必须进行临床试验。
临床试验对于保障新药上市后人们的用药安全是至关重要的。
按照美国国家卫生研究院(NIH)的观点,精心设计、实施的临床试验是寻找有效治疗措施的最快和最安全的一种途径。
必须经国家药监局批准才能进行新药临床试验新药在上市前都必须进行临床试验,但是临床试验的实施不是随意的,不是谁想实施临床试验就能实施的,要进行临床试验,必须经过国家药监局(也就是现在的国家食品药品监督管理局,SFDA)的批准,取得新药的临床试验批准文件(文号)之后,才有可能实施。
取得批准文号是实施临床试验的法律前提。
因为涉及人体试验,为了保障参加试验的病人或者健康志愿者的人身安全与健康,批准进行临床试验的过程是非常严格的,研究新药的制药企业或者研究机构要进行大量的实验室研究,取得动物的疗效与安全性试验数据,以及其它的药学数据,所有这些研究工作,都有详细的技术指导原则来进行规范。
而得到国家药监局的批准,只是进行临床试验之前的第一步,要想实施临床试验,还必须通过独立伦理委员会的审核。
每个能够进行临床试验的医院,都有一个由医生与其它职业的人员(外单位)组成的独立伦理委员会,负责对在本医院进行的临床试验进行伦理审核,这是保障参加试验人员安全、权益的另一个有效措施。
伦理审核将对参加试验的受益(好处)与风险进行综合权衡,只有当参加试验的受益大于风险时,这个临床试验才会通过独立伦理委员会的审核,然后医生们才会按照要求进行临床试验。
新药的临床试验研究一、新药临床研究的重要性新药的临床研究十分重要。
一方面新药药效的评价,因试验的动物不同有所差异;在动物身上的反应和在人体上的反应有所不同。
2010心力衰竭的药物治疗新进展

万方数据
果已证实有较好的扩血管和抑制钠水潴留的作用, 与缬沙坦合用作用进一步增强,在心力衰竭患者取 得了较好的治疗效果,不良反应与ARB单一治疗 组及安慰剂组差异无统计学意义[1 4‘。2007年公布 的阿利吉仑治疗心力衰竭患者疗效(ALOFT)研究 共纳入302例NYHAⅡ~Ⅳ级的心力衰竭患者, 平均左心室射血分数31%,随机分为阿利吉仑组 (156例)和安慰剂组(146例),观察3个月,结果显 示阿利吉仑组的血浆肾素活性下降,与安慰剂组比 较,差异有统计学意义(P<O.05)[1 5‘。然而,口服 阿利吉仑能否改善心力衰竭患者的长期预后还需 要有进一步的循证医学证据。
万方数据
中华老年医学杂志2010年2月第29卷第2期Chin J Geriatr,February 2010,V01,应严密监测 血清钠水平以防止出现高钠血症;同时,要观察患 者是否出现继发性脱水征兆以及时应对。
二、重组B型钠尿肽 利钠肽是一种小蛋白,对抗RAAS,在调节水 钠排泄及血管舒张过程中起着重要作用,同时也参 与了内环境稳定、脂肪代谢和长骨生长的调节。利 钠肽系统有3个主要成员:心房肌分泌的心钠肽 (ANP)、主要由心室肌分泌的脑钠肽(BNP)和由 内皮细胞生成释放的C型利钠肽(CNP)。当心房 心室扩张,血压升高,缺氧或肾功能不全均可刺激 利钠肽的分泌[5]。BNP具有排钠利尿效应,还可 扩张血管,降低体循环血管阻力。由于BNP是在 心室壁压力增加时直接由心室肌快速分泌,其释放 量似乎与心室容积扩大和压力超负荷的程度成正 比,与心室功能状态呈负相关,故其血中浓度能直 接反映心功能损伤程度。有研究结果证实,血浆 BNP水平的增高与左心室射血分数受损程度密切 相关,所以BNP水平可作为发现无症状性左心室 收缩功能不全的一个指标[6]。奈西利肽是重组人 BNP,在2001年被美国药品食品管理局(FDA)批 准应用于急性失代偿性心力衰竭,因为其能够降低 肺毛细血管楔嵌压,因而明显改善劳力性呼吸困 难。亦有研究结果显示,人重组BNP治疗急性失 代偿性心力衰竭能促进水的排出,降低肺动脉压, 增加左心室射血分数。从而改善呼吸困难,指标优 于常规治疗组U-83。在奈利肽连续静脉滴注治疗心 力衰竭患者的随访(FUSION I)试验中,虽然慢性 失代偿性心力衰竭的患者应用奈西利肽结果为中 性,但对高危患者则表现出一定的益处。在这个基 础上,FUSIONⅡ试验入选了920例左心室射血分 数<40%,美国纽约心功能分级(NYHA)Ⅲ~Ⅳ 级的慢性失代偿性心力衰竭患者,院外随机双盲每 周应用1~2次重组BNP的序贯疗法或安慰剂共 12周,随访24周,以观察奈西利肽治疗心力衰竭 的安全性及有效性。结果显示,奈西利肽组(605 例)与安慰剂组(306例)比较,无论全因病死率 (9.5%与9.6%)还是心肾原因所致住院率 (32.9%与33.9%),差异均无统计学意义(P> 0.05)。提示重组BNP的序贯疗法不适合慢性心 力衰竭患者b]。 三、钙增敏剂 主要代表药物为左西孟旦。左西孟旦具有独 特的药理作用和药物代谢动力学特点,能够增加心
肿瘤疗效评价标准

癌症化疗新的疗效评价标准中国医学科学院肿瘤医院冯奉仪一. 实体瘤的疗效评价标准( Response Evaluation Criteria in Solid Tumors 、RECIST ) 细胞毒化疗药是通过肿瘤缩小量耒评价其抗肿瘤作用,1979年WHO ( World Health Organization ) 确定了实体瘤双径测量的疗效评价标准。
20多年来,这个标准被国内外的研究者和研究组普遍采用,但WHO的标准存在如下问题:(1)由WHO确定可评价的和可测量大小病灶的改变混为一体,来判断疗效在各研究组间各不相同。
(2)最小病灶的大小及病灶的数量亦无明确的规定。
(3)PD 的定义在涉及单个病灶还是全部肿瘤( 可测量肿瘤病灶的总和)不明确。
(4)新的诊断病变范围的影像学方法,如CT和MRI己被广泛的应用。
因此,多年来造成了对于单个药物、联合化疗方案及治疗方法各研究组之间疗效评价存在差异而难以比较,往往导致不正确的结论。
针对以上问题,1994年EORTC ( European Organization for Research and Treatment of Cancer )、美国NCI ( National Cancer Institute ) 和加拿大NCI在回顾普遍使用的WHO疗效评价的基础上,进行了充分的交流和讨论,以后又相继的召开了多次会议,讨论和完成尚未解决的问题,直至1998年10月在包括学术界、企业、官方当局的会议上取得了一致的意见。
在WHO疗效评价标准的基础上进行了必要的修改和补充,采用简易精确的单径测量代替传统的双径测量方法,保留了WHO标准中的CP、PR、SD、PD。
RECIST首次在1999年美国的ASCO会议上介绍,并于同年的JNCI杂志上正式发表。
抗癌药物的疗效评价至少包括三个不同的目的:(1)在早期临床试验中,客观肿瘤疗效是试验药物或方案的预期目的,其结果是决定该药物或方案是否值得进一步研究的依据,体现在II期临床研究中。
阿尔茨海默病靶向Aβ疾病修饰治疗

阿尔茨海默病靶向Aβ疾病修饰治疗阿尔茨海默病(AD)是老年期最常见的神经系统退行性疾病,目前临床对其发病机制认识仍较为局限、治疗药物研发滞后。
β-淀粉样蛋白(Aβ)级联瀑布假说仍然是目前AD 发病机制的主流学说,也是AD疾病修饰药物研发的重要理论基础。
近年来,靶向Aβ的免疫治疗药物相继通过美国食品药品监督管理局批准上市或突破性疗法认定,为AD的疾病修饰治疗带来了曙光。
文中评述了近年来靶向Aβ疾病修饰治疗临床试验的研究进展,分析总结了以往靶向Aβ疾病修饰治疗临床试验失败的原因。
虽然目前靶向Aβ疾病修饰疗法不是十分成熟,但其俨然已成为极具前景的AD药物研发策略。
阿尔茨海默病(Alzheimer′s disease,AD)以经过长期的前驱症状后出现进行性加重的认知和行为障碍为临床特征,以β-淀粉样蛋白(amyloid-β,Aβ)在脑内沉积为特征性病理表现,是临床上最为常见的中枢神经系统退行性疾病。
随着AD患病率的升高和日益严重的公共卫生危机,通过治疗手段来实现AD预防、延缓发病、减缓疾病进展和改善症状迫在眉睫。
目前AD的治疗方法有限,以对症治疗为主。
近年来,AD疾病修饰治疗的研发工作取得了可喜的进展。
DMT 是指对导致神经元死亡的潜在病理生理过程进行医疗干预,以期达到改善AD临床进展轨迹的治疗方法。
靶向Aβ的DMT药物阿杜那单抗和仑卡奈单抗相继通过美国食品药品监督管理局批准上市,为患者和家属带来新的希望,也为AD靶向药物开发带来曙光。
一、AD靶向Aβ的DMT种类淀粉样蛋白级联假说认为Aβ在大脑中的沉积是疾病病理发生的始发和核心事件。
靶向Aβ药物根据其作用机制不同可以分为:减少Aβ产生和聚集的生物制剂,以及促进A β清除的抗Aβ生物制剂。
(一)减少Aβ产生和聚集的生物制剂首个靶向Aβ的DMT的主要目的在于通过Aβ酶抑制剂来减少Aβ产生。
β-分泌酶1和γ-分泌酶是Aβ生成的关键酶。
BACE1 抑制剂曾经是治疗或预防AD 的主要研究方向,然而,许多针对症状性AD 或轻度认知障碍的研究产生临床未预料到的不良反应。
药物临床试验现场核查关注点探讨

药物临床试验现场核查关注点探讨肺癌靶向治疗和免疫治疗药物临床试验具有抗肿瘤药物临床试验的一般特点,如受试者预后差、耐受性差、试验设计和风险控制参差不齐等。
同时也具有自身特点,如不同病理和基因分型直接影响药物应答导致入排标准更为严格,不同类型疗效终点数据的获取方式不同,不良反应较多且存在免疫相关不良事件等。
现场核查时在执行统一核查标准的前提下,如何结合以上特点开展更具有针对性的核查工作,是本文探讨的重点。
1受试者筛选入组方面受试者筛选入组是开展临床试验的首要环节。
现场核查时需逐一核对每一条入选排除标准的符合性证据,查看入组试验的受试者是否均符合试验方案规定的入选与排除标准。
1.1病理和基因检测病理检测和基因检测是肺癌靶向治疗药物临床试验的关键入选标准。
试验设计通常只针对腺癌、鳞癌或非鳞癌中的一种,病理组织类型为混合型的患者一般不能入组试验。
NSC1C发病机制与多种基因突变密切相关,存在EGFR>A1K、MET、RET等特定基因改变通常是肺癌靶向药物临床试验的重要入选标准。
另外,受试者在入选临床试验时通常需要排除其他已知激活突变。
现场核查时需重点关注病理报告、基因检测结果与试验入选标准的符合性,同时关注其他已知突变的检测结果。
在基因检测结果的确认方面,需关注检测机构是否与试验方案中的规定一致,如由各研究单位检测或中心实验室检测。
1.2肺癌伴脑转移肺癌有多发脑转移的特点,24%-40%的晚期NSC1C患者会出现脑转移。
出于对受试者安全的考虑,肺癌晚期患者较易因发生脑转移而被临床试验排除或经放疗后再行评估入组。
在评估时,需关注所采用的评估方式,如按照症状评估的无症状脑转移、放疗后病变稳定脑转移和有症状脑转移,或按照转移数量评估的脑转移。
现场核查时需注意对颅脑磁共振成像(magneticresonanceimaging,MRI)的检查结果、既往治疗记录进行溯源,关注受试者在入组时是否存在脑转移或经过放疗的情况,脑转移的评估是否采用方案规定的评估方式等。
在临床试验设计中设对照组的重要性

在临床试验设计中设对照组的重要性中国医学论坛报社名誉总编辑照日格图Drazen教授在这一次来访期间,参观访问了北京的3家医院和1个专科疾病研治中心,与不少专家教授和科研人员进行了面对面的交流。
他反复强调,他此次北京之行的目的是为推动中国临床医学科研的发展。
在言谈话语中提到的最多的是临床研究的设计问题。
在临床研究的设计方面,提到和讨论的最多的是设对照组的问题。
这个问题看起来似乎很简单,我们在国内开展的和在国内期刊上刊登的许多临床研究文章都设有对照组。
大家对设对照组的目的和方法也并不陌生。
但实际上这个问题并不简单。
有不少已经设的对照组实际上并不合适,因此研究的结果和结论缺乏说服力。
目前还有不少研究没有设对照组。
有些临床工作者认为,一些临床研究,特别是有创性治疗方法的临床研究,似乎不能设对照组。
例如,Drazen教授在参观了北京西山医院神经疾病研治中心黄红云医师用细胞移植方法治疗肌萎缩性侧索硬化症(ALS)及脊髓损伤的病房后,建议其开展一项设有假手术(shamsurgery)对照的临床研究。
黄红云医师则提出设假手术对照组完全是不可行的,其原因有多种,包括伦理学、假手术本身对病人可能造成伤害、病人不可能接受、有关管理部门不会批准,等等。
黄医师还提出了极具挑战性的反问:“为什么心脏移植、肾脏移植和肝脏移植这样全世界在普遍开展的手术治疗的疗效没有经过随机对照的研究去证实?”Drazen教授对这些问题的意见如何?我们究竟应当如何看待这些问题?以下介绍Drazen教授发表的意见和与这些问题相关的文献资料中有关的一些内容。
有创性疗法临床研究中设假手术对照的先例当今世界上对病人进行新疗法临床研究时,有没有设立假手术对照组的先例呢?回答是有的,虽然不太多。
我们以“shamsurgery”为主要检索词,年代限制设为1995年3月至2005年3月,文献类型设为“临床试验”,未加其他限制条件情况下,检索Medline结果得到111篇文章。
肿瘤退缩分级标准的对比及其在直肠癌诊治中的应用进展

肿瘤退缩分级标准的对比及其在直肠癌诊治中的应用进展中华结直肠疾病电子杂志2016 年12 月第5 卷第6 期Chin J Colorec Dis ( Electronic Edition ) , December 2016,V ol.5, No.6 ·458··述评·肿瘤退缩分级标准的对比及其在直肠癌诊治中的应用进展谭伊诺陈海燕丁克峰丁克峰教授、主任医师、博士生导师。
现任浙江大学医学院附属第二医院院长助理、肿瘤外科副主任、大肠外科病区主任、浙江大学肿瘤研究所副所长。
中国抗癌协会理事,中国抗癌协会大肠癌专业委员会副主任委员,中国医师协会结直肠外科委员会常委兼副秘书长,浙江省抗癌协会肿瘤转移专业委员会主任委员等职务。
长期从事大肠癌综合诊治和大肠癌腹腔镜微创治疗方面的工作,率先在国内提出并开展腹腔镜辅助结直肠癌快速康复治疗模式,规范MDT 模式,协助制定全国《大肠癌诊治规范》,推动我院成为“全国结直肠癌多学科综合治疗先进技术示范推广工程”首批示范医院之一。
主要从事肿瘤转移和肿瘤耐药机制研究,主持并完成国家自然科学基金课题5 项,卫生部课题1 项,省部级课题10 余项,主持临床研究3 项,发表SCI 及国内核心期刊论文50 余篇。
DOI:10.3877/cma.j.issn.2095-3224.2016.06.001基金项目:2014 年中国国家卫生行业公益性基金项目(No.201402015);国家自然科学基金项目(No.81272455,81472664);浙江省重点研发项目(No.2016CG1360721)作者单位:310009杭州,浙江大学医学院附属第二医院肿瘤外科通讯作者:丁克峰,Email:dingkefeng@【摘要】随着新辅助治疗在直肠癌中的规范化推广,肿瘤退缩分级(TRG)标准逐渐引起广泛关注和重视。
多项研究证实TRG 与直肠癌患者新辅助治疗反应、生存预后有一定相关性,在患者生存预测、随访和临床诊疗策略等方面均有应用前景,甚至也有报道考虑将其纳入临床试验替代终点。
ICH-E10-临床试验中对照组的选择和相关问题-20000720

ICHE10现行第四阶段版本2000年7月20日该指导原则由相应的ICH专家工作组制定,按照ICH 进程,已通过药品监管机构讨论。
在ICH进程第四阶段,最终草案被推荐给欧盟、日本和美国的管理机构采纳。
E10首次编码历史日期重新编码2005年11月E10 由指导委员会根据第二阶段程序批准,并公开征求意见。
1999年5月7日E10E10 由大会根据第四阶段程序批准,并建议三个ICH监管实体采纳。
2000年7月20日E10ICH本指导原则经2000年7月20日召开的ICH指导委员会会议上确认已达到ICH进程第四阶段,建议ICH三个监管机构采纳本指导原则。
目录1.引言 (1)1.1指南的总体框架和目的 (1)1.2对照组的目的 (3)1.2.1随机化 (4)1.2.2盲法 (4)1.3 对照的类型 (5)1.3.1安慰剂平行对照 (6)1.3.2无治疗平行对照 (7)1.3.3剂量-效应平行对照 (7)1.3.4活性(阳性)平行对照 (8)1.3.5外部对照(包括历史对照) (8)1.3.6多个对照组 (9)1.4临床试验的目的及相关问题 (9)1.4.1有效性的证据 (9)1.4.2可比的有效性和安全性 (10)1.4.3 比较的公平性 (11)1.4.3.1剂量 (11)1.4.3.2病人人群 (11)1.4.3.3终点选择和时间确定 (12)1.5检测灵敏度 (13)1.5.1非劣效/等效性试验的检测灵敏度 (14)1.5.1.1药效灵敏度的历史性证据和非劣效界值的选择 (15)1.5.1.2试验良好的实施 (19)1.5.2 优效性临床试验的检测灵敏度 (21)2.对照类型的详细考虑 (21)2.1安慰剂对照 (21)2.1.1描述(参阅1.3.1) (21)2.1.2减少偏倚的能力 (23)2.1.3伦理学问题 (23)2.1.4 安慰剂对照研究的用途和在特定情况下推断的正确性 (24)2.1.5设计方案的修改以及结合其他对照药解决伦理的、实践的或推断问题 (25)2.1.5.1其他对照组 (26)2.1.5.1.1三臂研究;安慰剂和活性对照 .. 262.1.5.1.2 其他剂量 (26)2.1.5.1.3析因/联合研究 (26)2.1.5.2研究设计的其他改变 (27)2.1.6安慰剂对照试验的优点 (31)2.1.6.1能可靠地证明药物的有效性 (31)2.1.6.3高效率 (32)2.1.6.4最小化受试者和研究者期望值的影响. 322.1.7安慰剂对照试验的缺点 (32)2.1.7.1伦理方面的问题(见2.1.3和2.1.4).. 322.1.7.2病人和医师的顾虑 (33)2.1.7.3通用性 (33)2.1.7.4无比较信息 (34)2.2无治疗平行对照(见1.3.2) (34)2.3剂量-效应平行对照(见1.3.3) (34)2.3.1描述 (34)2.3.2减少偏倚的能力 (36)2.3.3伦理学问题 (36)2.3.4剂量-效应研究的用途和特定情况下推断的正确性 (36)2.3.5修改设计和结合其他对照解决伦理的、实践的或推断问题 (36)2.3.6剂量-效应试验的优点 (37)2.3.6.1高效率 (37)2.3.6.2可能的伦理方面优点 (37)2.3.7剂量-效应研究的缺点 (37)2.4活性药物对照(见1.3.4) (38)2.4.1描述 (38)2.4.2减少偏倚的能力 (39)2.4.3伦理学问题 (39)2.4.4活性对照试验的应用及特定情况下推断的正确性 (40)2.4.5修改设计和结合其他对照解决伦理的、实践的问题或推断问题 (40)2.4.6活性对照试验的优点 (41)2.4.6.1伦理和实践的优点 (41)2.4.6.2信息含量 (41)2.4.7活性对照试验的缺点 (42)2.4.7.1信息含量 (42)2.4.7.2样本量大 (42)2.5外部对照(包括历史对照,见1.3.5部分) (42)2.5.1描述 (42)2.5.2减少偏倚的能力 (43)2.5.3伦理学问题 (45)2.5.4外部对照试验的用途以及在特定情况下推断的正确性 (46)2.5.5修改设计和结合其他对照解决伦理的、实践的问题或推断问题 (47)2.5.6外部对照试验的优点 (47)2.5.7外部对照试验的缺点 (48)3.选择平行对照组 (48)表1 不同情况下各种平行对照类型的用途 (50)图1 为描述有效性选择平行对照 (51)1.引言在设计临床试验时,选择对照组一直是一个关键性的决定。
0期临床试验研究进展:零期临床试验与一期临床试验的区别

0期临床试验研究进展1.零期临床试验概念的提出创新药物是指具有自主知识产权专利的药物,从实验室发现新的分子或化合物开始,需经过动物实验了解其安全性以及毒性反应、在动物体内的代谢过程、作用部位和效果,再通过首次人体试验和各期临床试验,证实安全有效及质量可控制之后才可以获得药物监管机构的批准,通常经历10到15年的时间,耗资可达数十亿美元。
近年来,为适应市场需要,国内外医药公司和科研工作者致力于开发以抗肿瘤药物、大分子药物为主导的创新药物,但据文献报道大约只有10%创新药物能最后进入市场,而在研究越早期阶段停止问题药物的研发,越能最大程度地降低损失。
如何早期从一组候选化合物中确定最有价值的先导化合物进行后续的研发?如何提高临床前试验结果的预测价值?这些在动物体内安全的药物在人体一定安全吗?它们在人体的组织分布特点如何,是否能在人体内有效地与靶点结合?按照传统模式,回答上述问题需要至少完成传统的Ⅰ 期所有试验,甚至需要Ⅰ 和Ⅰ 期的确认,不仅需要大量受试者暴露于试验药物之中,还需要花费大量时间和金钱,而临床试验过程中一旦药物出现安全性问题,后果更是不堪设想。
为了引导创新药物的快速开发、控制新药研发过程中的临床风险,美国食品和药物管理局(FDA)于2006年颁布了“探索性新药研究”指导原则,提出在进行传统的Ⅰ 期临床试验之前,开展小规模人体“微剂量”研究的思路,即零期临床试验的概念。
2.零期临床试验的定义零期临床试验是指活性化合物在完成临床前试验后未正式进入临床试验之前,研制者使用微剂量在少量健康志愿者或者病人(通常为6~15人)进行的药物试验,收集必要的有关药物安全及药代动力学的试验数据,以评估研发药物是否具有进一步开发为新药或生物制剂的可能性,是从临床前试验过渡到Ⅰ期临床试验的中间环节。
其特点在于“3 个有限”,即有限的受试人数、有限的剂量范围、有限的研究周期。
3.零期临床试验使用范围不是所有类型的候选化合物都适合零期试验,eIND 研究多数集中在肿瘤、艾滋病、心血管疾病、神经系统疾病等个别研究领域。
JAMA美国医学会杂志投稿须知

3500字 50 75参考 ≤4个表格和/或数字 结构化摘要 关键点 小标题应该包括“一个元分析” 遵循EQUATOR报告指南,包括PRISMA和MOOSE
https:///journals/jama/pages/instructions-for-authors
1/29
关爱危重病人
这些手稿是原创的研究报告,最好是临床试验或系统评价(参见上文对文章类别的稿件提交要求分类),几乎涵盖了从预防和分类,复苏和急性治疗到危重病的任何方面,姑 息关怀。手稿为危重病人的诊断,预后和治疗以及探索危重病医学的病理生理,技术,伦理或其他相关方面提供了新的见解。遵循EQUATOR报告指南。对于原始数据和系统评 价的报告,需要一个结构化的摘要; 请参阅准备原始数据报告摘要的说明或摘要评论。需要3个要点清单(见关于准备要点的指导)。最大长度:不超过5个表格和/或数字的文 字(不包括摘要,表格,数字,致谢,参考文献和在线材料)的3000字。
临床试验
https:///journals/jama/pages/instructions-for-authors
回到顶部
3/29
2018/1/10
关于作者的说明| JAMA | JAMA网络
ICMJE将临床试验定义为任何研究项目,前瞻性地将人类参与者分配到干预组或比较组,以研究干预和健康结果之间的因果关系。干预包括但不限于药物,手术程序,装置,行 为治疗,教育程序,饮食干预,质量改善干预,护理过程改变等。所有报告临床试验的手稿,包括那些仅限于对试验结果进行二次探索性或事后分析的手稿,都必须包括一份 试验方案的副本,包括完整的统计分析计划(见方案),CONSORT流程图(图) CONSORT 清单。所有临床试验必须在适当的在线公共登记处进行登记(见试验注册要求)。 有关准备报告集群试验,非劣效性和等效试验以及实用试验的附加指导,请参见CONSORT声明的扩展。每份手稿应清楚地陈述客观或假设; 设计和方法(包括研究设置和日 期,患者或参与者纳入和排除标准,或数据来源,以及如何选择这些研究); 任何干预措施的基本特征; 主要成果措施; 研究的主要成果; 讨论部分将结果与发表的文献结合起 来,解决研究限制; 和结论。遵循EQUATOR报告指南。 必须提供结构化的摘要,并在摘要末尾列出试用注册信息(注册名称,试用ID和URL); 有关更多信息,请参阅准备原始数据报告摘要的说明。需要3个要点清单(见关于准备 要点的指导)。最大长度:不超过5个表格和/或数字的文字(不包括摘要,表格,数字,致谢,参考文献和在线材料)的3000字。小标题应包括“随机临床试验”一词。
IMpower150研究最新OS和亚组分析结果出炉
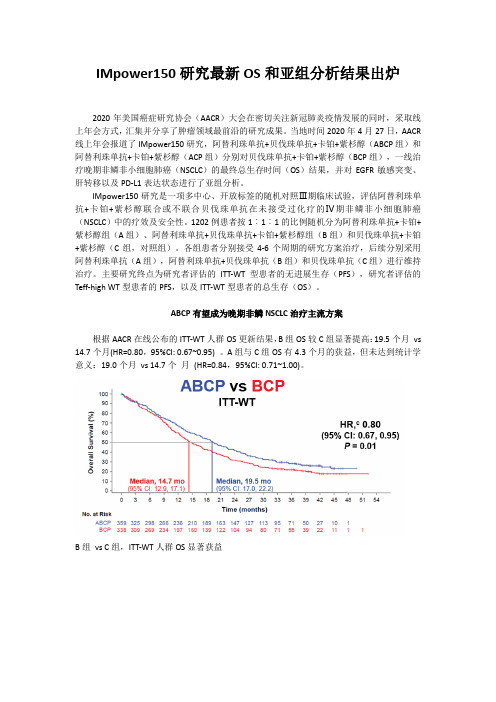
IMpower150研究最新OS和亚组分析结果出炉2020年美国癌症研究协会(AACR)大会在密切关注新冠肺炎疫情发展的同时,采取线上年会方式,汇集并分享了肿瘤领域最前沿的研究成果。
当地时间2020年4月27日,AACR 线上年会报道了IMpower150研究,阿替利珠单抗+贝伐珠单抗+卡铂+紫杉醇(ABCP组)和阿替利珠单抗+卡铂+紫杉醇(ACP组)分别对贝伐珠单抗+卡铂+紫杉醇(BCP组),一线治疗晚期非鳞非小细胞肺癌(NSCLC)的最终总生存时间(OS)结果,并对EGFR敏感突变、肝转移以及PD-L1表达状态进行了亚组分析。
IMpower150研究是一项多中心、开放标签的随机对照Ⅲ期临床试验,评估阿替利珠单抗+卡铂+紫杉醇联合或不联合贝伐珠单抗在未接受过化疗的Ⅳ期非鳞非小细胞肺癌(NSCLC)中的疗效及安全性。
1202例患者按1︰1︰1的比例随机分为阿替利珠单抗+卡铂+紫杉醇组(A组)、阿替利珠单抗+贝伐珠单抗+卡铂+紫杉醇组(B组)和贝伐珠单抗+卡铂+紫杉醇(C组,对照组)。
各组患者分别接受4-6个周期的研究方案治疗,后续分别采用阿替利珠单抗(A组),阿替利珠单抗+贝伐珠单抗(B组)和贝伐珠单抗(C组)进行维持治疗。
主要研究终点为研究者评估的ITT-WT型患者的无进展生存(PFS),研究者评估的Teff-high WT型患者的PFS,以及ITT-WT型患者的总生存(OS)。
ABCP有望成为晚期非鳞NSCLC治疗主流方案根据AACR在线公布的ITT-WT人群OS更新结果,B组OS较C组显著提高:19.5个月vs 14.7个月(HR=0.80,95%CI: 0.67~0.95) 。
A组与C组OS有4.3个月的获益,但未达到统计学意义:19.0个月vs 14.7个月(HR=0.84,95%CI: 0.71~1.00)。
B组vs C组,ITT-WT人群OS显著获益A组vs C组的ITT-WT人群OS结果对于ITT人群A组vs C组的OS结果,需要慎重解读,从OS的绝对数值来看,A组和B 组都达到了19个月以上,而2组都包含了免疫治疗,再次确认了阿替利珠单抗作为免疫药物在方案中带来的价值。
卵巢癌的临床研究进展

0.56
0.34-0.86
Paracetቤተ መጻሕፍቲ ባይዱmol (7+pills) 0.32
0.09-1.10
paracetamol 的应用可降低卵巢癌的发病风险
(Cancer Epidemiol Biomarkers Prev ,2001;10(8):903-6)
非甾体消炎药的应用
Akhmedkhanov(2001)进行病例对照研究,发现
Petricoin(2002)利用已经建立的标准方法测定:卵巢癌患者55例, 均为阳性
➢测定对照组66例 63例结果符合
➢敏感性100% 95% CI 93%-100%
➢特异性95%
95% CI 87%-99%
➢阳性预测值94% 95% CI 84%-89%
卵巢癌筛查新技术——
血清蛋白质组学(proteome)
但人们一直在怀疑肿瘤细胞歼灭术本身对患者生存期延长发挥的作用到 底有多大。
卵巢癌最重要的预后因素
——肿瘤细胞减灭术
Covens(2000)认为卵巢癌患者的预后、生存期、细胞减灭术 能否成功是肿瘤本身内在生物学特性所决定的,并非由手术所左 右。 (Gynecol Oncol,2000;78:269-274)
OR
95%CI
paracetamol 0.90
0.60-1.20
Paracetamol对卵巢癌发病无保护作用
(Int J Cancer, 2002 ;97(1):96-101)
卵巢癌筛查新技术——
血清蛋白质组学(proteome)
➢ 蛋白质组 (proteome)是由澳大利亚学者Wilkins于1994年首 次提出的一个术语,指一个细胞或一个组织的基因组所表达的 全部蛋白质
美国研究型医院:NIH临床中心

美国研究型医院:NIH临床中心李林;蒋义;曹秀堂;刘丽华;田鸥【摘要】为更加深刻认识研究型医院概念内涵与国外研究型医院先进案例建设情况,从愿景使命、工作人员、设施设备、财务预算、历史沿革等方面介绍了美国国立卫生研究院临床中心基本情况,分析了该临床中心患者来源、医学培训、临床工作和科学研究模式,剖析了其治理结构和组织架构,为国内研究型医院建设提供参考借鉴.【期刊名称】《中国医院》【年(卷),期】2016(020)012【总页数】3页(P78-80)【关键词】研究型医院;NIH临床中心;组织结构【作者】李林;蒋义;曹秀堂;刘丽华;田鸥【作者单位】解放军总医院,100853 北京市海淀区复兴路28号;解放军总医院,100853 北京市海淀区复兴路28号;解放军总医院,100853 北京市海淀区复兴路28号;解放军总医院,100853 北京市海淀区复兴路28号;解放军总医院,100853 北京市海淀区复兴路28号【正文语种】中文研究型医院是以新医学知识和新医疗技术的产生与传播为使命,坚持临床与科研并举的医院发展模式。
我国创建研究型医院的目的就是要把医院建设成为医疗卫生战略的科技创新基地,通过深化拓展医院的功能定位、职责使命、目标任务,为人才成长、技术创新、学科进步、成果涌现创造支撑条件,使我国卫生事业步入世界发展前列,为实现中国梦、健康梦提供有力支撑。
创建研究型医院对于我国是一项崭新的事业,缺乏现成的经验遵循。
本研究就美国国立卫生研究院(NIH)临床中心的基本情况、设施设备、历史沿革、临床工作、科学研究、管理架构等方面展开介绍,以期为我国研究型医院建设提供参考借鉴。
1.1 基本情况NIH隶属于美国健康与人类服务部(department of health and human services,DHHS),是其5个公共卫生服务机构之一。
NIH临床中心是一所专门开展临床研究的医院,是NIH的研究型医院,其功能是为NIH的其他机构和员工开展的临床研究提供支撑。
NCCN淋巴瘤治疗指南【42页】

TdT+(除非它们是BL或ALL-L3 )
• 细胞遗传学: Burkitt淋巴瘤8号异常t(8;22)
R R
t(2;8),t整(8理课;1件4)
38
Burkitt 淋巴瘤
低 正常LDH
危险判断
完全切除腹部病灶
或单个腹部外病灶
高
注意预防肿瘤溶解综合症
R
R
整理课件
39
诱导治疗c
随访
复发
低危
临床试验或 CR—每2个月×1年
R
整理课件
12
最初措施
否
治疗指征 ⅠⅡ期复发 Ⅱ期巨大腹
部病变
是
Ⅲ 、 Ⅳ期
R R
观察
每3m体检一次×1年
以后3-6m一次
基于临床表现—形象学
临床试验
或
CR
局部RT
PR 进展
或
单因子或联合化疗±干扰素
或
无反应
抗体为基础的治疗±化疗
进展
整理课件
13
疾病进展
疾病进展应排除转化(组织学证据)
尤在下列情况:LDH
或选择病例继观 考虑其他抗生素治疗
HP(+)淋巴瘤(+) 局部RT(若既往未治 )
考虑其他抗生素
R
R
整理课件
25
随访内窥镜
• 最佳时间不清楚
• 结果——CR——观察——内镜随访
RT后复发——参阅滤泡性淋巴瘤
治疗指征
抗生素治疗后复发:系统性:同上
局灶性:RT
R
R
整理课件
26
套细胞淋巴瘤
• 免疫表型:CD5+ CD10± CD20+ CD23-
3.5%利多卡因眼用凝胶的研究进展

3.5%利多卡因眼用凝胶的研究进展林通;龚岚【摘要】局部麻醉药在眼科的各类操作和手术中应用广泛,目前临床上使用的主要是表面麻醉药滴眼液。
3.5%利多卡因眼用凝胶是眼科表面麻醉用新品,与表面麻醉药滴眼液相比,具有麻醉效果更好、有效麻醉作用时间更长、术中重复给药次数减少、角膜上皮毒性降低并能保护眼表等特性。
本文介绍3.5%利多卡因眼用凝胶及其药代动力学、临床疗效和安全性方面的研究进展。
%Topical anesthetics play an important role in both ophthalmic procedures and surgeries. Topical anesthetic eye drops are primarily used for ocular anesthesia in the current practice while 3.5% lidocaine ophthalmic gel is a novel ocular anesthesia. Compared with topical anesthesia eye drops, this ophthalmic gel has some advantages, including better anesthetic effect and longer duration, less times for intraoperative repeated administration, lower corneal epithelial toxicity and better ocular surface protection. The history of ophthalmic topical anesthetics is brielfy reviewed and the characteristics, pharmacokinetics, clinical efifcacy and safely of 3.5% lidocaine ophthalmic gel are also introduced in this article.【期刊名称】《上海医药》【年(卷),期】2016(037)021【总页数】4页(P6-9)【关键词】利多卡因;眼用凝胶;麻醉药【作者】林通;龚岚【作者单位】复旦大学附属眼耳鼻喉科医院眼科、卫生部近视重点实验室上海200031;复旦大学附属眼耳鼻喉科医院眼科、卫生部近视重点实验室上海 200031【正文语种】中文【中图分类】R988.1局部麻醉在眼科的各类操作和手术中应用广泛,较全身麻醉或不麻醉更加安全、有效。
nsabp研究综述

美国乳腺与肠道外科辅助治疗研究组(NSABP) 乳腺癌临床试验进展(复旦大学附属肿瘤医院乳腺外科,上海200032)美国乳腺与肠道外科辅助治疗研究组(National Surgical Adjuvant Breast and Bowel Project , 简称NSABP) 成立于1971 年,是一个进行大规模乳腺癌和结、直肠癌临床试验的合作团体。
NSABP的成员包括了美国、加拿大和澳大利亚的近300 个医学中心,6 000 多名工作人员。
NSABP 有关乳腺癌的临床试验包括治疗性试验和预防性试验 1958 年一、治疗性试验(一) NSABP B-04 开始于1971 年7 月和B-06 试验NSABP B-04 试验开始于1971 年7 月,NSABP B-04 试验拟解决以下问题:1. 对于腋淋巴结临床阴性的乳腺癌患者:①行乳房单纯切除术后,待发展为淋巴结临床阳性后再行腋淋巴结清扫术(简称TM) ;②行乳房单纯切除术后, 再加行腋淋巴结放疗( 简称TMR) 。
以上两种方法与乳腺癌根治术(简称RM)比较,效果是否相同。
2. 对于腋淋巴结阳性的患者,TMR 与RM 的效果是否相同。
该试验共征集到1 665名有条件行乳腺癌根治术的患者参与该试验,腋淋巴结阴性的患者随机分为TM、TMR 和RM 三组,腋淋巴结阳性的患者随机分为TMR 和RM 两组。
经过10 年的随访,NSABP 于1985 年公布结果如下:在腋淋巴结临床阴性的乳腺癌患者中,RM、TMR 和TM 三组的无瘤生存率(简称DFS) 、无远处转移生存率(简称DDFS) 和总生存率(简称OS)无显著性差异;在腋淋巴结阳性的乳腺癌患者中,RM 和TMR 的DFS、DDFS 和OS 也无显著性差异。
同时,乳腺肿瘤的发生位置不影响病人的预后,肿瘤位于乳头内侧病人行内乳区淋巴结的放疗也不提高生存率。
在试验中还发现,在腋淋巴结阴性的RM组病人中,有40 %的病人术后病理发现腋淋巴结为阳性,因为该试验为随机分组,故推测TM 组中同样有40 %病人腋淋巴结病理阳性,但10 年内腋淋巴结发展为临床阳性的病人仅18 % ,同时其他复发和转移的发生率并没有增加。
DMD研究进展概述问题-TREAT-NMD

DMD研究进展概述——2015年8月更新翻译:柏林夏丽特医学院,赛福地非盈利组织丁灿医生审校:华中科技大学同济医学院附属同济医院王伟医生、潘邓记医生天津医科大学尹海芳教授香港大学陈凯珊医生本概述旨在于为患者及其家人提供杜兴氏肌营养不良症(以下缩写为DMD)治疗方法的研究进展,描述每种治疗方法的优缺点,并列出这些新治疗方法从实验室进入有患者参与的临床实验过程中需要克服的各种障碍。
备注:研发DMD治疗是一个飞速发展,包含广泛内容的大领域。
在一篇简要概述中不可能囊括每一方面的内容。
因此,本概述提供主要的治疗方法全面的发展。
一份由Gȕnter Scheuerbrandt博士翻译成几种语言杜氏疗法的补充概述,也可以在DMD本节找到。
链接线可以在这个页面的左侧底部找到。
问题杜兴氏肌营养不良症以首次报导该病的法国医师杜兴(Duchenne)命名,缩写为DMD (人类门德尔遗传病库OMIM 编号310200),发病率为每3600-6000个男孩中就有一人患此病。
本病是由dystrophin基因突变导致,该基因位于X染色体短臂 Xp21.2基因座。
在肌肉细胞中,dystrophin基因被翻译成dystrophin蛋白。
该蛋白的主要功能是将肌纤维骨架与肌纤维外的保护层连接起来,在肌肉收缩时 (运动) 这联机有稳定肌纤的作用。
如图1所示,我们可以把肌纤维骨架比作船锚,肌纤维外保护层可以比作船,那么Dystrophin蛋白就是连接两者的绳子。
功能丧失,船失去了船锚。
因此,DMD患者的肌纤维非常容易受损,肌肉在-般运动过程中都有可能受损伤。
因翻译出只具有部分功能的蛋白(如图3,船与船锚仍然可以相连,但是明显短一些)。
这样的一类基因突变会导致临床症状较轻的贝克型肌营养不良。
正在发展的治疗方法目前有许多种治疗方法正在研究中,其中的大多数均需要遵循相同的过程,由临床前期研究开始而发展到后临床研究。
首先会在取自患者的细胞中测试该治疗方法,之后在疾病动物模型中测试(通常为mdx小鼠模型)—若在细胞和动物实验中均得出信服的结果—可继续执行有患者参与的试验(临床试验)。