富马酸泰诺福韦酯标准 USP MC Tenofovir Disoporoxil Fumarate - 2012-07-25
富马酸替诺福韦二吡呋酯片说明书
核准日期:年月日修改日期:年月日富马酸替诺福韦二吡呋酯片说明书请仔细阅读说明书并在医师指导下使用【药品名称】通用名称:富马酸替诺福韦二吡呋酯片英文名称:Tenofovir Disoproxil Fumarate Tablets汉语拼音:FumasuanTinuofuwei’erbifuzhi Pian【成份】本品主要成份为富马酸替诺福韦二吡呋酯。
化学名称:9-[(R)-2[[双[[(异丙氧基羰基)氧基]甲氧基]氧膦基]甲氧基]-丙基]腺嘌呤富马酸盐(1:1)。
化学结构式:分子式:C19H30N5O10P·C4H4O4分子量:635.52【性状】本品为薄膜衣片,除去包衣后显白色。
【适应症】HIV-1感染富马酸替诺福韦二吡呋酯适用于与其他抗反转录病毒药物联用,治疗成人HIV-1感染。
使用富马酸替诺福韦二吡呋酯开始治疗HIV-1感染时,应考虑以下几点:富马酸替诺福韦二吡呋酯不应与含有替诺福韦的固定剂量复方制剂联用,包括:●依非韦伦/恩曲他滨/富马酸替诺福韦二吡呋酯、●利匹韦林/恩曲他滨/富马酸替诺福韦二吡呋酯、●恩曲他滨/丙酚替诺福韦、●艾维雷韦/考比司他/恩曲他滨/丙酚替诺福韦、●恩曲他滨/利匹韦林/丙酚替诺福韦、●艾维雷韦/考比司他/恩曲他滨/富马酸替诺福韦二吡呋酯、●恩曲他滨替诺福韦、●丙酚替诺福韦慢性乙型肝炎富马酸替诺福韦二吡呋酯适用于治疗慢性乙肝成人和≥12岁的儿童患者。
在开始使用富马酸替诺福韦二吡呋酯治疗HBV感染时,应考虑到以下要点:•成人患者中该适应症的确立基于从初次接受核苷治疗的受试者和既往接受过治疗且证实拉米夫定耐药的受试者中获得的安全性和疗效数据。
受试者为肝功能代偿的HBeAg阳性和HBeAg阴性慢性乙肝成人受试者。
•富马酸替诺福韦二吡呋酯在数量有限的患有失代偿期肝病的慢性乙肝受试者中进行过评价。
•临床试验中基线时存在阿德福韦相关突变的受试者数量太少,因此尚无法对疗效下结论。
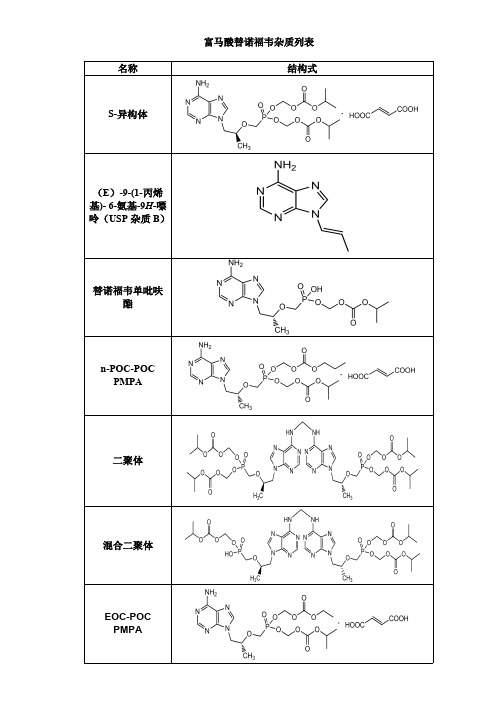
富马酸替诺福韦杂质列表
深圳菲斯是专业进口标准品代理商,主要品牌有 TRC、TLC、Molcan、EP、BP、 USP、Sinco 等品牌标准品。 1.替卡格雷杂质 2.利伐沙班杂质 3.阿考替胺杂质 4.普拉克索杂质 5.阿哌沙班杂 质 6.氨氯地平杂质 7.硼替佐米杂质 8.比索洛尔杂质 9.阿比特龙杂质 10.非布司 他杂质 11.安立生坦杂质 12.依折麦布杂质 13.厄洛替尼杂质 14.索拉非尼杂质 15. 维格列汀杂质 16.阿伐那非杂质 17.托法替尼杂质 18.米格列奈杂质 19.沃替西汀 杂质 20 尼贝地平杂质 21.艾帕列净杂质 22.阿普斯特杂质 23.门冬氨酸缩合物 24. 依托考昔杂质 25.达格列净杂质 26.尼达尼布杂质 27.托匹司他杂质 28.坎格列净 杂质 29.帕泊昔利布杂质 30.依鲁替尼杂质 31.盐酸氨溴索杂质 32.达比加群酯杂 质等库存 库存新产品,当天发货!
富马酸替诺福韦杂质列表 名称 结构式
S-异构体
(E)-9-(1-丙烯 基)- 6-氨基-9H-嘌 呤(USP 杂质 B)
替诺福韦单吡呋 酯
n-POC-POC PMPA
二聚体
混合二聚体
EOC-POC PMPA
DEC-POC PMPA
Des-Methyl TDF (去甲基替诺福 韦二吡呋酯)
MOC-POC PMPA
富马酸替诺福韦酯--------印度药典

NOTE - Prepare the solutions immediately before use. Test solution. Dissolve 100 mg of the substance under examination in 50 ml of methanol. Reference solution (a). A 0.2 per cent w/v solution of tenofovir disoproxil jitmarate RS in methanol. Reference solution (b). Dilute 1.0 ml ofreference solution (a) to 100.0 ml with methanol. Reference solution (c). Dissolve 10 mg ofthejUmaric acid in 50 ml of methanol.
TELMISARTAN TABLETS
IP 2010
Solvent mixture. 80 volumes of buffer solution prepared by diluting 5.0 ml of triethylamine to 2000ml with water and 20 volumes of methanol. Test solution. Weigh and powder 20 tablets. Disperse a quantity ofpowder containing about 100 mg ofTelmisartan in 100.0 ml ofsolvent mixture, sonicate for 45 minutes and filter. Reference solution. A 0.0005 per cent w/v solution of telmisartan RS in the solvent mixture.
富马酸替诺福韦酯杂质

富马酸替诺福韦酯杂质富马酸替诺福韦酯(Tenofovir disoproxil fumarate,CAS:202138-50-9)中文名:富马酸替诺福韦酯杂质A英文名:Tenofovir disoproxil fumarate impurity A规格:10mg-100mg纯度:>95%用途:实验室分析及新药研究中文名:富马酸替诺福韦酯杂质B英文名:Tenofovir disoproxil fumarate impurity B规格:10mg-100mg纯度:>95%用途:实验室分析及新药研究中文名:富马酸替诺福韦酯杂质C英文名:Tenofovir disoproxil fumarate impurity C规格:10mg-100mg纯度:>95%用途:实验室分析及新药研究中文名:富马酸替诺福韦酯杂质D英文名:Tenofovir disoproxil fumarate impurity D规格:10mg-100mg纯度:>95%用途:实验室分析及新药研究中文名称:富马酸替诺福韦酯中文别名:(R)-9-(2-磷酸甲氧基丙基)腺嘌呤二(异丙氧羰基氧甲基)酯富马酸盐;富马酸泰诺福韦酯;富马酸替诺福韦二吡呋酯英文名称:Tenofovir disoproxil fumarate英文别名:9-((R)-2-((Bis(((isopropoxycarbonyl)oxy)methoxy)phosphinyl)methoxy)propyl)adenine fumarate; bis({[(1-methylethoxy)carbonyl]oxy}methyl){[(1R)-2-(6-amino-9H-purin-9-yl)-1-methylethoxy]methyl}phosphonate(2E)-but-2-enedioate CAS号:202138-50-9该产品信息由广州优瓦仪器有限公司整理上传,相关杂质还有新利司他杂质、沃替西汀杂质、巴多昔芬杂质、醋酸巴多昔芬杂质、非马沙坦杂质、芬戈莫德杂质、枸橼酸托法替尼杂质、瑞替加滨杂质等,品牌有CATO、USP、Reagecon、EP、BP、TRC、TLC020-********。
富马酸泰诺福韦酯片说明书(英文)

HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use VIREAD safely and effectively. See full prescribing informationfor VIREAD.VIREAD® (tenofovir disoproxil fumarate) tabletsInitial U.S. Approval: 2001WARNINGS: LACTIC ACIDOSIS/SEVERE HEPATOMEGALY WITH STEATOSIS and POST TREATMENT EXACERBATION OFHEPATITISSee full prescribing information for complete boxed warning. • Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use ofnucleoside analogs, including VIREAD. (5.1)• Severe acute exacerbations of hepatitis have been reported in HBV-infected patients who have discontinued anti-hepatitis B therapy, including VIREAD. Hepatic functionshould be monitored closely in these patients. Ifappropriate, resumption of anti-hepatitis B therapy may bewarranted. (5.2)---------------------------RECENT MAJOR CHANGES--------------------------- Indications and Usage (1.2) 10/2010 Dosage and Administration (2.1, 2.2, 2.3) 10/2010 Warnings and PrecautionsNew Onset or Worsening Renal Impairment (5.3) 10/2009Decreases in Bone Mineral Density (5.6) 03/2010----------------------------INDICATIONS AND USAGE--------------------------- VIREAD is a nucleotide analog HIV-1 reverse transcriptase inhibitor and an HBV reverse transcriptase inhibitor.VIREAD is indicated in combination with other antiretroviral agents for the treatment of HIV-1 infection in adults and pediatric patients 12 years of age and older. (1)VIREAD is indicated for the treatment of chronic hepatitis B in adults. (1)---------------------------DOSAGE AND ADMINISTRATION-------------------- • Recommended dose for the treatment of HIV-1 or chronic hepatitis B in adults: 300 mg once daily taken orally withoutregard to food. (2.1)• Recommended dose for the treatment of HIV-1 in pediatric patients (≥12 years of age and ≥35 kg): 300 mg once daily taken orally without regard to food. (2.2)• Dose recommended in renal impairment in adults:Creatinine clearance 30-49 mL/min: 300 mg every 48 hours.(2.3)Creatinine clearance 10-29 mL/min: 300 mg every 72 to 96 hours.(2.3)Hemodialysis: 300 mg every 7 days or after approximately 12hours of dialysis. (2.3)-----------------------DOSAGE FORMS AND STRENGTHS-------------------- Tablets: 300 mg. (3)--------------------------------CONTRAINDICATIONS------------------------------ None. (4)--------------------------WARNINGS AND PRECAUTIONS--------------------- • New onset or worsening renal impairment: Can include acute renal failure and Fanconi syndrome. Assess creatinine clearance (CrCl) before initiating treatment with VIREAD. Monitor CrCl and serum phosphorus in patients at risk. Avoid administeringVIREAD with concurrent or recent use of nephrotoxic drugs. (5.3) • Coadministration with Other Products: Do not use with other tenofovir-containing products (e.g., ATRIPLA and TRUVADA). Do not administer in combination with HEPSERA. (5.4) • HIV testing: HIV antibody testing should be offered to all HBV-infected patients before initiating therapy with VIREAD. VIREAD should only be used as part of an appropriate antiretroviralcombination regimen in HIV-infected patients with or without HBV coinfection. (5.5)• Decreases in bone mineral density (BMD): Observed in HIV-infected patients. Consider assessment of BMD in patients with a history of pathologic fracture or other risk factors for osteoporosis or bone loss. (5.6)• Redistribution/accumulation of body fat: Observed in HIV-infected patients receiving antiretroviral combination therapy. (5.7)• Immune reconstitution syndrome: Observed in HIV-infected patients. May necessitate further evaluation and treatment. (5.8)• Triple nucleoside-only regimens: Early virologic failure has been reported in HIV-infected patients. Monitor carefully and considertreatment modification. (5.9)-----------------------------ADVERSE REACTIONS--------------------------------In HIV-infected subjects: Most common adverse reactions (incidence ≥10%, Grades 2 - 4) are rash, diarrhea, headache, pain, depression, asthenia, and nausea. (6)In HBV-infected subjects with compensated liver disease: most common adverse reaction (all grades) was nausea (9%). (6)In HBV-infected subjects with decompensated liver disease: most common adverse reactions (incidence ≥10%, all grades) were abdominal pain, nausea, insomnia, pruritus, vomiting, dizziness, and pyrexia. (6)To report SUSPECTED ADVERSE REACTIONS, contact Gilead Sciences, Inc. at 1-800-GILEAD-5 or FDA at 1-800-FDA-1088 or /medwatch------------------------------DRUG INTERACTIONS--------------------------------• Didanosine:Coadministration increases didanosineconcentrations. Use with caution and monitor for evidence ofdidanosine toxicity (e.g., pancreatitis, neuropathy). Considerdose reductions or discontinuations of didanosine if warranted.(7.1)• Atazanavir:Coadministration decreases atazanavirconcentrations and increases tenofovir concentrations. Useatazanavir with VIREAD only with additional ritonavir; monitor for evidence of tenofovir toxicity. (7.2)• Lopinavir/ritonavir:Coadministration increases tenofovir concentrations. Monitor for evidence of tenofovir toxicity. (7.3)----------------------------USE IN SPECIFIC POPULATIONS-------------------• Pregnancy: There is a registry available. Enroll patients by calling 1-800-258-4263.• Nursing mothers: Women infected with HIV should be instructed not to breast feed. (8.3)• Safety and efficacy not established in patients less than 12 years of age. (8.4)See 17 for PATIENT COUNSELING INFORMATION and FDA-Approved Patient LabelingRevised: October 2010 FULL PRESCRIBING INFORMATION: CONTENTS*WARNINGS: LACTIC ACIDOSIS/SEVERE HEPATOMEGALY WITH STEATOSIS and POST TREATMENT EXACERBATION OF HEPATITIS1 INDICATIONS AND USAGE1.1 HIV-1 Infection1.2 Chronic Hepatitis B2 DOSAGE AND ADMINISTRATION2.1 Recommended Dose in Adults2.2 Recommended Dose in Pediatric Patients (≥12 Years ofAge and ≥35 kg)2.3 Dose Adjustment for Renal Impairment in Adults3 DOSAGE FORMS AND STRENGTHS4 CONTRAINDICATIONS5 WARNINGS AND PRECAUTIONS5.1 Lactic Acidosis/Severe Hepatomegaly with Steatosis5.2 Exacerbation of Hepatitis after Discontinuation ofTreatment5.3 New Onset or Worsening Renal Impairment5.4 Coadministration with Other Products5.5 Patients Coinfected with HIV-1 and HBV5.6 Decreases in Bone Mineral Density5.7 Fat Redistribution5.8 Immune Reconstitution Syndrome5.9 Early Virologic Failure6 ADVERSE REACTIONS6.1 Adverse Reactions from Clinical Trials Experience6.2 Postmarketing Experience7 DRUG INTERACTIONS7.1 Didanosine7.2 Atazanavir7.3 Lopinavir/Ritonavir7.4 Drugs Affecting Renal Function8 USE IN SPECIFIC POPULATIONS8.1 Pregnancy8.3 Nursing Mothers8.4 Pediatric Use8.5 Geriatric Use8.6 Patients with Impaired Renal Function10 OVERDOSAGE11 DESCRIPTION12 CLINICAL PHARMACOLOGY12.1 Mechanism of Action12.3 Pharmacokinetics12.4 Microbiology13 NONCLINICAL TOXICOLOGY13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility13.2 Animal Toxicology and/or Pharmacology14 CLINICAL STUDIES14.1 Clinical Efficacy in Patients with HIV-1 Infection14.2 Clinical Efficacy in Patients with Chronic Hepatitis B16 HOW SUPPLIED/STORAGE AND HANDLING17 PATIENT COUNSELING INFORMATION AND FDAAPPROVED PATIENT LABELING* Sections or subsections omitted from the full prescribing information are not listedFULL PRESCRIBING INFORMATIONWARNINGS: LACTIC ACIDOSIS/SEVERE HEPATOMEGALY WITHSTEATOSIS and POST TREATMENT EXACERBATION OF HEPATITISLactic acidosis and severe hepatomegaly with steatosis, including fatalcases, have been reported with the use of nucleoside analogs, including VIREAD, in combination with other antiretrovirals [See Warnings and Precautions (5.1)].Severe acute exacerbations of hepatitis have been reported in HBV-infected patients who have discontinued anti-hepatitis B therapy, including VIREAD. Hepatic function should be monitored closely with both clinical andlaboratory follow-up for at least several months in patients who discontinueanti-hepatitis B therapy, including VIREAD. If appropriate, resumption ofanti-hepatitis B therapy may be warranted [See Warnings and Precautions(5.2)].1 INDICATIONS AND USAGEInfection1.1 HIV-1VIREAD® is indicated in combination with other antiretroviral agents for the treatment of HIV-1 infection in adults and pediatric patients 12 years of age and older.The following points should be considered when initiating therapy with VIREAD for the treatment of HIV-1 infection:• VIREAD should not be used in combination with TRUVADA® or ATRIPLA® [See Warnings and Precautions (5.4)].1.2 Chronic Hepatitis BVIREAD is indicated for the treatment of chronic hepatitis B in adults.The following points should be considered when initiating therapy with VIREAD for the treatment of HBV infection:• This indication is based primarily on data from treatment of subjects who were nucleoside-treatment-naïve and a smaller number of subjects who had previously received lamivudine or adefovir dipivoxil. Subjects were adults with HBeAgpositive and HBeAg-negative chronic hepatitis B with compensated liver disease [See Clinical Studies (14.2)].• VIREAD was evaluated in a limited number of subjects with chronic hepatitis B and decompensated liver disease. [See Adverse Reactions (6.1), Clinical Studies(14.2)]• The numbers of subjects in clinical trials who had lamivudine- or adefovirassociated substitutions at baseline were too small to reach conclusions ofefficacy [See Microbiology (12.4), Clinical Studies (14.2)].2 DOSAGE AND ADMINISTRATION2.1 Recommended Dose in AdultsFor the treatment of HIV-1 or chronic hepatitis B: The dose is one 300 mg VIREAD tablet once daily taken orally, without regard to food.In the treatment of chronic hepatitis B, the optimal duration of treatment is unknown.2.2 Recommended Dose in Pediatric Patients (≥12 Years of Age and ≥35 kg) For the treatment of HIV-1 in pediatric patients 12 years of age and older with body weight ≥35 kg (≥77 lb): The dose is one 300 mg VIREAD tablet once daily taken orally, without regard to food.2.3 Dose Adjustment for Renal Impairment in AdultsSignificantly increased drug exposures occurred when VIREAD was administered to subjects with moderate to severe renal impairment [See Clinical Pharmacology (12.3)]. Therefore, the dosing interval of VIREAD should be adjusted in patients with baseline creatinine clearance <50 mL/min using the recommendations in Table 1. These dosing interval recommendations are based on modeling of single-dose pharmacokinetic data in non-HIV and non-HBV infected subjects with varying degrees of renal impairment, including end-stage renal disease requiring hemodialysis. The safety and effectiveness of these dosing interval adjustment recommendations have not been clinically evaluated in patients with moderate or severe renal impairment, therefore clinical response to treatment and renal function should be closely monitored in these patients [See Warnings and Precautions (5.3)].No dose adjustment is necessary for patients with mild renal impairment (creatinine clearance 50–80 mL/min). Routine monitoring of calculated creatinine clearance and serum phosphorus should be performed in patients with mild renal impairment [See Warnings and Precautions (5.3)].Table 1 Dosage Adjustment for Patients with Altered Creatinine ClearanceCreatinine Clearance(mL/min)aHemodialysis Patients≥50 30–49 10–29Recommended 300 mg Dosing Interval Every 24hoursEvery 48hoursEvery 72 to96 hoursEvery 7 days or after a total ofapproximately 12 hours ofdialysis ba. Calculated using ideal (lean) body weight.b. Generally once weekly assuming three hemodialysis sessions a week of approximately 4 hours duration.VIREAD should be administered following completion of dialysis.The pharmacokinetics of tenofovir have not been evaluated in non-hemodialysis patients with creatinine clearance <10 mL/min; therefore, no dosing recommendation is available for these patients.No data are available to make dose recommendations in pediatric patients 12 years of age and older with renal impairment.3 DOSAGE FORMS AND STRENGTHSVIREAD is available as tablets. Each tablet contains 300 mg of tenofovir disoproxil fumarate, which is equivalent to 245 mg of tenofovir disoproxil. The tablets are almond-shaped, light blue, film-coated, and debossed with “GILEAD” and “4331” on one side and with “300” on the other side.4 CONTRAINDICATIONSNone.5 WARNINGS AND PRECAUTIONS5.1 Lactic Acidosis/Severe Hepatomegaly with SteatosisLactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogs, including VIREAD, in combination with other antiretrovirals. A majority of these cases have been in women. Obesity and prolonged nucleoside exposure may be risk factors. Particular caution should be exercised when administering nucleoside analogs to any patient with known risk factors for liver disease; however, cases have also been reported in patients with no known risk factors. Treatment with VIREAD should be suspended in any patient who develops clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity (which may include hepatomegaly and steatosis even in the absence of marked transaminase elevations).5.2 Exacerbation of Hepatitis after Discontinuation of Treatment Discontinuation of anti-HBV therapy, including VIREAD, may be associated with severe acute exacerbations of hepatitis. Patients infected with HBV who discontinue VIREAD should be closely monitored with both clinical and laboratory follow-up for at least several months after stopping treatment. If appropriate, resumption of anti-hepatitis B therapy may be warranted.5.3 New Onset or Worsening Renal ImpairmentTenofovir is principally eliminated by the kidney. Renal impairment, including cases of acute renal failure and Fanconi syndrome (renal tubular injury with severe hypophosphatemia), has been reported with the use of VIREAD [See Adverse Reactions (6.2)].It is recommended that creatinine clearance be calculated in all patients prior to initiating therapy and as clinically appropriate during therapy with VIREAD. Routine monitoring of calculated creatinine clearance and serum phosphorus should be performed in patients at risk for renal impairment, including patients who have previously experienced renal events while receiving HEPSERA®.Dosing interval adjustment of VIREAD and close monitoring of renal function are recommended in all patients with creatinine clearance <50 mL/min [See Dosage and Administration (2.3)]. No safety or efficacy data are available in patients with renal impairment who received VIREAD using these dosing guidelines, so the potential benefit of VIREAD therapy should be assessed against the potential risk of renal toxicity.VIREAD should be avoided with concurrent or recent use of a nephrotoxic agent.5.4 Coadministration with Other ProductsVIREAD should not be used in combination with the fixed-dose combination products TRUVADA or ATRIPLA since tenofovir disoproxil fumarate is a component of these products.VIREAD should not be administered in combination with HEPSERA (adefovir dipivoxil) [See Drug Interactions (7.4)].Coinfected with HIV-1 and HBV5.5 PatientsDue to the risk of development of HIV-1 resistance, VIREAD should only be used in HIV-1 and HBV coinfected patients as part of an appropriate antiretroviral combination regimen.HIV-1 antibody testing should be offered to all HBV-infected patients before initiating therapy with VIREAD. It is also recommended that all patients with HIV-1 be tested for the presence of chronic hepatitis B before initiating treatment with VIREAD.5.6 Decreases in Bone Mineral DensityAssessment of bone mineral density (BMD) should be considered for adults and pediatric patients 12 years of age and older who have a history of pathologic bone fracture or other risk factors for osteoporosis or bone loss. Although the effect of supplementation with calcium and vitamin D was not studied, such supplementation may be beneficial for all patients. If bone abnormalities are suspected then appropriate consultation should be obtained.In HIV-1 infected adult subjects treated with VIREAD in Study 903 through 144 weeks, decreases from baseline in BMD were seen at the lumbar spine and hip in both arms of the study. At Week 144, there was a significantly greater mean percentage decrease from baseline in BMD at the lumbar spine in subjects receiving VIREAD + lamivudine + efavirenz (-2.2% ± 3.9) compared with subjects receiving stavudine + lamivudine + efavirenz (-1.0% ± 4.6). Changes in BMD at the hip were similar between the two treatment groups (-2.8% ± 3.5 in the VIREAD group vs. -2.4% ± 4.5 in the stavudine group). In both groups, the majority of the reduction in BMD occurred in the first 24–48 weeks of the study and this reduction was sustained through Week 144. Twenty-eightpercent of VIREAD-treated subjects vs. 21% of the stavudine-treated subjects lost at least 5% of BMD at the spine or 7% of BMD at the hip. Clinically relevant fractures (excluding fingers and toes) were reported in 4 subjects in the VIREAD group and 6 subjects in the stavudine group. In addition, there were significant increases in biochemical markers of bone metabolism (serum bone-specific alkaline phosphatase, serum osteocalcin, serum C-telopeptide, and urinary N-telopeptide) in the VIREAD group relative to the stavudine group, suggesting increased bone turnover. Serum parathyroid hormone levels and 1,25 Vitamin D levels were also higher in the VIREAD group. Except for bone specific alkaline phosphatase, these changes resulted in values that remained within the normal range.In a clinical study of HIV-1 infected pediatric subjects 12 years of age and older (Study 321), bone effects were similar to adult subjects. Under normal circumstances BMD increases rapidly in this age group. In this study, the mean rate of bone gain was less in the VIREAD-treated group compared to the placebo group. Six VIREAD treated subjects and one placebo treated subject had significant (>4%) lumbar spine BMD loss in 48 weeks. Among 28 subjects receiving 96 weeks of VIREAD, Z-scores declined by -0.341 for lumbar spine and -0.458 for total body. Skeletal growth (height) appeared to be unaffected. Markers of bone turnover in VIREAD-treated pediatric subjects 12 years of age and older suggest increased bone turnover, consistent with the effects observed in adults.The effects of VIREAD-associated changes in BMD and biochemical markers on long-term bone health and future fracture risk are unknown.Cases of osteomalacia (associated with proximal renal tubulopathy and which may contribute to fractures) have been reported in association with the use of VIREAD [See Adverse Reactions (6.2)].The bone effects of VIREAD have not been studied in patients with chronic HBV infection.Redistribution5.7 FatIn HIV-infected patients redistribution/accumulation of body fat including central obesity, dorsocervical fat enlargement (buffalo hump), peripheral wasting, facial wasting, breast enlargement, and "cushingoid appearance" have been observed in patients receiving combination antiretroviral therapy. The mechanism and long-term consequences of these events are currently unknown. A causal relationship has not been established. 5.8 Immune Reconstitution SyndromeImmune reconstitution syndrome has been reported in HIV-infected patients treated with combination antiretroviral therapy, including VIREAD. During the initial phase of combination antiretroviral treatment, patients whose immune system responds may develop an inflammatory response to indolent or residual opportunistic infections [such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jirovecii pneumonia (PCP), or tuberculosis], which may necessitate further evaluation and treatment.5.9 Early Virologic FailureClinical studies in HIV-infected subjects have demonstrated that certain regimens that only contain three nucleoside reverse transcriptase inhibitors (NRTI) are generally less effective than triple drug regimens containing two NRTIs in combination with either a non-nucleoside reverse transcriptase inhibitor or a HIV-1 protease inhibitor. In particular, early virological failure and high rates of resistance substitutions have been reported. Triple nucleoside regimens should therefore be used with caution. Patients on a therapy utilizing a triple nucleoside-only regimen should be carefully monitored and considered for treatment modification.6 ADVERSEREACTIONSThe following adverse reactions are discussed in other sections of the labeling:• Lactic Acidosis/Severe Hepatomegaly with Steatosis [See Boxed Warning, Warnings and Precautions (5.1)].• Severe Acute Exacerbation of Hepatitis [See Boxed Warning, Warnings and Precautions (5.2)].• New Onset or Worsening Renal Impairment [See Warnings and Precautions (5.3)]. • Decreases in Bone Mineral Density [See Warnings and Precautions (5.6)].• Immune Reconstitution Syndrome [See Warnings and Precautions (5.8)].6.1 Adverse Reactions from Clinical Trials ExperienceBecause clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.Clinical Trials in Adult Patients with HIV-1 InfectionMore than 12,000 subjects have been treated with VIREAD alone or in combination with other antiretroviral medicinal products for periods of 28 days to 215 weeks in clinical trials and expanded access studies. A total of 1,544 subjects have received VIREAD 300 mg once daily in clinical trials; over 11,000 subjects have received VIREAD in expanded access studies.The most common adverse reactions (incidence ≥10%, Grades 2–4) identified from any of the 3 large controlled clinical trials include rash, diarrhea, headache, pain, depression, asthenia, and nausea.Treatment-Naïve PatientsStudy 903 - Treatment-Emergent Adverse Reactions: The most common adverse reactions seen in a double-blind comparative controlled study in which 600 treatment-naïve subjects received VIREAD (N=299) or stavudine (N=301) in combination with lamivudine and efavirenz for 144 weeks (Study 903) were mild to moderate gastrointestinal events and dizziness.Mild adverse reactions (Grade 1) were common with a similar incidence in both arms, and included dizziness, diarrhea, and nausea. Selected treatment-emergent moderate to severe adverse reactions are summarized in Table 2.Table 2Selected Treatment-Emergent Adverse Reactions a (Grades 2–4) Reported in≥5% in Any Treatment Group in Study 903 (0–144 Weeks)VIREAD + 3TC + EFVd4T + 3TC + EFV N=299 N=301 Body as a WholeHeadache Pain Fever Abdominal pain Back painAsthenia 14%13% 8% 7% 9% 6% 17% 12% 7% 12% 8% 7% Digestive SystemDiarrhea Nausea Dyspepsia Vomiting 11% 8% 4% 5% 13% 9% 5% 9% Metabolic Disorders Lipodystrophy b1% 8%Musculoskeletal Arthralgia Myalgia 5% 3% 7% 5% Nervous SystemDepression InsomniaDizziness Peripheral neuropathy cAnxiety 11%5% 3% 1% 6% 10% 8% 6% 5%6% Respiratory Pneumonia 5% 5% Skin and Appendages Rash event d18% 12%a. Frequencies of adverse reactions are based on all treatment-emergent adverse events, regardless of relationshipto study drug.b. Lipodystrophy represents a variety of investigator-described adverse events not a protocol-defined syndrome.c. Peripheral neuropathy includes peripheral neuritis and neuropathy.d. Rash event includes rash, pruritus, maculopapular rash, urticaria, vesiculobullous rash, and pustular rash.Laboratory Abnormalities: With the exception of fasting cholesterol and fastingtriglyceride elevations that were more common in the stavudine group (40% and 9%)compared with VIREAD (19% and 1%) respectively, laboratory abnormalities observedin this study occurred with similar frequency in the VIREAD and stavudine treatmentarms. A summary of Grade 3 and 4 laboratory abnormalities is provided in Table 3.Table 3 Grade 3/4 Laboratory Abnormalities Reported in ≥1% of VIREAD-TreatedSubjects in Study 903 (0–144 Weeks)VIREAD + 3TC + EFV d4T + 3TC + EFVN=299 N=301Any ≥ Grade 3 Laboratory Abnormality 36% 42%Fasting Cholesterol (>240 mg/dL) 19% 40%Creatine Kinase (M: >990 U/L; F: >845 U/L) 12% 12%Serum Amylase (>175 U/L) 9% 8%AST (M: >180 U/L; F: >170 U/L) 5% 7%ALT (M: >215 U/L; F: >170 U/L) 4% 5%Hematuria (>100 RBC/HPF) 7% 7%Neutrophils (<750/mm3) 3%1% Fasting Triglycerides (>750 mg/dL) 1% 9%Study 934 - Treatment Emergent Adverse Reactions: In Study 934, 511 antiretroviralnaïve subjects received either VIREAD + EMTRIVA® administered in combination withefavirenz (N=257) or zidovudine/lamivudine administered in combination with efavirenz(N=254). Adverse reactions observed in this study were generally consistent with thoseseen in previous studies in treatment-experienced or treatment-naïve subjects(Table 4).Table 4Selected Treatment-Emergent Adverse Reactions a (Grades 2–4) Reported in≥5% in Any Treatment Group in Study 934 (0–144 Weeks) VIREAD b + FTC + EFV AZT/3TC + EFVN=257 N=254 Gastrointestinal Disorder Diarrhea Nausea Vomiting9% 9% 2% 5% 7% 5% General Disorders and Administration Site Condition Fatigue9% 8% Infections and Infestations SinusitisUpper respiratory tract infectionsNasopharyngitis 8% 8%5% 4% 5%3% Nervous System Disorders Headache Dizziness 6% 8% 5% 7%Psychiatric Disorders Depression Insomnia9% 5% 7% 7% Skin and Subcutaneous TissueDisorders Rash eventc7%9%a. Frequencies of adverse reactions are based on all treatment-emergent adverse events, regardless of relationshipto study drug.b. From Weeks 96 to 144 of the study, subjects received TRUVADA with efavirenz in place of VIREAD + EMTRIVAwith efavirenz. c. Rash event includes rash, exfoliative rash, rash generalized, rash macular, rash maculopapular, rash pruritic, andrash vesicular. Laboratory Abnormalities: Laboratory abnormalities observed in this study were generally consistent with those seen in previous studies (Table 5).Table 5 Significant Laboratory Abnormalities Reported in ≥1% of Subjects in AnyTreatment Group in Study 934 (0–144 Weeks)VIREAD a + FTC + EFV AZT/3TC + EFVN=257 N=254Any ≥ Grade 3 Laboratory Abnormality 30% 26%Fasting Cholesterol (>240 mg/dL) 22% 24%Creatine Kinase (M: >990 U/L; F: >845 U/L) 9% 7%Serum Amylase (>175 U/L) 8% 4%Alkaline Phosphatase (>550 U/L) 1% 0%AST (M: >180 U/L; F: >170 U/L) 3% 3%ALT (M: >215 U/L; F: >170 U/L) 2% 3%Hemoglobin (<8.0 mg/dL) 0% 4%Hyperglycemia (>250 mg/dL) 2% 1%Hematuria (>75 RBC/HPF) 3% 2%Glycosuria (≥3+) <1% 1%Neutrophils (<750/mm3) 3%5% Fasting Triglycerides (>750 mg/dL) 4% 2%a. From Weeks 96 to 144 of the study, subjects received TRUVADA with efavirenz in place of VIREAD + EMTRIVAwith efavirenz.Treatment-Experienced PatientsTreatment-Emergent Adverse Reactions: The adverse reactions seen in treatmentexperienced subjects were generally consistent with those seen in treatment naïvesubjects including mild to moderate gastrointestinal events, such as nausea, diarrhea,vomiting, and flatulence. Less than 1% of subjects discontinued participation in theclinical studies due to gastrointestinal adverse reactions (Study 907).A summary of moderate to severe, treatment-emergent adverse reactions that occurredduring the first 48 weeks of Study 907 is provided in Table 6.。
WHO-富马酸替诺福韦Tenofovir_disoproxil_fumarate质量标准

Monographs: Pharmaceutical substances: Tenofoviri disoproxili fumaras - Tenofovir disoproxil fumarateC19H30N5O10P,C4H4O4Relative molecular mass. 635.5Chemical name. 1,1'-bis(1-methylethyl)1,1'-[({[(1R)-2-(6-amino-9H-purin-9-yl)-1-methylethoxy]methyl}phosphonoyldioxy)dimet hyl] dicarbonate (ester) hydrogen (2E)-but-2-enedioate (salt); CAS Reg.No. 202138-50-9.Description. White to almost-white, crystalline powder.Solubility. Slightly soluble in water, soluble in methanol, very slightly soluble in dichloromethane.Category. Antiretroviral (Nucleotide Reverse Transcriptase Inhibitor).Storage. Tenofovir disoproxil fumarate should be kept in a tightly closed container, protected from light, and stored at a temperature between 2 and 8°C.Additional information. Tenofovir disoproxil fumarate may exhibit polymorphism.RequirementsDefinition. Tenofovir disoproxil fumarate contains not less than 98.5 percent and not more than 101.0 percent of tenofovir disoproxil fumarate (C19H30N5O10P,C4H4O4), calculated with reference to the anhydrous substance.Manufacture. The production method is validated to ensure that the substance, if tested, would comply with:- a limit of not more than 5 ppm for the mutagenic impurity9-(prop-1-enyl)-9H-purin-6-amine (impurity K), which may be a synthesis related substance, using a suitable method, and- a limit of not more than 1.0% for the tenofovir disoproxil (S)-enantiomer (impurity G), using a suitable chiral chromatographic method.Identity tests• Either tests A, B and C or test D may be applied.A. Carry out test A.1 or, where UV detection is not available, test A.2.A.1 Carry out the test as described under 1.14.1 Thin-layer chromatography, using silica gel R6 as the coating substance and a mixture of 67 volumes of dichloromethane R, 20 volumes of acetonitrile R, 10 volumes of methanol R and 3 volumes of ammonia (~260 g/l) TS as the mobile phase. Apply separately to the plate 5 μl of each of 2 solutions in methanol containing (A) 10 mg of the test substance per ml and (B) 10 mg of tenofovir disoproxil fumarate RS per ml. After removing the plate from the chromatographic chamber, allow it to dry exhaustively in air or in a current of air. Examine the chromatogram in ultraviolet light (254 nm).The principal spot obtained with solution A corresponds in position, appearance, and intensity with that obtained with solution B.A.2 Carry out the test as described under 1.14.1 Thin-layer chromatography, using the conditions described above under test A.1 but using silica gel R5 as the coating substance. Stain the plate with iodine vapour and examine the chromatogram in daylight.The principal spot obtained with solution A corresponds in position, appearance, and intensity with that obtained with solution B.B. Carry out test B.1 or, where UV detection is not available, test B.2.B.1 Carry out the test as described under 1.14.1 Thin-layer chromatography, using silica gel R6 as the coating substance and a mixture of 50 volumes of heptane R, 30 volumes of glacial acetic acid R and 20 volumes of dichloromethane R as the mobile phase. Apply separately to the plate 5 μl of each of the following 2 solutions in ethanol R. For solution (A) use 10 mg of the test substance per ml and for solution (B) use 2 mg of fumaric acid R per ml. Develop the plate in an unsaturated tank over a path of 10 cm. After removing the plate from the chromatographic chamber, allow it to dry exhaustively in air or in a current of air. Examine the chromatogram in ultraviolet light (254 nm).One of the principal spots obtained with solution A corresponds in position, appearance, and intensity with that obtained with solution B.B.2 Carry out the test as described under 1.14.1 Thin-layer chromatography, using the conditions described above under test B.1 but using silica gel R5 as the coating substance. Spray lightly with a 16 g/l solution of potassium permanganate R and examine the chromatogram in daylight.The principal spot obtained with solution A corresponds in position, appearance, and intensity with that obtained with solution B.C. The absorption spectrum (1.6) of a 25 µg/ml solution, when observed between 220 nmand 320 nm, exhibits a maximum at about 261 nm; the specific absorbance () is 230 to 250.D. Carry out the examination as described under 1.7 Spectrophotometry in the infrared region. The infrared absorption spectrum is concordant with the spectrum obtained from tenofovir disoproxil fumarate RS or with the reference spectrum of tenofovir disoproxil fumarate. If the spectra thus obtained are not concordant, repeat the test using the residues obtained by separately dissolving the test substance and tenofovir disoproxil fumarate RS in a small amount of methanol R and evaporating to dryness. The infrared absorption spectrum is concordant with the spectrum obtained from tenofovir disoproxil fumarate RS.Specific optical rotation (1.4). Use a 10.0 mg/ml solution in hydrochloric acid (0.1 mol/l)VSand calculate with reference to the anhydrous substance; = -20° to -26°.Water. Determine as described under 2.8 Determination of water by the Karl Fischer method, Method A. Use about 1.0 g of the substance; the water content is not more than 10 mg/g.Heavy metals. Use 1.0 g in 30 ml of methanol R for the preparation of the test solution as described under 2.2.3 Limit test for heavy metals, Procedure 2; determine the heavy metals content accordi ng to Method A; not more than 20 μg/g.Sulfated ash (2.3). Not more than 1.0 mg/g.Related substances. Carry out the test as described under 1.14.4 High-performance liquid chromatography, using a stainless steel column (25 cm x 4.6 mm) packed withbase-deactivated particles of silica gel the surface of which has been modified with chemically bonded oct adecylsilyl groups (5 μm).The mobile phases for the gradient elution consist of a mixture of Mobile phase A and Mobile phase B, using the following conditions:Mobile phase A: 2 volumes of acetonitrile R, 20 volumes of phosphate buffer pH 6.0 and 78 volumes of water R.Mobile phase B: 65 volumes of acetonitrile R, 20 volumes of phosphate buffer pH 6.0 and 15 volumes of water R.Prepare the phosphate buffer pH 6.0 by dissolving 3.50 g of potassium dihydrogen phosphate R and 1.70 g of tetrabutyl ammonium hydrogen sulfate R in 800 ml of water R, adjust the pH to 6.0 by adding sodium hydroxide (1 mol/l) VS and dilute to 1000 ml with water R.After preparation, keep the solutions at about 6°C, or use an injector with cooling.Prepare the following solutions using water R as diluent. For solution (1) use 1.0 mg of the test substance per ml. For solution (2) dilute a suitable volume of solution (1) to obtain a concentration of 5 µg of tenofovir disoproxil fumarate per ml. For solution (3) use 0.2 mg of fumaric acid R per ml.For the system suitability test: prepare solution (4) by heating solution (1) carefully in a boiling water-bath for 20 minutes.Operate with a flow rate of 1.0 ml per minute. As a detector use an ultraviolet spectrophotometer set at a wavelength of 260 nm.Maintain the column temperature at 30 °C.Inject 20 μl of solution (4). The test is not valid unless the resolution between the principal peak (retention time about 40 minutes) and the peak due to the tenofovir monoester (with a relative retention of about 0.5) is not less than 25.Inject alternat ively 20 μl each of solutions (1) and (2) and (3). In the chromatogram obtained with solution (1), the following peak is eluted at the following relative retention, with reference to tenofovir (retention time about 40 minutes): fumarate about 0.15.In the chromatogram obtained with solution (1), the area of any peak due to the tenofovir monosoproxil (impurity A) is not greater than twice the area of the principal peak obtained with solution (2) (1.0%); the area of any other impurity peak is not greater than the area of the principal peak obtained with solution (2) (0.5%) and the areas of not more than two such peaks are greater than 0.4 times the area of the principal peak obtained with solution (2) (0.2%). The sum of the areas of all peaks, other than the principal peak, is not greater than 5 times the area of the principal peak obtained with solution (2) (2.5%). Disregard any peak corresponding to the peak obtained in the chromatogram with solution (3) and any peak with an area less than 0.1 times the area of the principal peak in the chromatogram obtained with solution (2) (0.05%).AssayDissolve 0.40 g, accurately weighed, in 30 ml of glacial acetic acid R1 and titrate with perchloric acid (0.1 mol/l) VS, determine the end point potentiometrically as described under 2.6 Non-aqueous titration Method A. Each ml of perchloric acid (0.1 mol/l) VS is equivalent to 63.55 mg of tenofovir disoproxil fumarate (C19H30N5O10P,C4H4O4).ImpuritiesA. (1-methylethyl)(8R)-9-(6-amino-9H-purin-9-yl)-5-hydroxy-8-methyl-5-oxo-2,4,7-trioxa-5-λ5-phosphanon anoate (tenofovir monosoproxil),B. (1-methylethyl)(5RS,8R)-9-(6-amino-9H-purin-9-yl)-5-methoxy-8-methyl-5-oxo-2,4,7-trioxa-5-λ5-phosph anonanoate,B. methyl (1-methylethyl)(5RS)-5-{[(1R)-2-(6-amino-9H-purin-9-yl)-1-methylethoxy]methyl}-5-oxo-2,4,6,8-tetrao xa-5-λ5-phosphanonanedioate,D. (1-methylethyl)(5RS,8R)-9-(6-amino-9H-purin-9-yl)-8-methyl-5-(1-methylethoxy)-5-oxo-2,4,7-trioxa-5-λ5-phosphanonanoate,E. (1-methylethyl)(8R)-5-hydroxy-8-methyl-9-(6-{[(1-methylethoxy)carbonyl]amino}-9H-purin-9-yl)-5-oxo-2,4,7-trioxa-5-λ5-phosphanonanoate,F. bis(1-methylethyl)9,9'-[methylenebis(imino-9H-purine-6,9-diyl)]bis[(8R)-5-hydroxy-8-methyl-5-oxo-2,4,7-tri oxa-5-λ5-phosphanonanoate] (tenofovir monosoproxil dimer),G. bis(1-methylethyl)5-{[(1S)-2-(6-amino-9H-purin-9-yl)-1-methylethoxy]methyl}-5-oxo-2,4,6,8-tetraoxa-5-λ5 -phosphanonanedioate (tenofovir disoproxil (S)-enantiomer) [see under Manufacture],H. 1-methylethyl propyl(5RS)-5-{[(1R)-2-(6-amino-9H-purin-9-yl)-1-methylethoxy]methyl}-5-oxo-2,4,6,8-tetrao xa-5-λ5-phosphanonanedioate,I. bis(1-methylethyl)5-{[(1R)-2-(6-{[({9-[(2R)-5-hydroxy-2,11-dimethyl-5,9-dioxo-3,6,8,10-tetraoxa-5-λ5-pho sphadodecyl]-9H-purin-6-yl}amino)methyl]amino}-9H-purin-9-yl)-1-methylethoxy]methyl }-5-oxo-2,4,6,8-tetraoxa-5-λ5-phosphanonanedioate (tenofovir di- and monosoproxil heterodimer),J. tetrakis(1-methylethyl)5,5'-(methylenebis{imino-9H-purine-6,9-diyl[(2R)-propane-1,2-diyl]oxymethylene})bis[5-oxo-2,4,6,8-tetraoxa-5-λ5-phosphanonanedioate] (tenofovir disoproxil dimer),K. 9-(prop-1-enyl)-9H-purin-6-amine, [see under Manufacture],L. (1-methylethyl)(5RS)-5-{[(1R)-2-(6-amino-9H-purin-9-yl)-1-methylethoxy]methyl}-10-methyl-5,9-dioxo-2,4,6,8-tetraoxa-10-aza-5-λ5-phosphaundecanoate,M. ethyl 1-methylethyl(5RS)-5-{[(1R)-2-(6-amino-9H-purin-9-yl)-1-methylethoxy]methyl}-5-oxo-2,4,6,8-tetrao xa-5-λ5-phosphanonanedioate.。
中国富马酸替诺福韦酯行业市场环境分析

中国富马酸替诺福韦酯行业市场环境分析1. 市场背景富马酸替诺福韦酯是一种新型的抗病毒药物,广泛应用于治疗乙肝、艾滋病等病毒感染。
近年来,随着人们对健康的重视和疾病的不断发展,富马酸替诺福韦酯市场需求不断增长。
本文将对富马酸替诺福韦酯市场环境进行分析。
2. 市场规模富马酸替诺福韦酯市场规模在过去几年持续增长。
据统计数据显示,全球富马酸替诺福韦酯市场规模预计从20XX年的XX亿美元增长到20XX年的XX亿美元。
这主要得益于对抗病毒药物需求的增加以及富马酸替诺福韦酯疗效的持续验证。
3. 市场竞争富马酸替诺福韦酯市场竞争激烈。
市场上存在着多家制药企业生产和销售富马酸替诺福韦酯药品,包括国内外知名企业。
这些企业在市场占有率、产品质量、研发能力、生产能力等方面存在差异。
由于市场需求不断增长,各企业都在加大研发投入,提高生产能力来满足市场需求。
4. 市场发展趋势4.1 新药研发随着科技的不断进步,新型抗病毒药物研发不断推进。
富马酸替诺福韦酯市场将面临来自新药的竞争。
因此,企业需加强科研力量,提高研发效率,推动创新药物的开发,以保持市场竞争力。
4.2 健康需求增长随着人们对健康和生活质量的追求,对于抗病毒药物的需求将继续增长。
富马酸替诺福韦酯作为一种重要的抗病毒药物,将会受益于这一趋势。
4.3 医疗条件改善随着医疗条件的改善,病毒感染的诊断和治疗水平将提高。
这将有助于富马酸替诺福韦酯市场的扩大,增加患者数量和药物使用量。
4.4 市场监管加强随着相关法规政策的不断完善,对富马酸替诺福韦酯市场的监管力度将加强。
这将促使企业更加规范经营,提高产品质量,并增强市场透明度。
5. 市场机遇与挑战5.1 市场机遇富马酸替诺福韦酯市场的市场机遇主要来自于病毒感染的不断增加以及新药的需求。
市场规模持续扩大为企业提供了更大的发展空间。
5.2 市场挑战市场竞争激烈和监管加强是富马酸替诺福韦酯市场面临的挑战。
企业需要加强研发能力、提高生产效率、控制成本并确保产品质量,以应对竞争压力和监管要求。
富马酸泰诺福韦酯合成工艺改进

富马酸泰诺福韦酯合成工艺改进发布时间:2021-03-03T01:27:33.603Z 来源:《中国科技人才》2021年第3期作者:静桂兰胡文俊[导读] 近年来随着艾滋病感染者数量的不断上升,昂贵的治疗费用使患者们无门可依,这已经成为严重的社会问题,所以,抗HIV的治疗已经不仅仅局限于医学范畴了,而实验研究证明泰诺福韦酯具有良好的抗HIV活性,其用于对其他药物的HIV效果已得到验证,另外其抗HBV的活性也相当高,所以富马酸泰诺福韦酯极具市场潜力。
静桂兰胡文俊浙江嘉福新材料科技有限公司浙江省嘉兴市 314102摘要:近年来随着艾滋病感染者数量的不断上升,昂贵的治疗费用使患者们无门可依,这已经成为严重的社会问题,所以,抗HIV的治疗已经不仅仅局限于医学范畴了,而实验研究证明泰诺福韦酯具有良好的抗HIV活性,其用于对其他药物的HIV效果已得到验证,另外其抗HBV的活性也相当高,所以富马酸泰诺福韦酯极具市场潜力。
但传统的合成工艺比较复杂,杂质残留较大,质量不高,我们要探寻新的简单易操作的富马酸泰诺福韦酯合成工艺。
关键词:富马酸泰诺福韦酯;合成工艺;简单易操作富马酸泰诺福韦酯化学名:9- [(R)-2-[[双[[(异丙氧擬基)氧基]甲氧基]磷酸基]甲氧基]丙基]腺嘌呤富马酸盐,是美国Gilead公司研制的核苷酸逆转录酶抑制剂。
于2001年批准其上市用于治疗艾滋病,2008年又批准其用于治疗慢性乙型肝炎。
富马酸泰诺福韦酯本身具有天然的良好性能,值得开发。
一、富马酸泰诺福韦酯物化性质1.1富马酸泰诺福韦酯图1-1为富马酸泰诺福韦酯分子结构图分子式:C19H30N5O10P分子量:635.51性状:白色结晶粉末熔点:113-115℃沸点:642.7℃at760mmHg闪点:342.5℃水溶性:在二甲基甲酰胺中易溶,在甲醇中溶解;在水、乙腈中微溶。
1.2富马酸泰诺福韦酯用途富马酸泰诺福韦酯是一种抗病毒药物,针对这款药物的特性来看,作用特点与替诺韦福类似,具有非常好的抗病毒性药物,但本药的细胞毒性更强,化学及酶学的稳定性也更强不过本药品的活性代谢的产物PMPA在细胞内迅速蓄积,因而在细胞内的重摄取也迅速增快,进而发挥强效抗逆转录病毒作用。
富马酸试剂级标准

富马酸试剂级标准
一、外观
试剂级的富马酸应为无色至浅黄色的透明液体,不应含有悬浮物、沉淀物或颜色。
二、鉴别
富马酸应与硝酸、硫酸、高氯酸作用分别产生乙酸、甲酸和乙二酸的气味。
三、熔点
富马酸的熔点应不低于128℃。
四、水分
富马酸的水分应不大于0.2%。
五、灼烧残渣
灼烧残渣应不大于0.1%。
六、氯化物
氯化物的限量应不大于0.01%。
七、硫酸盐
硫酸盐的限量应不大于0.01%。
八、还原物质
还原物质限量应不大于0.03%。
九、游离酸度
游离酸度(以H2SO4计)应不大于0.15%。
十、砷盐
砷盐的限量应不大于0.0001%。
十一、重金属
重金属的限量应不大于0.001%。
十二、微生物限度
微生物限度应符合以下要求:细菌数应不大于100个/ml;霉菌和酵母菌数应不大于10个/ml;不得检出大肠杆菌和铜绿假单胞菌。
富马酸质量标准

富马酸的质量标准主要涉及到以下方面:外观:富马酸应为白色或类白色颗粒或结晶性粉末,在乙醇中溶解,在水或乙醚中微溶,在二氯甲烷中几乎不溶。
含量测定:药典参考方法可取本品约1.0g,精密称定,加甲醇50ml,在热水浴中缓缓加热使溶解,加酚酞指示液数滴,用氢氧化钠滴定液(0.5mol/L)滴定,每1ml氢氧化钠滴定液(0.5mol/L)相当于29.02mg的C4H4O4。
此外,富马酸还具有多种用途,例如作为精细化工产品的原料和医药原料,以及作为食品加工行业中的酸度调节剂、抗热氧化助剂、防霉剂、防腐剂、腌制促进剂等。
富马酸替诺福韦酯杂质结构方式杂质标准品

33
Tenofovir Diphosphate
166403-66-3
34
Tenofovir Trimer Impurity 1
35
Tenofovir Trimer Impurity 2
如需咨询,欢送联络广州尤瓦化工产品:4000-868-328
富马酸替诺福韦酯分子构造式
序号
名称
构造
1
替诺福韦单异丙氧碳酸甲基酯
Tenofovir Disoproxil Related Compound E
CAS:211364-69-1
2
替诺福韦酯异丙氧碳酸甲基甲基酯
CAS:1246812-16-7
3
替诺福韦异丙氧碳酸甲基甲氧碳酸甲基酯
Mono-POC MethylTenofovir
CAS:1246812-43-0
4
Tenofovir ImpurityQ
替诺福韦异丙氧碳酸甲基异丙基酯
Tenofovir Disoproxil Related Compound G
CAS:1422284-15-8
6
替诺福韦-N-异丙氧羰基异丙氧碳酸甲基酯
Tenofovir Impurity E
CAS:1244022-56-7
(N6-CH2OH-POC PMPA)
25
Tenofovir Related Compound 1
342631-41-8
26
Tenofovir Related Compound 2
379270-35-6
27
Tenofovir Related Compound 3
52364-31-5
28
Tenofovir Related Compound 4
富马酸替诺福韦酯翻译

主题物质【中文名称】富马酸替诺福韦酯【分子式】C19H30N5O10P.C4H4O4【相对分子量】635.5【化学名称】(R)-[[2-(6-氨基-9H-嘌呤-9-基)- 1-甲基乙氧基]甲基]膦酸二异丙氧羰氧基甲酯富马酸盐【CAS号】202138-50-9【理化性质】本品为白色或类白色结晶性粉末。
【溶解度】在水中微溶,在甲醇中溶解,在二氯甲烷中不溶。
【类别】核苷酸逆转录酶抑制剂。
【贮藏】富马酸替诺福韦酯必须存储在密闭容器中,避光,存储温度为2-8度。
【其他信息】富马酸替诺福韦酯可能呈多态性。
【要求】【定义】富马酸替诺福韦酯的含量范围在98.5%-101.0%,测定时应先干燥到无水。
【产物】生产方法验证了这种物质的存在,如果被检测到,将遵守一个极限不超过5ppm的杂质K,它可能是一种合成的相关物质,用合适的方法,得到一个不超过1.0%的杂质G。
【薄层检验】测试方法A,B,C,D可能会被用到。
A.用方法A.1,如果不能被紫外检测到,换用方法A.2A.1在1.14.1薄层色谱法下进行这项试验,用硅胶R-16板,用含67%体积的二氯甲烷,20%体积的乙腈,10%体积的甲醇和3%的氨水作为流动相。
取10mg/ml的测试物质A和10mg/ml的富马酸替诺福韦酯B各5微升。
把薄层板从色谱室中拿出来后,让它在空气中彻底干燥。
在紫外线为254nm下检查色谱。
从溶液A对应的位置看到清晰的点,对应溶液B中的外光和强度。
A.2在1.14.1薄层色谱法下进行这项检查,在测试方法A.1下描述现象。
但要用硅胶R5作为涂料物质。
用碘试液在薄层板上染色并在日光下检验色谱。
从溶液A对应的位置看到清晰的点,对应溶液B中的外光和强度。
B.用方法B.1进行检测,如果用紫外得不到,改用方法B.2。
B.1在1.14.1薄层色谱法下进行检测,用硅胶R6作为涂料物质,并且以50%体积的庚烷,30%体积的冰醋酸和20%体积的二氯甲烷混合体作为流动准备好之后,保持溶液在大约6度,或用注射剂加冷却剂保持在6度。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
IDENTIFICATION • A. Infrared Absorption <197K> • B. Fumaric Acid Content
Sample solution: 5 mg/mL of Tenofovir Disoproxil Fumarate in water Titrimetric system (See Titrimetry <541>.) Mode: Direct titration Titrant: 0.1 M sodium hydroxide Endpoint detection: Potentiometric
Tenofovir Disoproxil Fumarate is a white to off-white, crystalline powder.
Performance-Based Monograph (Contains tests, procedures and acceptance criteria for the material under test. It also includes the criteria-based procedures to demonstrate that an Acceptable Procedure is equivalent to the Reference Procedures.)
IMPURITIES • Residue on Ignition <281>: NMT 0.1%
• Elemental Impurities <232>: Proceed as directed in the chapter. • Residual Solvents <467>: Proceed as directed in the chapter. • Organic Impurities Standard solution: USP Tenofovir Disoproxil Fumarate CRM and all appropriate USP Impurity RSs, at concentrations corresponding to the Acceptance criteria of the impurity, in an appropriate diluent Sample solution: Tenofovir Disoproxil Fumarate in an appropriate diluent Analytical system: Use a procedure validated as described in MC general chapter Assessing Validation Parameters for Reference and Acceptable Procedures <10>.
USP Fumaric acid RS USP Tenofovir Disoproxil Fumarate CRM
USP Tenofovir Disoproxil Impurity A RS (Use USP Tenofovir Disoproxil Related Compound A RS) S-Isomer of tenofovir disoproxil (S)-5-[[2-(6-Amino-9H-purin-9-yl)-1-methylethoxy]methyl]-2,4,6,8-tetraoxa-5-phosphanonanedioic acid, bis(1-methylethyl) ester, 5-oxide, (E)-2-butenedioate (1:1).
USP Tenofovir Disoproxil Impurity B RS (E)-9-(Prop-1-enyl)-9H-purin-6-amine.
USP Tenofovir Disoproxil Impurity C RS (R)-(1-(6-Amino-9H-purin-9-yl)propan-2-yloxy)methylphosphonic acid.
Analysis: Each mL of titrant is equivalent to 5.804 mg of fumaric acid (C4H4O4) on the anhydrous basis. Acceptance criteria: 17.5%–19.0% fumaric acid is present. • C. The response for Tenofovir Disoproxil from the Sample solution corresponds to that of the Standard solution, as obtained in the test for Performance-Based Monograph, Organic Impurities.
USP Tenofovir Disoproxil Impurity E RS ({[(R)-1-(6-Amino-9H-purin-9-yl)propan-2-yloxy]methyl}(hydroxy)phosphoryloxy)methyl isopropyl carbonate.
USP Tenofovir Disoproxil Impurity F RS O-(Ethoxycarbonyloxymethyl)-O-(isopropoxycarbonyloxymethyl)-{(R)-[1-(6-amino-9H-purin-9-yl)propan-2-yloxy]}methyl phosphonate.
Bis(hydroxymethyl) [[(R)-2-(6-amino-9H-purin-9-yl)-1-methylethoxy]methyl]phosphonate, bis(isopropyl carbonate) (ester), fumarate (1:1);
9-[(R)-2-[[Bis[[(isopropoxycarbonyl)oxy]methoxy] phosphinyl]methoxy]propyl]adenine fumarate (1:1) [202138-50-9].
Result = (rU/rS) × (CS/CU) × F × 100
rU = response of each impurity from the Sample solution rS = response of each USP Impurity RS from the Standard solution. [Note—If no USP Impurity RSs are available, use the peak response of tenofovir disoproxil.] CS = concentration of standard material in the Standard solution CU = concentration of Tenofovir Disoproxil Fumarate in the Sample solution F = dilution factor(s) Acceptance criteria Tenofovir disoproxil impurity A: NMT 1.0% Tenofovir disoproxil impurity B: NMT 0.0005% (5 µg/g) Any other individual impurity: NMT 0.10% Total impurities: NMT 3.0%
SPECIFIC TESTS • Water Determination, Method I <921>
Acceptance criteria: NMT 1.0%
ADDITIONAL REQUIREMENTS • Reference Standards <11>
USP Adenine RS 9H-Purin-6-amine.
Analysis
Samples: Standard solution and Sample solution Calculate the percentage of tenofovir disoproxil fumarate (C19H30N5O10P · C4H4O4) in the Sample solution:
Result = (rU/rS) × (CS/CU) × F × 100
rU = peak response from the Sample solution rS = peak response from the Standard solution CS = concentration of the Standard solution CU = concentration of the Sample solution F = dilution factor(s) Acceptance criteria: 98.0%–102.0% on the anhydrous basis
DEFINITION Tenofovir Disoproxil Fumarate contains NLT 98.0% and NMT 102.0% of tenofovir disoproxil fumarate (C19H30N5O10P · C4H4O4), calculated on the anhydrous basis
ASSAY • Procedure
Standard solution: USP Tenofovir Disoproxil Fumarate CRM in an appropriate diluent Sample solution: Tenofovir Disoproxil Fumarate in an appropriate diluent Analytical system: Use a procedure validated as described in MC general chapter Assessing Validation Parameters for Reference and Acceptable Procedures <10>.