人、兽药典中部分干扰素产品总结归纳
干扰素在猪治疗病毒性疾病中的应用分析

干扰素在猪治疗病毒性疾病中的应用分析干扰素(interferon)是一种由宿主细胞在病毒感染或其他刺激下产生的一类蛋白质。
它具有多种生物学活性,能够调节宿主免疫反应,抑制病毒复制和传播,对病毒性疾病的治疗具有重要的作用。
在猪的病毒性疾病治疗中,干扰素通常通过注射或饲料添加等方式给予。
猪是重要的畜牧养殖动物,也是许多病毒感染的易感宿主。
常见的猪病毒性疾病包括猪瘟热、猪繁殖与呼吸综合征病毒、猪流行性腹泻病毒等。
干扰素在猪的病毒性疾病治疗中的应用有以下几个方面:1. 抗病毒作用:干扰素能够刺激猪体内的免疫系统,增加机体抵抗病毒的能力。
研究表明,干扰素可以抑制猪瘟病毒和猪繁殖与呼吸综合征病毒的复制,减轻病程和病情,降低死亡率。
干扰素还可以抑制猪流行性腹泻病毒的感染,减少病毒在肠道中的复制和传播,缓解腹泻症状。
2. 免疫调节作用:干扰素能够促进猪体内的免疫系统产生免疫应答,增强机体的抗病能力。
研究发现,干扰素可以调节猪体内多种免疫细胞的活性和功能,如增加巨噬细胞的吞噬能力,增强T细胞的杀伤作用,促进B细胞的抗体生成等。
通过调节免疫系统的功能,干扰素可以加强猪体对病毒的抵抗能力,减轻病毒感染的症状和病程。
3. 抗炎作用:病毒感染通常会引起机体的炎症反应,导致组织损伤和病情恶化。
干扰素具有抗炎作用,能够调节炎症反应,减轻组织损伤。
研究发现,干扰素可以抑制炎症介质的生成和释放,减少炎症细胞的浸润和激活,从而减轻病毒感染引起的炎症反应和组织损伤。
干扰素在猪的病毒性疾病治疗中也存在一些限制和挑战。
干扰素的治疗效果受到多种因素的影响,如病程、病毒株的变异和免疫状态等。
干扰素治疗可能会引起一些副作用,如发热、食欲减退和免疫抑制等。
干扰素的治疗成本较高,限制了其在养猪业中的广泛应用。
干扰素在猪的病毒性疾病治疗中具有重要的应用价值。
通过抗病毒作用、免疫调节作用和抗炎作用,干扰素能够减轻病毒感染的严重程度和病情,提高猪的存活率和生产性能。
动物治疗中干扰素的使用及其副作用

胞表面的干扰素具有一定的关联和影响,两者相互
作 用 可 以激 发新 的细胞 的产生 。干 扰 素可 以对 多种 病 毒 产生 抗 体 ,能 够 抵御 病 毒 的入 侵 。一 些 实 验表 明, 干 扰素 还能 抑制 多种致 癌性 的病 毒 。干扰 素 的运
的数量 , 那么就需停药 , 并全面仔细 的观察 , 对症治 疗, 另 外 还要 注意 是否会 出血 。如果 一切症 状恢 复 正 常 的话就 可 以继续 治疗 。 炎症 , 多 在 注射 后 2 — 4个小 时 出现 , 伴 随 的会 有
些 并发 症状 的产 生 。例 如 , 恶心 、 食 欲 不振 、 腹泻 及
呕吐。如果是感 冒等症状的话 , 可以采取一些止痛降
温 的方法 来减 轻疼 痛 。治 疗炎症 需 要对症 处理 , 还 要
按时服药 ,注射 的安排尽量根据患畜 的身体状况而
定。
了病毒信息 的转录,从 而也抑制 了病毒相关蛋 白质 的产生 , 阻止了病毒在宿主细胞 内的繁殖 , 使得病毒 无法 表 现相 应 的特征 性状 。机体 内 的相关 免 疫 系 统
毒 以外 , 细菌 、 真菌 、 原虫 、 立克次 氏体 、 植物血凝素
以及某 些 人 工合 成 的核 苷 酸多 聚 物 ( 如 聚肌 胞 ) 等
菌药 、 抗病毒药 , 达到标本 兼治 、 缩短病程 、 降低损
失。
都能刺激机体产生干扰素。凡能刺激机体产生干扰 素 的物质统称为干扰素诱生剂 。干扰素 的主要成分 是 糖蛋 白 , 按 其 抗原 性 不 同可 分 为 d、 p 和 三种
中起 着 十分 重 要 的 使 得 病 毒生 长 受 到
自身免疫 , 如 甲状腺炎 、 风湿性关节炎 、 血管炎
干扰素在兽医临床上的科学应用

2020第6期 121干 扰 素 在 兽 医 临 床 上 的 科 学 应 用万遂如(中国畜牧兽医学会动物传染病学分会,吉林 长春 130122) 中图分类号:S858.28 文献标志码:A 文章编号:1002-1957(2020)06-0121-03收稿日期:2018-08-31作者简介:万遂如(1938-),男,江西南昌人,教授,主要从事家畜传染病的教学、科研及临床工作,现任中国畜牧兽医学会动物传染病学分会名誉秘书长.干扰素(IFN)是干扰素诱生剂作用于有关生物细胞所产生的一类具有高度生物学活性、多功能的小分子糖蛋白,由于它们能干扰病毒RNA和DNA 的合成而抑制病毒的复制,故称之为干扰素(抗病毒蛋白)。
干扰素是细胞因子中的一个家族,目前已发现有20多种干扰素家族的细胞因子。
干扰素无抗原性,不被免疫血清中和,也不被核酸酶灭活,具有抗病毒、抗细胞增殖与分化、抗肿瘤和免疫调节等作用。
本文就当前畜牧业生产中禁止滥用抗生素,保障动物性食品安全、公共卫生安全和生态环境安全,促进健康养殖的持续发展,替代抗生素,在兽医临床上如何科学应用干扰素问题,谈点个人意见,仅供参考。
1 干扰素的分类1980年国际干扰素命名委员会建议将各种干扰素先根据动物的来源确定分类系统,再按干扰素的抗原特异性和分子结构分成不同的型别。
哺乳动物的干扰素分为两类:Ⅰ型干扰素和Ⅱ型干扰素。
Ⅰ型干扰素(耐酸型)主要包括IFN-α、IFN-β、IFN-ω、IFN-К、IFN-ξ、IFN-δ等类型。
主要是由单核-巨噬细胞、B细胞和成纤维细胞产生的,在耐酸环境中稳定。
IFN-α和IFN-β二者结合相同的受体,分布较为广泛,包括单核-巨噬细胞、多形核白细胞、B细胞、T细胞、血小板、上皮细胞、内皮细胞和肿瘤细胞等。
Ⅱ型干扰素即IFN-γ(酸敏感型),主要是由T淋巴细胞受到有丝分裂原或特异抗原刺激所产生,自然杀伤细胞(NK细胞)也可产生,对酸不稳定,结合的受体与Ⅰ型干扰素不同,但IFN-γ的免疫刺激活性在两类干扰素中最强。
干扰素的种类、用法及优缺点比较

临床上使用的干扰素主要分为:普通干扰素和长效干扰素,现从种类、用法、价格、疗效等方面比较如下:
普通干扰素有:
1.干扰素α-2a ,商品名:罗荛愫因特芬贝尔芬;
2.干扰素α-1b ,商品名:赛若金运德素;
3.干扰素α-2b ,商品名:甘乐能安达芬英特龙;
用法用量:1周3次或隔日1次,肌肉注射或皮下注射。
价格:1针几十元。
长效干扰素有:
1.聚乙二醇干扰素α-2a,商品名:派罗欣;
2.聚乙二醇干扰素α-2b,商品名:佩乐能。
用法用量:1周1次,肌肉注射或皮下注射。
价格:1针大概在1000~1400元之间。
普通干扰素的缺点是显而易见的,由于吸收快,注射几个小时后,血药浓度达到最高,这时副作用的反应强烈,抑制病毒的作用也达到最高,但是由于排泄比较快,造成在下次注射前,血药浓度的偏低,病毒可能会恢复复制。
而长效干扰素,很好的解决了这个问题,血药浓度基本可以保持恒定,不会存在病毒重新复制的机会,作用时间延长。
因此,相对普通干扰素,长效干扰素的优点是避免了频繁的注射,效果也较普通干扰素为好,但是价格比较昂贵,建议经济条件好的患者首选长效干扰素。
关于多肽、植物血凝素、干扰素、白介素、聚肌胞、转移因子等

关于多肽、植物血凝素、干扰素、白介素、聚肌胞、转移因子等_兽药配方的空间18:42作者按:现在好多人问我关于多肽、植物血凝素、干扰素、白介素、聚肌胞、转移因子等产品在畜牧兽药的应用事宜。
本人特整理了以下内容,供大家参考!干扰素 IFN什么是干扰素干扰素是一组具有多种功能的活性蛋白质(主要是糖蛋白),是一种由单核细胞和淋巴细胞产生的细胞因子。
它们在同种细胞上具有广谱的抗病毒、影响细胞生长,以及分化、调节免疫功能等多种生物活性。
干扰素的分类根据干扰素蛋白质的氨基酸结构、抗原性和细胞来源,可将其分为:IFN-α、IFN-β、IFN-γ。
IFN-ω属于IFN-α家族,其结构和大小与其它IFN-α稍有差异,但抗原性有较大的不同。
现在公认IFN-β和IFN-γ只有一个亚型,而IFN-α有约二十余个亚型。
干扰素的临床应用1、疫苗免疫失败后的临床补救治疗。
当错误使用毒力较强的疫苗免疫,或免疫时未察觉鸡群有潜在性疾病感染等情况时,导致疫苗免疫后鸡群出现病症,此时可使用干扰素进行紧急补救治疗,减少损失。
2、家禽家畜各种病毒性疾病,如猪的口蹄疫、蓝耳病、鸡的新城疫、禽流感、传染性支气管炎等疾病的预防和治疗。
即当发生上述疾病时,在使用其他抗病毒的同时配合使用干扰素,可提高疗效,加速康复。
临床用药指导:一、猪1.在病毒性传染病流行期、易感日龄、引种后以及病毒性传染病发病初期使用效果极好。
2.对猪传染性胃肠炎,流行性腹泻,轮状病毒病的治疗:按以上用量肌肉注射本品,每日一次,连用3日;同时注射抗病毒水针,每日1次连用3-5天,电解多维饮水;痊愈后,建议饲料里添加益生素,以重建和维持肠道微生态平衡,提高增重速度,挽回或减少动物因生病期间消瘦带来的损失。
3.对于无名高热,混合感染,病情较重,死亡率较高的疾病如:蓝耳病,圆环病毒感染,非典型猪瘟,伪狂犬,细小病毒病等紧急治疗时,使用本品同时配合分点注射退烧药和抗生素。
猪恢复采食后,用敏感抗生素拌料,连用3-5天.注意事项1.本品应在有经验的临床兽医师指导下按规定剂量、疗程和投药途径使用。
干扰素

干扰素据了解基层有不少兽医和养殖户都是在家禽发病的中后期才使用干扰素,或者是在使用几天抗病毒的药无效的情况下才使用干扰素.其结果是病情得不到抑制,得出的结论是干扰素效果不好.其实是错过了干扰素的最佳使用时机,应当在发病的早期,越早越好,到中后期机体出现败血症或毒血症时,为时已晚。
1.干扰素必须在病毒感染的早期,即体内病毒尚末扩期或引起严重病变之前临床上使用才能凑效2.如果机体组织损伤严重,已处于败血症或毒血症阶段,此时使用干扰素,起不到决定性的作用,为时已晚.3.干扰素以注射,滴口,饮水方式进行,预防和治疗病毒性疾病一次或者两次即可.但是在各种疾病混合感染的情况下可加量2~3倍,连用2~3天.饮水前要断水2~3个小时,以确保鸡群都能饮到干扰素.4.干扰素对活毒疫苗有干扰作用,因此在正常冻干苗免疫72小时前不得使用干扰素.灭活油苗不受干扰素的影响,并且在使用油苗的时候联合使用干扰素可以预防在油苗产生免疫力前野毒的感染。
5。
使用基因工程干扰素既不会促进抗体的生成也不削弱或中和抗体,因此在使用干扰素4天后,可以结合相应传染病的免疫特点并接种相应疫苗,使在发病后使用干扰素得到控制后进一获得免疫,以防止其它疫病的侵害。
6。
在发病期间使用干扰素可以联合使用转移因子,以提高干扰素使用效果并能缩短病程`降低疫病造成的损失。
7。
使用干扰素可以与其它抗病毒的药物联合使用,也可以和卵黄抗体联合使用。
8。
使用干扰素期间可联合使用抗生素,防止细菌性疾病的混合感染,添加电解多维以促进疾病的康复。
9。
在治疗时,只用干扰素不注意配合其它药物使用,把干扰素当成神药,其结果是耽误了病情。
10。
在中后期,不采用滴口或注射方式,为省事采用饮水方式,病情严重的家禽已经不吃不喝,同样达不到好的治疗效果。
干扰素控制禽类疾病效果好干扰素作用机理病毒感染细胞导致干扰素的产生,并随被感染细胞死亡、崩解而释出。
干扰素分子向附近扩散,随血液循环至全身。
干扰素简介

在MDCK 细胞中北极狐γ -干扰素抗病毒活性测定 A: the control MDCK cells; B: severe CPE in the control; C: cell disposed by over 10 −5 renatured recombined arctic fFox gamma interferon
16
参考文献及致谢
干扰素发现的艰难历程 大众卫生报/ 2004 年/ 01 月/ 21 日/ 第T00 版 贺进朝 科技日报 / 2003 年/ 06 月/ 23 日/ / 干扰素是一种天然的抗病毒蛋白质 中科院上海生命科学院生化与细胞所研究员中科院院 士刘新垣 我国基因工程干扰素的研究概况 郑永霞凌善峰 中国免疫学杂志 2006年第 22 干扰素及其最新研究进展 卷 孙亚萍 王英明 乔守怡 复旦大学生命科学院遗传 学研究所遗传工程国家重 点实验室 CNKI学术图片知识库 聊城大学学报自然科学版
※干扰素受体与信号传导
干扰素产生以后,并不直接发挥抗病毒作用,而 是结合在临近的同种细胞的受体上,使该细胞产生 多种蛋白,包括抗病毒调节物和转录调节因子,从而 发挥抗病毒作用。三个干扰素家族结合不同的受 体,通过类似的信号传导途径,发挥生物学效应。 Ⅰ型干扰素受体 Ⅰ型干扰素受体由两条链组成 , IFNAR1和IFNAR2,都是糖蛋白。 Ⅱ型干扰素受体 Ⅱ型干扰素受体( IFN-γ R) 也由两种不同的亚基组成, IFNGR1和IFNGR2。 IFN-λ 受体 IFN-λ 的受体是一种异源二聚体, 其中一个亚基IFNLR1,或者IL228Ra。
干扰素的信号传导
三族干扰素都经JAK- STAT途径发挥活性
基因工程大量生产
1987年, 我国科学家侯云德 等成功获得基因工程干扰素 。
畜禽干扰素应用及体会

清洗干净 , 避免药物残 留对精子 的伤害 。公猪爬跨上假母猪后 , 41 .精液分装完后若不能及时用掉 , 应在 室温下避光冷却 1 h以
重悬 殊 造 成 的配 种 难 问 题 , 服 时 间 和 区域 上 的差 异 , 克 特别 是 对 伸出空拳 1 1左右 , " O1 1 或不让其伸出拳外 , 用中指 、 无名指和小指
农牧 民散养猪配种更为方便 , 打一个电话配种员即可登 门输精 , 锁定 龟头 , 意不要抓 握阴茎体部 E 注 螺旋部 分)以防造成公猪 , 减少以往赶猪 的不便和不良影响 ,让猪 场问的种猪精液交 流更 不适而从假母猪上退下 。然后顺其 向前冲力 , 将阴茎的… ’ S状弯 方便 , 还能防止生殖道疾病和寄生虫病 的传播。随着种公猪精液 曲尽 可 能 地 拉 直 , 紧 阴茎 龟 头 防止 其 旋转 , 猪 即 可 安静 下 来 握 公
能充分发挥 , 笔者近几年在基层推广猪的人工授精技术 , 现总结 尿 。当射出乳 白色的液体时 , 即为浓精液 , 就要用采精杯收集起 以下五点关键技术环节供初学者参考。 来。有些公 猪射精的过程 中, 可射出 2 个乳 白色 的浓份精液 , ~3
应 全 部 接取 。
2 . 4采精过程中 , 如果仍有包皮 中的尿 液顺 阴茎流出 , 可先将龟 1 调教公猪 帕跨假母猪时 ,采精员的耐心是调教公猪成功的关 头抬 高 , . 1 然后在射精 间隙或 只射 出清亮液体时 , 放下集精杯 , 不 键。 不要期望一次调教就能成功 , 不要强迫公猪爬跨假母猪 , 不要 收集 , 左手拿纸 巾将其 吸附, 避免流入集精杯中。
可训 练பைடு நூலகம்1 , 1 最好 不要 少 于 3次 , 至爬 跨成 功 。调教 时 间 稀释液 的温度 , 次 但 周 直 不可逆操作。在稀释时要把等温的稀释液沿温度
干扰素学习资料

近年来,畜禽疾病出现了新病增多、老病新发、混感广泛等特点,治疗难度不断加大。
畜牧兽医工作者尝试了很多的方法和药物,对畜禽疾病的防控起到了一定的积极作用。
干扰素(interferon,IFN) 作为一种高效的抗病毒生物活性物质和具有广泛免疫调节作用的淋巴因子被广泛应用于畜禽疾病防控,本文就IFN的性质、应用等做一下详细、全面的介绍。
一、IFN的理化性质及分类:干扰素本质是蛋白质,是机体感染病毒时宿主细胞通过抗病毒应答反应而产生的一组结构相似、功能相近的低分子糖蛋白。
按细胞结构、来源分:分为两型。
Ⅰ型干扰素包括α-IFN和β-IFN等,是由白细胞和成纤维细胞等产生;α-IFN又称白细胞干扰素,具有较强的抗病毒作用,可以分为23种亚型,亚型相互之间有较高的同源性;β-IFN有一个亚型,有与α-IFN相似效果,但它在肌肉组织中易被灭活;α/β-IFN二者结合相同受体,分布广泛,包括单核-巨噬细胞、多形核白细胞、B细胞、T细胞、上皮细胞、内皮细胞与肿瘤细胞等。
Ⅱ型干扰素,又称γ-IFN(免疫干扰素)是由有丝分裂原刺激T淋巴细胞产生,与α、β干扰素之间没有明显的同源性。
按来源:可分为天然和非天然干扰素,非天然即以DNA重组技术生产的基因工程干扰素。
基因工程干扰素具有与天然干扰素完全相同的生物学活性。
按疗效:分为普通干扰素和长效干扰素。
普通干扰素分子小、作用时间短,一般情况下12小时后基本完全排出体外,因而需要多次用药;长效干扰素一般指聚乙二醇化干扰素,半衰期长。
二、IFN的作用:1、抗病毒作用:干扰素在抗病毒感染中发挥着重要的、必不可少的作用,主要在早期的病毒感染期间即可抑制病毒的生长和增殖。
其作用机理为:病毒进入细胞→病毒RNA附着于宿主细胞核糖体→使形成干扰素mRNA的宿主细胞DNA顺反子去抑制→干扰素mRNA刺激干扰素产生→干扰素进入细胞→使形成翻译抑制蛋白质mRNA的细胞DNA顺反子去抑制→TIP形成并结合到核糖体→TIP阻止病毒RNA结合到核糖体。
干扰素及其类似兽用药品的应用

干扰素及其类似兽用药品的应用一、应用现状及其作用原理目前市场上供应的干扰素及其类似兽用药品按其功能分类,有干扰素、白细胞介素、抗体、卵黄、血清。
产品剂型有冻干制品、液态制品和干粉剂。
干扰素是能够透过细胞膜同病毒、细菌的单位膜上特殊位点直接结合的小分子黏蛋白,其最大特点是能够干扰病毒的复制或细菌的增殖,使新产生的病毒或细菌发生结构上的改变,从而失去其对特定组织和器官的侵袭能力,值得注意的是干扰素并不能直接杀死病毒或消灭细菌。
常见的有α、β、γ三种,前一种为酸性,后两种为碱性。
白细胞介素是一种白细胞增殖过程中产生的多糖类蛋白质,能够激活B-细胞和T-细胞,干预免疫细胞的代谢,增强体液免疫能力,应用的目的不是直接杀死病毒和细菌,而是在于增强机体抵抗力,从而提高动物体战胜病毒和细菌的能力。
市场上供应的多数为白细胞介素-2。
抗体是一种具有专一性免疫能力的球蛋白,它可以与特定的病毒和细菌相结合,从而形成病毒、细菌和特定球蛋白的复合体,一方面体积增大使其不能通过特定组织和器官的各种屏障,如体液免疫屏障、血脑屏障、细胞膜屏障等;另一方面由于其结构的改变失去致病性。
常用的有猪瘟抗体、鸡新城疫抗体、法氏囊抗体以及新城疫-法氏囊二联抗体。
卵黄属于一种抗体产品。
最常见为鸡新城疫卵黄抗体、新城疫-法氏囊二联卵黄抗体。
严格讲,卵黄抗体是抗体生产过程中的一种初级产品。
2007年以来,流行性腹泻、传染性胃肠炎、圆环病毒等卵黄抗体也在生产中开始运用。
血清同卵黄一样属于一种抗体产品,最常见为猪瘟血清,也是抗体生产过程中的一种初级产品。
除了前述产品之外,市场上还有“转移因子”、“植物血凝素”、“黄芪多糖”、“多聚寡糖”、“反义多聚糖”、“金丝桃素”等据说有抗病毒作用的药品,这些产品在基层兽医的临床实际运用中,或多或少有一些抗病毒或抑制病毒的作用,因为这些产品有的就是干扰素,有的是白细胞介素,有的是抗病毒药品,如利巴韦林、金刚烷胺等,有的则是尚未标明成分的特殊产品。
在养猪生产中干扰素应用简介

在养猪生产中干扰素的应用简介近两年,由于畜禽免疫抑制性疾病的危害日趋严重,免疫增强剂在畜牧生产中被广泛应用。
动物受到疫病威胁或处于应激状态,特别是畜禽处于免疫抑制时使用免疫增强剂是有效的。
随着科学技术的进步及现代畜牧业的需要,免疫增强剂的预防、保健作用越来越受到重视,特性确定、高效、稳定、无毒的理想免疫增强剂将是未来防治畜禽疾病的主要物质,猪用免疫增强剂主要包括干扰素、猪用转移因子、免疫球蛋白、低聚糖、黄芪多糖等,本文主要介绍干扰素的用法、功效和注意事项等。
1 免疫增强剂的概念与分类免疫增强剂是指单独使用或者与抗原联用均能增强机体免疫应答的物质,具体讲就是能够通过增强巨噬细胞强活性和非特异性物质的免疫原性的稳定物质。
免疫增强剂分为两大类,即主动性免疫增强剂和被动性免疫增强剂。
主动性免疫增强剂又分为特异性和非特异性免疫增强剂。
常用的特异性免疫增强剂有灭活的细菌、病毒,或由其提取的抗原成分,如大家很熟悉的猪瘟、猪伪狂犬、猪乙型脑炎等各种疫苗,机体受这些免疫刺激剂的作用后,会产生一系列的免疫反应和相应的抗体,可防止相应的疾病的发生。
这类免疫增强剂特异性强、疗效肯定且作用持久,已广泛应用于临床。
非特异性免疫增强剂作用于机体,可促进机体的非特异性免疫反应,如使白细胞、巨噬细胞的噬菌功能增强。
我们平常所说的黄芪多糖、左旋咪唑等属于非特异性免疫增强剂,当与疫苗一起使用时也具有特异性免疫增强剂的作用。
2 干扰素(Interferon, IFN)1957年,英国病毒生物学家Alick Isaacs和瑞士研究人员Jean Lindenmann,在利用鸡胚绒毛尿囊膜研究流感干扰现象时了解到,病毒感染的细胞能产生一种因子,后者作用于其他细胞,干扰病毒的复制,故将其命名为干扰素。
1966-1971年,Friedman发现了干扰素的抗病毒机制,引起了人们对干扰素抗病毒作用的关注,而后,干扰素的免疫调控及抗病毒作用、抗增殖作用以及抗肿瘤作用逐渐被人们认识。
干扰素的不同亚型与临床应用
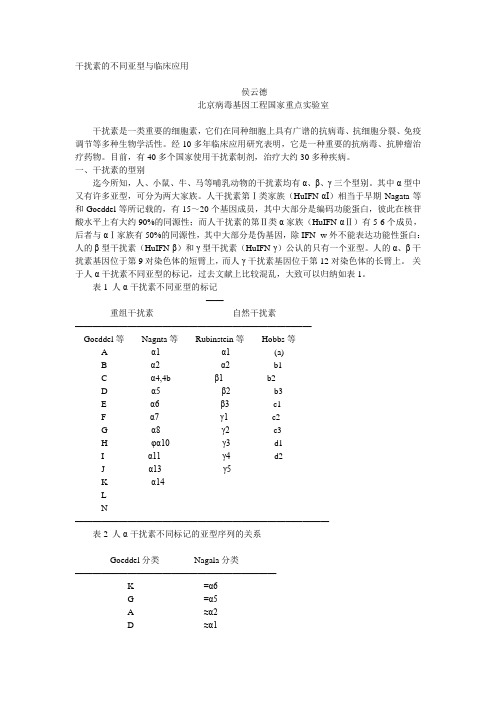
干扰素的不同亚型与临床应用侯云德北京病毒基因工程国家重点实验室干扰素是一类重要的细胞素,它们在同种细胞上具有广谱的抗病毒、抗细胞分裂、免疫调节等多种生物学活性。
经10多年临床应用研究表明,它是一种重要的抗病毒、抗肿瘤治疗药物。
目前,有40多个国家使用干扰素制剂,治疗大约30多种疾病。
一、干扰素的型别迄今所知,人、小鼠、牛、马等哺乳动物的干扰素均有α、β、γ三个型别。
其中α型中又有许多亚型,可分为两大家族。
人干扰素第Ⅰ类家族(HuIFN-αI)相当于早期Nagata等和Goeddel等所记载的,有15~20个基因成员,其中大部分是编码功能蛋白,彼此在核苷酸水平上有大约90%的同源性;而人干扰素的第Ⅱ类α家族(HuIFN-αⅡ)有5-6个成员,后者与αⅠ家族有50%的同源性,其中大部分是伪基因,除IFN- w外不能表达功能性蛋白:人的β型干扰素(HuIFN-β)和γ型干扰素(HuIFN-γ)公认的只有一个亚型。
人的α、β干扰素基因位于第9对染色体的短臂上,而人γ干扰素基因位于第12对染色体的长臂上。
关于人α干扰素不同亚型的标记,过去文献上比较混乱,大致可以归纳如表1。
表1 人α干扰素不同亚型的标记---------------------------------------------——--重组干扰素自然干扰素———————————————————————————Goeddel等Nagnta等Rubinstein等Hobbs等Aα1α1(a)Bα2α2b1Cα4,4bβ1b2Dα5β2b3Eα6β3c1Fα7γ1c2Gα8γ2c3Hφα10γ3d1Iα11γ4d2Jα13γ5Kα14LN—————————————————————————————表2 人α干扰素不同标记的亚型序列的关系-------------------------------------------Goeddel分类Nagala分类———————————————————————K=α6G=α5A≈α2D≈α1C≈L= φα10J1≈J2~α7B≈B2= α8---------------------------------------------表3 α1和α2型干扰素的等位基因----------------------------------------------亚型等位基因氨基酸序列114158 IFN- α11a Va1Leu1b Ala Leu1c Ala Val—————————————————————————2334 IFN- α22a Lys His 2b Arg His2c Arg Arg-----------------------------------------------在重组干扰素中,根据核苷酸和氨基酸序列的比较,人α干扰素不同标记亚型之间的关系,如表2所示。
干扰素在猪治疗病毒性疾病中的应用分析

干扰素在猪治疗病毒性疾病中的应用分析干扰素是一类重要的免疫调节蛋白,具有广泛的生物学活性和抗病毒能力。
在病毒感染和病毒性疾病治疗中,干扰素的应用具有重要的意义。
本文主要对干扰素在猪治疗病毒性疾病中的应用进行分析。
一、干扰素的作用机制及种类干扰素主要通过调节免疫反应和拮抗病毒复制来发挥其抗病毒活性。
干扰素能够调节和增强机体的免疫应答,包括促进自然杀伤细胞的活性、增强B细胞的抗体产生、加强T 细胞的活性等。
干扰素能够抑制病毒的复制和传播,包括抑制病毒基因的表达、抑制病毒蛋白的合成、抑制病毒核酸的复制等。
根据其作用机制和效应,干扰素可以分为三类:干扰素α、干扰素β和干扰素γ。
干扰素α和干扰素β主要由细胞产生,对于大多数病毒感染具有广谱的抗病毒活性。
干扰素γ主要由T细胞和自然杀伤细胞产生,对于一些特定的病毒具有特异的抗病毒活性。
二、干扰素在猪治疗病毒性疾病中的应用1. 干扰素的产生与病毒感染猪作为重要的养殖动物,面临着多种病毒感染的威胁,如猪繁殖与呼吸综合征病毒(PRRSV)、猪肺疫病毒(PPV)、猪流感病毒(PIv)等。
这些病毒感染会导致猪的免疫系统受损,引发疾病。
病毒感染后,猪体内会产生干扰素作为免疫应答的一部分。
猪产生的干扰素主要为干扰素α和干扰素γ。
干扰素的产生能够增强机体的免疫应答,促进病毒的清除和康复。
在猪流感病毒感染和治疗中,干扰素α和干扰素γ的应用也具有一定的疗效。
研究表明,干扰素α和干扰素γ能够抑制猪流感病毒的复制和传播,减轻疾病症状,促进康复。
干扰素α和干扰素γ还可以增强机体的免疫应答,提高病毒清除能力。
三、干扰素在猪治疗病毒性疾病中存在的问题和展望干扰素在猪治疗病毒性疾病中的应用虽然取得了一定的疗效,但仍然存在一些问题和挑战。
干扰素的治疗效果存在一定的差异性,可能受到病毒株的差异、动物个体的差异等因素的影响。
干扰素的应用需要注射给药,具有一定的副作用和毒性。
干扰素的生产成本较高,限制了其广泛的应用。
干扰素及类似兽药的注意事项

2020年第02期干扰素及其类似兽用药品是治疗病毒性疫病的常用药品,在临床治疗中得到了广泛应用,具有良好的防治效果。
但是在应用这类药品的过程中,需要注意的内容比较多,否则,达不到治疗效果,本文重点介绍了干扰素及类似药品应用的注意事项,供参考。
1确定干扰素及其类似兽用药品的应用时机其作用机理是干扰动物体内病毒的复制,但没有清理病毒的作用,还需要动物自身进行抵抗,若动物的抵抗能力比较差,即使病毒不在特定的靶组织或靶器官发生病变,病毒增多也还会对动物身体产生影响。
通常,干扰素比较常用于体质强壮动物的发病早期,对病情进行控制,对病毒引起的器官病变还需要使用专门的治疗方法。
动物病症减轻后还需要进行针对性治疗,接种免疫疫苗,提高动物的自身免疫力和抵抗能力。
另外,对早期病例,使用干扰素治疗后也需要经过一段时间后才能完全治愈,无法立即恢复。
主要是直接作用于病毒,与病毒形成复合体,能够对病毒的浓度进行控制,从而缓解病情并逐渐治愈。
但是在实际应用之前需要对具体病症进行明确,针对性的选择抗体,否则容易出现变态反应,不利于疾病的治疗,还容易导致病情加重。
2药品的用量和使用方法干扰素产品类型比较多,每一种干扰素都有具体的含量、使用量、保存方法和使用期限,在实际应用干扰素的过程中,一定要结合动物的发病情况来确定具体的用量。
通常情况下,干扰素要在短时间内使用,保存时间越长,干扰素的效价越低。
在使用的过程中也需要根据保存时间的长短来对干扰素的用量进行调节,尽量现用现买,同时保证保存环境的稳定性,避免干扰素反复冻融影响其生物活性。
其次,干扰素冻干制剂与水剂相比,若出现溶解情况很容易被发现,便于购买者的保存。
再次,要对解冻的时间进行有效控制,因为干扰素解冻会后长时间放置会影响其活性。
在解冻的过程中需要保证温度的适宜性,在解冻之后待温度达到26~30℃后进行应用。
稀释干扰素需要专门的稀释液,因为干扰素对稀释液的渗透压,所以不能使用饮用水进行解冻和稀释,避免影响干扰素活性。
干扰素、转移因子、免疫球蛋白在猪病防治中重要作用

现代生物工程制剂(猪用干扰素、转移因子、免疫球蛋白)在猪病防治中的应用近年来,基因工程生物制剂在畜禽疾病防治上的研究有着突飞猛进的进展,因为无药残、不产生耐药性、发挥效果快等原因被广泛推广应用,发挥着常规化学药品难以达到的效果,专业从事生物制剂研究的大连三仪动物药品有限公司研发出的猪用基因工程干扰素、猪用转移因子、排疫肽(猪浓缩免疫球蛋白)等系列生物工程制剂通过大量临床证实,对猪病具有良好的预防与治疗效果。
现简介如下,供有关单位参考。
一、猪用干扰素(IFN)干扰素是机体受病毒或其他干扰素诱生剂刺激巨噬细胞、淋巴细胞及体细胞产生的具有高活性的多功能糖蛋白,在正常机体的脾脏、肝脏、肾脏、外周血淋巴细胞和骨髓中都可以检出。
通过基因工程手段获得的猪用干扰素无论是含量或是活性,均高于机体受病毒或其他干扰素诱生剂刺激产生的干扰素。
(一)作用机理1、抗病毒作用:干扰素作用于动物机体细胞内的干扰素受体,经信号传导等一系列的生物化学过程,启动基因合成抗病毒蛋白,抑制病毒多肽链的合成,阻断了病毒的繁殖,使病毒不能在动物机体内生长与繁殖,从而起到抗病毒的作用。
2、免疫调节作用:猪干扰素能增强免疫器官巨噬细胞的吞噬作用和消毁能力,从而达到调节免疫自稳功能,降低应激反应等目的。
(二)猪用干扰素在猪病防治中的应用主要用于猪流行性腹泻、传染性胃肠炎、轮状病毒感染、猪瘟、兰耳病、圆环病毒II型感染、伪狂犬病、猪流感、水疱病、细小病毒病等的防治,特别是和猪用转移因子协同使用时,对猪的一些免疫抑制性疾病的防治有更好的效果。
1、使用方法:每40公斤体重肌肉注射1毫升,每日1 次,连用3天,重症加量2~3倍。
与猪用转移因子联合使用,应用灭菌注射用水或生理盐水稀释,混合后肌肉注射。
2、预防与治疗方案(1)猪传染性胃肠炎、流行性腹泻、轮状病毒病的治疗:按使用治疗量肌注干扰素,每日1次,连用3天,同时肌注复方穿心连注射液或双黄连注射液或痢菌净,每日1~2次,连用4天,并改饮电解质多维或口服补液盐7天。
干扰素基础知识

干扰素干扰素(IFN)是一种广谱抗病毒剂,并不直接杀伤或抑制病毒,而主要是通过细胞表面受体作用使细胞产生抗病毒蛋白,从而抑制乙肝病毒的复制;同时还可增强自然杀伤细胞(NK细胞)、巨噬细胞和T淋巴细胞的活力,从而起到免疫调节作用,并增强抗病毒能力。
干扰素是一组具有多种功能的活性蛋白质(主要是糖蛋白),是一种由单核细胞和淋巴细胞产生的细胞因子。
它们在同种细胞上具有广谱的抗病毒、影响细胞生长,以及分化、调节免疫功能等多种生物活性。
目录:一、干扰素二、干扰素简介三、干扰素多少钱四、发现五、什么叫干扰素(IFN)六、品种及价位七、作用机制①间接性②广谱性③种属特异性④发挥作用迅速八、分类九、干扰素制剂如何分类十、临床上常用的干扰素有哪些制剂1自然干扰素2人体白细胞重组干扰素3复合干扰素十一、干扰素适应症十二、干扰素有哪些不良反应十三、如何应对干扰素的不良反应十四、干扰素研究、应用历程十五、病毒的克星——干扰素十六、哪些人不宜使用干扰素治疗十七、什么是长效干扰素十八、普通干扰素和长效干扰素的区别十九、干扰素治疗的禁忌证二十、用途及用法二十一、干扰素治疗乙肝效果一、干扰素药物类别:抗肿瘤药,抗病毒药;所属类别:生物反应调节剂药物名称:干扰素英文名称:Interferon药物别名:序号中文别名英文别名一.α干扰素制剂/规格:序号制剂规格1.注射剂5×10。
单位(1 ml);1×106。
单位(1 ml);2.冻干剂l×10。
单位成份/化学结构:序号成份化学结构药理作用:1.抗病毒作用:其抗病毒活性不是杀灭而是抑制病毒,它一般为广谱病毒抑制剂,对RNA和DNA病毒都有抑制作用。
当病毒感染的恢复期可见干扰素的存在,另一方面用外源性干扰素亦可缓解感染。
2.抑制细胞增殖干扰素抑制细胞分裂的活性有明显的选择性,对肿瘤细胞的活性比正常细胞大500~1000倍。
干扰素抗肿瘤效果可以是直接抑制肿瘤细胞增殖,或通过宿主机体的免疫防御机制限制肿瘤的生长。
干扰素在猪治疗病毒性疾病中的应用分析

干扰素在猪治疗病毒性疾病中的应用分析病毒性疾病在猪养殖中常常给养殖业者带来巨大的损失。
干扰素是抗病毒免疫响应中发挥关键作用的细胞因子。
其主要功能是在细胞感染病毒时增强细胞免疫能力,通过诱导细胞抗病毒蛋白的表达来抵御病毒感染。
因此,干扰素在农业养殖中的应用已经引起广泛关注。
干扰素的种类有多种,按其来源可分为内源性和外源性干扰素。
内源性干扰素在免疫应答中由受到病毒感染的宿主细胞自身合成,它们共同发挥诱导抗病毒免疫反应、调节免疫应答等功能。
外源性干扰素由人工或生物合成制剂提供,其作用机制与内源性干扰素相似,但具有更强的抗病毒效果。
在猪的病毒性疾病治疗中,外源性干扰素可通过多种途径提供。
一种方法是使用合成品。
目前已经有多种类型的合成物被研制出来,如人重组干扰素、人纯化干扰素和猪重组干扰素等。
这些干扰素可用于注射、口服和残留在喂料中等方式给予权贵猪,以起到治疗疾病、预防疾病和促进免疫应答的作用。
另外,传统药材中也存在着多种干扰素的源头,如菊花、黄连、绿茶等,这些中药成分不仅能够促进机体抗病毒免疫反应,而且还能够减轻兽医制剂的负担,这是一种有前途的治疗病毒性疾病的方法。
其中,外源性重组干扰素是一种最常用的疗法。
通过对重组干扰素进行检测和筛选,可以找到一种在治疗猪白痢方面表现出最佳效果的品种。
研究表明,注射外源性重组干扰素可有效促进猪体内免疫细胞的生长和发育,增强免疫细胞的抵御病毒感染的能力,从而提高猪的免疫能力和生产能力。
此外,干扰素也可用于病毒性疾病的预防。
研究表明,年轻的猪,在注射疫苗时,同时注射干扰素,可以有效增强疫苗的免疫效果,提高疫苗的保护能力,达到更好的预防控制效果。
通过提高疫苗的保护能力,同时加强猪的免疫能力,可以有效地预防猪的病毒性疾病,并减少疫苗的使用量和成本,提高养殖效益。
在病毒性疾病的治疗中,干扰素的应用需要灵活掌握,并根据不同情况进行选用。
干扰素剂量的选择需根据疾病具体症状、病情严重程度和养殖环境等方面进行分析和评估。
药理学知识点归纳免疫增强药之干扰素

干扰素(interferon,IFN)是一类糖蛋白,它具有高度的种属特异性,故动物的IFN对人无效。
干扰素具有抗病毒、抑制细胞增殖、调节免疫及抗肿瘤作用。
临床应用:
在抗病毒方面,它是一个广谱抗病毒药,其机制可能是作用于蛋白质合成阶段,临床可用于病毒感染性疾病,如疱疹性角膜炎、病毒性眼病、带状疱疹等皮肤疾患、慢性乙型肝炎等。
其免疫调节作用在小剂量时对细胞免疫和体液免疫都有增强作用,大剂量则产生抑制作用。
IFN的抗肿瘤作用在于它既可直接抑制肿瘤细胞的生长,又可通过免疫调节发挥作用。
临床试验表明,它对肾细胞癌、卡波济肉瘤、多毛细胞白血病,某些类型的淋巴瘤、黑色素瘤、乳癌等有效;而对肺癌、胃肠道癌、及某些淋巴瘤无效。
不良反应:
在临床应用时常见的不良反应有发热和白细胞减少等,少数病人快速静注时可出现血压下降。
约5%的病人用后可产生IGN抗体。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
2.4 成品
2.4.1 分批 应符合“生物制品分批规程”规定。
2.4.2 分装与冻干 应符合“生物制品分装和冻干规程”及附录ⅠA 有关规定。
2.4.3 规格 应为经批准的规格。
2.4.4 包装
应符合“生物制品包装规程”及附录ⅠA有关规 定。
3 检定 3.1 原液检定 3.1.1 生物学活性 依法测定(附录Ⅹ C)。
2.1.3.2 染色镜检 应为典型的革兰氏阴性杆菌。
2.1.3.3 对抗生素的抗性 应与原始菌种相符。
2.1.3.4 电镜检查(工作种子批可免做) 应为典型大肠杆菌形态,无支原体、病毒样颗 粒及其他微生物污染。
2.1.3.5 生化反应 应符合大肠杆菌生化反应特性。
2.1.3.6 干扰素表达量 在摇床中培养,应不低于原始菌种的表达量。
重组人干扰素α1b注射液
本品系由高效表达人干扰素α1b基因的大肠杆 菌,经发酵、分离和高度纯化后获得的重组人 干扰素α1b制成。含适宜稳定剂,不含防腐剂和
抗生素。
1 基本要求 生产和检定用设施、原材料及辅料、水、器具 、动物等应符合“凡例”的有关要求。
2 制造 2.1 工程菌菌种
2.1.1 名称及来源 重组人干扰素α1b工程菌株系由带有人干扰素α 1b基因的重组质粒.1 分批 应符合“生物制品分批规程”规定。
2.4.2 分装 应符合“生物制品分装和冻干规程”及附录ⅠA 有关规定。
2.4.3 规格 应为经批准的规格。
2.4.4 包装
应符合“生物制品包装规程”及附录ⅠA有关规 定。
3 检定 3.1 原液检定 3.1.1 生物学活性 依法测定(附录Ⅹ C)。
3.1.12 紫外光谱 用水或生理氯化钠溶液将供试品稀释至约 100~500μg/ml,在光路1cm、波长230~360nm 下进行扫描,最大吸收峰波长应为278± 3nm (附录ⅡA)。
3.1.13 肽图 依法测定(附录ⅧE),应与对照品图形一致
3.1.14 N端氨基酸序列(至少每年测定1次) 用氨基酸序列分析仪测定。N端序列应为: (Met)-Cys-Asp-Leu-Pro-Glu-Thr-His-Ser-LeuAsp-Asn-Arg-Arg-Thr-Leu。
3.2 半成品检定 3.2.1 细菌内毒素检查 每30万IU应小于10EU(附录ⅫE,凝胶限度试 验)
3.2.2 无菌检查 依法检查(附录Ⅻ A),应符合规定。
3.3 成品检定 除水分测定、装量差异检查外,应按标示量加 人灭菌注射用水,复溶后进行其余各项检定。
3.3.1 鉴别试验 按免疫印迹法(附录ⅧA)或免疫斑点法(附录ⅧB) 测定,应为阳性。
2.2.2 发酵用培养基 采用适宜的不含抗生素的培养基。
2.2.3 种子液接种及发酵培养 2.2.3.1 在灭菌培养基中接种适量种子液。
2.2.3.2 在适宜的温度下进行发酵,应根据经批 准的发酵工艺进行,并确定相应的发酵条件, 如温度、pH值、溶解氧、补料、发酵时间等。 发酵液应定期进行质粒丢失率检查(附录Ⅸ G)。
2.1.1 名称及来源 重组人干扰素α1b工程菌株系由带有人干扰素α 1b基因的重组质粒转化的大肠杆菌菌株。
2.1.2 种子批的建立 应符合”生物制品生产检定用菌毒种管理规程” 的规定。
2.1.3 菌种检定 主种子批和工作种子批的菌种应进行以下各项 全面检定。
2.1.3.1 划种LB琼脂平板 应呈典型大肠杆菌集落形态,无其他杂菌生长 。
3.1.5 分子量 依法测定(附录ⅣC)。用还原型SDS-聚丙烯酰 胺凝胶电泳法,分离胶胶浓度为15%,加样量 应不低于1.0μg,制品的分子质量应为19.4kD± 1.9kD。
3.1.6 外源性DNA残留量 每1次人用剂量应不高于10ng(附录ⅨB)
3.1.7 鼠IgG残留量 如采用单克隆抗体亲和色谱法纯化,应进行本 项检定。每1次人用剂量鼠IgG残留量应不高于 100ng(附录ⅨL)
2.1.2 种子批的建立 应符合“生物制品生产检定用菌毒种管理规程”的 规定。
2.1.3 菌种检定 主种子批和工作种子批的菌种应进行以下各项 全面检定。
2.1.3.1 划种LB琼脂平板 应呈典型大肠杆菌集落形态,无其他杂菌生长 。
2.1.3.2 染色镜检 应为典型的革兰氏阴性杆菌。
2.1.3.3 对抗生素的抗性 应与原始菌种相符。
3.3.3.2 pH值 应为6.5~7.5 (附录ⅤA)
3.3.3.3 渗透压摩尔浓度 依法测定(附录ⅤH),应符合批准的要求。
3.3.4 生物学活性 应为标示量的80%~150%(附录ⅩC)
3.3.5 残余抗生素活性 依法测定〔附录ⅨA),不应有残余氨苄西林或 其他抗生素活性。
3.3.6 无菌检查 依法检查(附录ⅫA),应符合规定。
2.2.5 初步纯化 采用经批准的纯化工艺进行初步纯化,使其纯 度达到规定的要求。
2.2.6 高度纯化 经初步纯化后,采用经批准的纯化工艺进行高 度纯化,使其达到3.1项要求,既为重组人干扰 素α1b原液。加人适宜稳定刹,除菌过滤后于适 宜温度下保存,并规定其有效期。
2.2.7 原液检定 按3.1项进行。
2.2.3 种子液接种及发酵培养 2.2.3.1 在灭菌培养基中接种适量种子液。
2.2.3.2 在适宜的温度下进行发酵,应根据经批 准的发酵工艺进行,并确定相应的发酵条件, 如温度、pH值、溶解氧、补料、发酵时间等。 发酵液应定期进行质粒丢失率检查(附录Ⅸ G)。
2.2.4 发酵液处理 用适宜的方法收集、处理菌体。
3.3.7 细菌内毒素检查
每1支应小于10EU(附录ⅫE凝胶限度试验)。
3.3.8 异常毒性检查 依法检查(附录ⅫF小鼠试验法),应符合要求 。
4 保存、运输及有效期 于2~8℃避光保存和运输。自生产之日起,按批 准的有效期执行。
5 使用说明
应符合“生物制品包装规程”规定和批准的内容。
中华人民共和国药典2010年版
3.3.2 物理检查 3.3.2.1 外观 应为白色薄壳状疏松体,按标示量加入灭菌注 射用水后迅速复溶为澄明液体。
3.3.2.2 可见异物 依法检查(附录ⅤB),应符合规定。
3.3.2.3 装量差异 依法检查(附录ⅠA),应符合规定。
3.3.3 化学检定 3.3.3.1 水分 应不高于3.0%(附录ⅦD)
3.1.2 蛋白质含量 依法规定(附录Ⅵ B第二法)。
3.1.3 比活性 为生物学活性与蛋自质含量之比,每1mg蛋白 质应不低于1.0× 107 IU。
3.1.4 纯度 3.1.4.1 电泳法 依法测定(附录ⅣC)。用非还原型SDS-聚丙烯酰 胺凝胶电泳法,分离胶胶浓度为15%,加样量 应不低于10μg(考马斯亮蓝R250染色法)或5μ g(银染法)。经扫描仪扫描,纯度应不低于 95.0% 。
3.1.8 宿主菌蛋白质残留量 应不高于蛋白质总量的0.10%(附录ⅨC)
3.1.9 残余抗生素活性 依法测定(附录ⅨA),不应有残余氨苄西林或 其他抗生素活性。
3.1.10 细胞内毒素检查 每30万IU应小于10EU(附录Ⅻ E凝胶限度试 验)
3.1.11 等电点 主区带应为4.0~ 6.5,且供试品的等电点图谱应 与对照品的一致(附录ⅣD)
2.3 半成品
2.3.1 配制与除菌 2.3.1.1 稀释液配制
按经批准的配方配制稀释液。配制后应立即用 于稀释。
2.3.1.2 稀释与除菌 将检定合格加稳定剂的重组人干扰素α1b原液用 2.3.1.1项稀释液稀释至所需浓度。除菌过滤后 即为半成品,保存于2~8℃。
2.3.2 半成品检定 按3.2项进行。
2.1.3.4 电镜检查(工作种子批可免做) 应为典型大肠杆菌形态,无支原体、病毒样颗 粒及其他微生物污染。
2.1.3.5 生化反应 应符合大肠杆菌生化反应特性。
2.1.3.6 干扰素表达量 在摇床中培养,应不低于原始菌种的表达量。
2.1.3.7 表达的干扰素型别 应用抗α1b型干扰素血清做中和试验,证明型别 无误。
中华人民共和国药典2010年版
注射用人重组干扰素α1b
本品系由高效表达人干扰素α1b基因的大肠杆 菌,经发酵、分离和高度纯化后获得的重组人 干扰素α1b冻干制成。含适宜稳定剂,不含防腐
剂和抗生素。
1 基本要求 生产和检定用设施、原材料及辅料、水、器具 、动物等应符合“凡例”的有关要求。
2 制造 2.1 工程菌菌种
2.2.4 发酵液处理 用适宜的方法收集、处理菌体。
2.2.5 初步纯化 采用经批准的纯化工艺进行初步纯化,使其纯 度达到规定的要求。
2.2.6 高度纯化 经初步纯化后,采用经批准的纯化工艺进行高 度纯化,使其达到3.1项要求,既为重组人干扰 素α1b原液。加人适宜稳定刹,除菌过滤后于适 宜温度下保存,并规定其有效期。
3.1.4.2 高效液相色谱法 依法测定(附录ⅢB)。色谱柱以适合分离分子 质量为5~ 60kD蛋白质的色谱用凝胶为填充 剂;流动相为0.1mol/L磷酸盐-0.1 mol/L氯化钠 缓冲液,pH7.0;上样量应不低于20μg,在波 长280nm处检测,以干扰素色谱峰计算的理论
依法测定(附录ⅢB)。色谱柱以适合分离分子 质量为5~ 60kD蛋白质的色谱用凝胶为填充 剂;流动相为0.1mol/L磷酸盐-0.1 mol/L氯化钠 缓冲液,pH7.0;上样量应不低于20μg,在波 长280nm处检测,以干扰素色谱峰计算的理论 板数应不低于1000。按面积归一法计算,干扰 素主峰面积应不低于总面积的95.0%。
2.2.7 原液检定 按3.1项进行。
2.3 半成品
2.3.1 配制与除菌 2.3.1.1 稀释液配制
按经批准的配方配制稀释液。配制后应立即用 于稀释。
2.3.1.2 稀释与除菌 将检定合格加稳定剂的重组人干扰素α1b原液用 2.3.1.1项稀释液稀释至所需浓度。除菌过滤后 即为半成品,保存于2~8℃。