DNA序列比对同源性分析图解BLAST
如何运用BLAST进行序列比对、检验引物特异性

序列比对,绝大多数战友都会想到BLAST,但BLAST的使用确实又是一个很大的难题,因为他的功能比较强悍,里面涉及到的知识比较多,而且比对结束后输出的结果参数(指标)又很多.如果把BLAST的使用详细的都讲出来,我想我发帖发到明天也发不完,更何况我自己也不是完全懂得BLAST的使用。
所以我在这里也就“画龙点睛"——以比对核酸序列为例来给大家介绍一下BLAST的使用,也算是BLAST的入门课程吧。
请看帖的战友好好体会,如果你用心看,在看帖完毕之后BLAST的基本使用(包括其他序列的比对)应该没有问题了。
一、打开BLAST 页面,http://www。
ncbi.nlm.nih。
go/BLAST/ 打开后如图所示:(缩略图,点击图片链接看原图)对上面这个页面进行一下必要的介绍:BLAST的这个页面主体部分(左面)包括了三部分:BLAST Assembled Genomes、Basic BLAST、Specialized BLAST.相信大家可以看懂这三个短语的意思,我就不多说了;我要说的是,可以认为这是三种序列比对的方法,或者说是BLAST的三条途径。
第一部分BLAST Assembled Genomes就是让你选择你要比对的物种,点击相应物种之后即可进入比对页面.第二部分Basic BLAST包含了5个常用的BLAST,每一个都附有简短的介绍。
第三部分Specialized BLAST是一些特殊目的的BLAST,如IgBLAST、SNP等等,这个时候你就需要在Specialized BLAST部分做出适当的选择了。
总之,这是一个导航页面,它的目的是让你根据自己的比对目的选择相应的BLAST 途径。
下面以最基本的核酸序列比对来谈一下BLAST的使用,期间我也会含沙射影的说一下其他序列比对的方法.二、点击Basic BLAST部分的nucleotide blast链接到一个新的页面.打开后如图所示:screen.width-333)this。
生物序列的同源性搜索 -blast简介及其应用

分析过程(三)
6.限制条件,我们限制 在病毒里面找。
7.其他选项保持默认值
打分矩阵
30
分析过程(四)
8.输出格式选项保持 默认值
9.点击开始搜索
31
分析过程(五)
10.查询序列的一些 相关信息 在cdd库里面找到 两个保守区域, 点击可以进入
32
分析过程(六)
图形结果
33
分析过程(七)
15
本地WEB版的Blast
在NCBI的FTP上,在blast程序的目录 下,还提供了一种供用户在自己的服务器 上建立Blast网页服务的软件包(wwwblast)。 使用该软件包,用户可以建立一个简 易的进行Blast运算的网站供实验室人员使 用。用于搜索的数据库同样可以灵活的定 义。
16
Blast程序评价序列相似性的两个数据
39
单机版的Blast使用(三)
3.获取Blast数据库 a.直接从ncbi下载 ftp:///blast/db/ b.用Blast程序包提供的formatdb工具自己格 式化序列数据成数据库。 假设有一序列数据(sequence.fa,多序列,fasta 格式),欲自己做成Blast数据库,典型的命令 如下:
Score:使用打分矩阵对匹配的片段进行打分,这是
对各片段越长、 相似性越高则Score值越大。
E value:在相同长度的情况下,两个氨基酸残基(或
碱基)随机排列的序列进行打分,得到上述Score值的 概率的大小。E值越小表示随机情况下得到该Score值的 可能性越低。
2.其他站点:
/blast/ /ncbi_blast.html /blast/(果蝇)
…
12
Blast结果给出的信息
DNA序列比对同源性分析图解BLAST
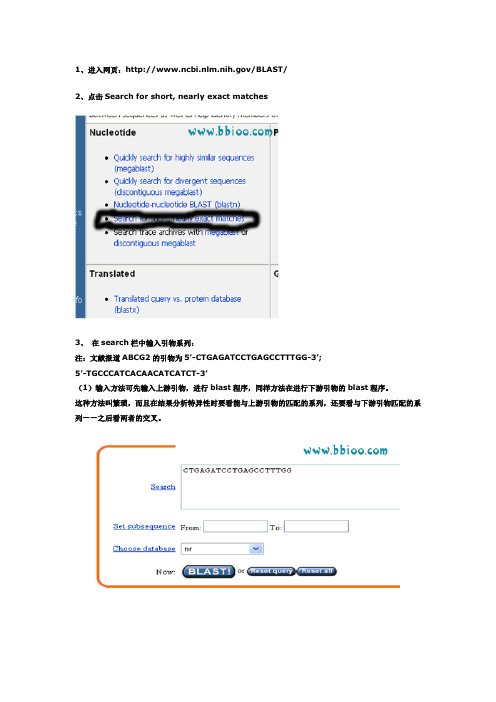
1、进入网页:/BLAST/2、点击Search for short, nearly exact matches3、在search栏中输入引物系列:注:文献报道ABCG2的引物为5’-CTGAGATCCTGAGCCTTTGG-3’;5’-TGCCCATCACAACATCATCT-3’(1)输入方法可先输入上游引物,进行blast程序,同样方法在进行下游引物的blast程序。
这种方法叫繁琐,而且在结果分析特异性时要看能与上游引物的匹配的系列,还要看与下游引物匹配的系列——之后看两者的交叉。
(2)简便的做法是同时输入上下游引物:有以下两种方法。
输入上下游引物系列都从5’——3’。
A、输入上游引物空格输入下游引物B、输入上游引物回车输入下游引物4、在options for advanced blasting中:select from 栏通过菜单选择Homo sapiensExpect后面的数字改为105、在format中:select from 栏通过菜单选择Homo sapiens Expect后面的数字填上0 106、点击网页中最下面的“BLAST!”7、出现新的网页,点击Format!8、等待若干秒之后,出现results of BLAST的网页。
该网页用三种形式来显示blast的结果。
(1)图形格式:图中①代表这些序列与上游引物匹配、并与下游引物互补的得分值都位于40~50分图中②代表这些序列与上游引物匹配的得分值位于40~50分,而与下游引物不互补图中③代表这些序列与下游引物互补的得分值小于40分,而与上游引物不匹配通过点击相应的bar可以得到匹配情况的详细信息。
(2)结果信息概要:从左到右分别为:A、数据库系列的身份证:点击之后可以获得该序列的信息B、系列的简单描述C、高比值片段对(high-scoring segment pairs, HSP)的字符得分。
按照得分的高低由大到小排列。
得分的计算公式=匹配的碱基×2+0.1。
生物信息学:第五讲BLAST序列比对PPT课件

7
精选ppt
8
精选ppt
9
精选ppt
10
精选ppt
11
精选ppt
12
精选ppt
13
精选ppt
14
例2:对给定的olfactory receptor基因进行blastn,目 标数据库为nt,物种为Eukaryota
精选ppt
15精Biblioteka ppt16精选ppt
17
精选ppt
18
精选ppt
19
精选ppt
20
精选ppt
21
精选ppt
22
作业:对给定的2个基因(test1,test2)分别进行 blastn, blastp, tblastn, tblastx, blastx
精选ppt
23
感谢亲观看此幻灯片,此课件部分内容来源于网络, 如有侵权请及时联系我们删除,谢谢配合!
24
对给定的2个基因test1test2分别进行blastnblastptblastntblastxblastx感谢亲观看此幻灯片此课件部分内容来源于网络如有侵权请及时联系我们删除谢谢配合
第五讲:BLAST 序列比对
精选ppt
1
1. Basic Local Alignment Search Tool
2. Compare a query sequence to all the sequences in a specified database
精选ppt
2
BLAST的常用种类(详细见教材): 1、blastn 2、blastp 3、tblastn 4、blastx 5、tblastx
精选ppt
3
例1:对给定的opsin蛋白进行blastp,目标数据库为 nt,物种为Eukaryota
生物序列的同源性搜索blast简介及其应用

序列同源性分析: 是将待研究序列加入到一组与之同源,但来自不同物种 的序列中进行多序列同时比较,以确定该序列与其它序 列间的同源性大小。这是理论分析方法中最关键的一步。 完成这一工作必须使用多序列比较算法。常用的程序包 有CLUSTAL等;
生物序列的同源性搜索blast简介及其应用
PPT文档演模板
2020/11/26
生物序列的同源性搜索blast简介及其 应用
•生物信息学常见的应用与软件
PPT文档演模板
序列数据的保存格式与相关数据库资源 在数据库中进行序列相似性搜索 多序列比对 进化树构建与分子进化分析 Motif的寻找与序列的模式识别 RNA二级结构,蛋白质二、三级结构的预测 基因芯片的数据分析
核酸序列6框翻译成蛋白质序列后和蛋白 质数据库中的序列逐一搜索。
蛋白质序列和核酸数据库中的核酸序列6 框翻译后的蛋白质序列逐一比对。
核酸序列6框翻译成蛋白质序列,再和核 酸数据库中的核酸序列6框翻译成的蛋 白质序列逐一进行比对。
PPT文档演模板
生物序列的同源性搜索blast简介及其 应用
Blast相关的问题
结果页面(一)
•图形示意结果
PPT文档演模板
生物序列的同源性搜索blast简介及其 应用
结果页面(二)
PPT文档演模板
•目标序列描述部分
•带有genbank的链接,点击可以进入
•匹配情况,分值,e
相应的genbank序列
生物序列的同源性搜索blast值简介及其
应用
结果页面(三)
PPT文档演模板
生物序列的相似性搜索NCBI_blast_使用教程

NCBI提供的Blast服务
登陆ncbi的 blast主页
核酸序列
蛋白序列
翻译序列
底下有其他一些针对 特殊数据库的和查看 以往的比对结果等
18
Blast任务提交表单(一)
序列范围 (默认全部)
1.序列信息部分
填入查询(query)的序列 选择搜索数据库 如果接受其他参数默认 设置,点击开始搜索
单机版的Blast程序包,把基本的blast分析, 包括blastn,blastp,blastx等都整合到了 blastall一个程序里面。
42Biblioteka 单机版的Blast使用(六)
以下是一个典型的blastn分析命令: (待分析序列seq.fa,数据库nt_db)
我们选上
29
分析过程(三)
6.限制条件,我们限制 在病毒里面找。
7.其他选项保持默认值
打分矩阵
30
分析过程(四)
8.输出格式选项保持 默认值
9.点击开始搜索
31
分析过程(五)
10.查询序列的一些 相关信息 在cdd库里面找到 两个保守区域, 点击可以进入
32
分析过程(六)
图形结果
33
分析过程(七)
序列同源性分析: 是将待研究序列加入到一组与之同源,但来自不同物种 的序列中进行多序列同时比较,以确定该序列与其它序 列间的同源性大小。这是理论分析方法中最关键的一步。 完成这一工作必须使用多序列比较算法。常用的程序包 有CLUSTAL等;
7
Blast简介(一)
BLAST 是由美国国立生物技术信息 中心(NCBI) 开发的一个基于序列相似性的数据库搜 索程序。
我们通过blast搜索来获取一些这个序列 的信息。
生物信息学-blast

筛选结果
点击开始搜索
其他一些显示格式参数
18
提交任务
返回查询号(request id)
修改完显示格式后点 击进入结果界面
可以修改显示结果格式
19
结果页面(一)
图形示意结果
20
结果页面(二)
目标序列描述部分
带有genbank的链接,点击可以进入 相应的genbank序列
匹配情况,分值,e值
21
结果页面(三)
匹配序列列表
31
分析过程(八)
具体匹配情况
32
单机版的Blast使用(一)
为什么使用单机版的Blast? 1.特殊的数据库要求。 2.涉及序列的隐私与价值。 3.批量处理 4.其他原因??
33
单机版的Blast使用(二)
单机版Blast的基本操作过程 1.下载单机版的Blast程序 ftp:///blast/executables/ 目录下,下载对应的操作系统版本。 2.解压程序包(blast.tar.gz) 命令是: $ tar zxvf blast.tar.gz
5
序列相似性比较和序列同源性分析
序列相似性比较: 就是将待研究序列与DNA或蛋白质序列库进行比较, 用于确定该序列的生物属性,也就是找出与此序列相似 的已知序列是什么。完成这一工作只需要使用两两序列 比较算法。常用的程序包有BLAST、FASTA等;
序列同源性分析: 是将待研究序列加入到一组与之同源,但来自不同物种 的序列中进行多序列同时比较,以确定该序列与其它序 列间的同源性大小。这是理论分析方法中最关键的一步。 完成这一工作必须使用多序列比较算法。常用的程序包 有CLUSTAL等;
6
Blast简介(一)
如何找同源基因并用DNAman比对序列

A
18
A
9
然后一直点击下一步
注意输入的序列是 DNA或者是蛋白质序 列,打勾相应的选项
A
10
选项不需要修 改,默认就行
A
11
选项不需要修 改,默认就行
A
12
选项不需要修 改,默认就行
A
13
序列前面一般会加上CREATED..FEATURESLCATINQUALIFIERS这一段标记
注意,由于是用了免费的版本,它 会在这里加上了一段标记,手工 去除它.
A
4
这是结果,得分越高说明同源性越高
A
5
点击第一个前面字母G,得到如图可以看到 CDS sequence和Peptide equence
A
6
把基因名和CDS序列如图复制到记事本,把 txt格式改为SEQ格式(直接更改扩展名)
A
7
接下来就是用DNAman分析了
选择输入序列
A
8
选择序列文件所 在位置
如何在phytozome中找物种同源基因 并用DNAman比对序列
Zakery 2016年5月26日
A
1
首先到phytozome网站上选择你要找的物种,比如 水稻Oryza sativa v7_JGI (Rice)
A
2
点击BLAST search
A
3
在target type中选择proteome,把氨基酸 序列复制到BLAST框中,点
点击这个按钮, 出现下拉菜单
A
16
保持EMF格式的文件即 可,然后用图片查看器打 开可以放大缩小而不改 变像素
A
17
前面软件演 示所用的序 列比对后做 出来的图
使用 NCBI 查找DNA引物设计BLAST序列比对

最近看到很多战友在论坛上询问如何查询基因序列、如何进行引物设计、如何使用BLAST 进行序列比对……,这些问题在 NCBI 上都可以方便的找到答案。
现在我就结合我自己使用 NCBI的一些经历(经验)跟大家交流一下 BCBI 的使用。
希望大家都能发表自己的使用心得,让我们共同进步!我分以下几个部分说一下 NCBI 的使用:Part one 如何查找基因序列、mRNA、PromoterPart two 如何查找连续的 mRNA、cDNA、蛋白序列Part three 运用 STS 查找已经公布的引物序列Part four 如何运用 BLAST 进行序列比对、检验引物特异性特别感谢本版版主,将这个帖子置顶!从发帖到现在,很多战友对该帖给与了积极的关注,在此向给我投票的(以及想给我投票却暂时不能投票的)各位战友表示真诚的感谢,谢谢各位战友!请大家对以下我发表的内容提出自己的意见。
关于NCBI 其他方面的使用也请水平较高的战友给予补充First of all,还是让我们从查找基因序列开始。
第一部分利用Map viewer 查找基因序列、mRNA 序列、启动子(Promoter)下面以人的 IL6(白细胞介素 6)为例讲述一下具体的操作步骤1.打开Map viewer 页面,网址为:/mapview/index.html 在 search 的下拉菜单里选择物种,for 后面填写你的目的基因。
操作完毕如图所示:2.点击“GO”出现如下页面:3.在步骤二图示的右下角有一个Quick Filter,下面是让你选择的几个复选框,在Gene 前面的小方框里打勾,然后点击Filter. 出现下图:说明一下:1、染色体的红色区域即为你的目的基因所处位置。
2、下面参考序列给出了三个,是不同的部门做出来的,经我验证,序列有微小的差异,但总体来说基本相同。
尽管你分别点击后,序列代码、序列代码等有所差异,但碱基基本一致,不影响大家研究分析序列。
NCBI中Blast序列比对结果解释

NCBI中Blast序列比对结果解释2011-07-26 20:30:12| 分类:生物信息学|字号大中小订阅NCBI中Blast可以用来进行序列比对、检验引物特异性Blast导航主页面主体包括三部分BLAST Assembled Genomes选择你要对比的物种,点击物种之后即可进入对比页面Basic BLAST包含5个常用的Blast,每一个都附有简单介绍Specialized BLAST是一些特殊目的的Blast,如Primer-BLAST、IgBLAST根据需要做出选择本人本学期学习了最基本的核苷酸序列的比对点击Basic BLAST部分的nucleotide链接到一个新的页面,打开后的页面特征:大体上包括三个部分Enter Query Sequence部分可以让我们输入序列,其中的Job Title部分可以为本次工作命一个名字Choose Search Set部分可以选择要与目的序列比对的物种或序列种类。
其中的Entrez Query可以对比对结果进行适当的限制。
Program Selection部分可以选择本次对比的精确度,种内种间等等。
其次Blast按钮下面有一个“Algorithm parameters”算法参数,可设置参数。
点击Blast后,出现的页面大体上包括四个部分一.所询问和比对序列的简单信息1.询问序列的简单信息——名称、描述、分子类型、序列长度2.所比对数据库的名称、描述和所用程序二.Graphic Summary——blast结果图形显示相似度颜色图(黑、蓝、绿、粉红、红,相似度由低到高)三.Descriptions——blast结果描述区1.到其他数据库的链接2.描述以表格的形式呈现(以匹配分值从大到小排序)(1)Accession下程序比对的序列名称,点击相应的可以进入更为详细的map viewer(2)Descriptions下是对所比对序列的简单描述接下来是5个结果数值:(3)Max score匹配分值,点击可进入第四部分相应序列的blast的详细比对结果(4)Total score总体分值(5)Query coverage覆盖率(6)E value——E(Expect)值,表示随机匹配的可能性。
实验三:利用Blast进行序列相似性比对(1)

3. 以大肠杆菌的胶原蛋白酶名称为pHK08_29的基因做为查 询序列 (1) 用Blastn能在nr/nt数据库中检索到多少条与之同源的序 列。其中大肠杆菌、弗累克斯讷氏杆菌、沙门氏菌各有多 少条序列。
(2) 换用megablast或discontiguous megablast,观察检索结 果的改变。 (3) 尝试修改Blastn的参数,观测对检索结果的影响。 (4) 使用Blastx预测在Refseq_ protein数据库中检索到多少 条与之同源的序列。 4. 用blast2分析YP_003683100与ADH70594、 YP_004926582、 YP_004925874、 YP_003273209、 YP_003646515、 YP_003514536、 ABP47302、 ADD45443、 ADW07065、 ADG78176、 ACY21316、 ABM16043、 EHP75935、 BAC74107、 YP_00407863之间的相似性.
实验三:利用Blast进行序列相似性比对
具体步骤
1.登陆blast主页 /BLAST/ 2.根据数据类型,选择合适的程序lo基因(Z83834)为查询序列 (1) 用Blastn能在nr/nt数据库中检索到多少条与之同源的序 列?有多少条是禾本科中的? (2) 换用megablast或discontiguous megablast,观察检索结 果的改变。 (3) 尝试修改Blastn的参数,观测对检索结果的影响。 (4) 找出Mlo基因的编码蛋白序列,用Blastp检索到的与 Mlo蛋白同源的序列与用PSI-Blast检索到的同源序列是否 有差别? (5) 使用BlastX预测Mlo基因的编码蛋白。 2. 用bl2seq分析大麦和小麦Mlo基因mRNA序列编码区和蛋白 质产物的同源性
序列相似性比较与同源性分析

序列相似性⽐较与同源性分析⾸先应该注意区分序列相似性与序列同源性的关系,序列相似不⼀定同源,但是判定同源性关系的时候有些算法(Maximum likelihood除外)要考虑到序列相似性。
序列相似性是将待研究序列与DNA或蛋⽩质序列库进⾏⽐较,⽤于确定该序列的⽣物属性,也就是找出与此序列相似的已知序列是什么,完成这⼀⼯作只需要⽤到两两序列⽐较算法,常⽤的程序包有BLAST,FASTA等。
同源性分析是将待研究序列加⼊到⼀组与之同源,但是来⾃不同物种的序列中进⾏多序列⽐对,以确定该序列与其它序列间的同源性⼤⼩。
多序列⽐较算法常⽤的程序包有CLUSTAL等。
1、序列⽐对,从数据库中寻找相似序列:⾸先打开NCBI的BLAST⽹站:,选择protein blast,然后将待⽐对序列粘贴进去,进⾏BLAST(⼀些参数的设置收藏夹或百度)。
等待⼀定时间后将会出现与所选数据库的⽐对结果,按照打分⾼低将top100(可以设置成其他数值)的序列显⽰出来,然后可以将该100条序列下载下来。
存成test.fasta⽂件。
这个⽂件就是在mega中进⾏多序列⽐对建树所⽤的⽂件。
2、多序列⽐对:打开mega,ALIGN-BUILDALIGNMENT-Create a new alignment-protein-open-retrieve sequences from file-no -test.fasta(或者直接拖动进去,或者双击打开test.fasta),然后点击Alignment——Align by ClustalW——OK——OK。
然后⽐对成功,选择Data——Export Alignment——MEGA format保存⽂件为test.meg,可以关闭Align会话框。
3、构建进化树:打开test.meg。
点击PHYLOGENY——选择最上⾯的ML⽅法,参数可以选择默认参数。
就出现了进化树。
当然⼀些参数最好还是⽤到,⽐如说可信度验证的次数设置最好要⼤于等于500次。
NCBI在线Blast的图文说明

NCBI在线Blast的图文说明Blast(Basic Local Alignment Search Tool)是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具。
BLAST程序能迅速与公开数据库进行相似性序列比较。
BLAST 结果中的得分是对一种对相似性的统计说明。
BLAST 采用一种局部的算法获得两个序列中具有相似性的序列。
Blast中常用的程序介绍:1、BLASTP 是蛋白序列到蛋白库中的一种查询。
库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。
2、BLASTX是核酸序列到蛋白库中的一种查询。
先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。
3、BLASTN 是核酸序列到核酸库中的一种查询。
库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。
4、TBLASTN是蛋白序列到核酸库中的一种查询。
与BLASTX相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对。
5、TBLASTX是核酸序列到核酸库中的一种查询。
此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6条可能的蛋白序列),这样每次比对会产生36种比对阵列。
NCBI的在线blast:/Blast.cgi1、进入在线blast界面,可以选择blast特定的物种(如人,小鼠,水稻等),也可以选择blast所有的核酸或蛋白序列。
不同的blast程序上面已经有了介绍。
这里以常用的核酸库作为例子。
NCBI在线blast页面2、粘贴fasta格式的序列。
选择一个要比对的数据库。
关于数据库的说明请看NCBI在线blast数据库的简要说明。
一般的话参数默认。
NCBI在线blast页面3、blast参数的设置。
注意显示的最大的结果数跟E值,E值是比较重要的。
筛选的标准。
最后会说明一下。
blast参数设置4、注意一下你输入的序列长度。
生物序列的相似性搜索NCBI_blast_使用教程
本地WEB版的Blast
在NCBI的FTP上,在blast程序的目录 下,还提供了一种供用户在自己的服务器 上建立Blast网页服务的软件包(wwwblast)。
使用该软件包,用户可以建立一个简 易的进行Blast运算的网站供实验室人员使 用。用于搜索的数据库同样可以灵活的定 义。
16
Blast程序评价序列相似性的两个数据
匹配情况,分值,e值
24
结果页面(三)
详细的比对上的序列的排列情况
25
一个具体的例子(blastp)
假设以下为一未知蛋白序列
>query_seq
MSDNGPQSNQRSAPRITFGGPTDSTDNNQNGGRNGARPKQRRPQGLPNNTASWFTALTQHGKEELRFPRGQGV PINTNSGPDDQIGYYRRATRRVRGGDGKMKELSPRWYFYYLGTGPEASLPYGANKEGIVWVATEGALNTPKDH IGTRNPNNNAATVLQLPQGTTLPKGFYAEGSRGGSQASSRSSSRSRGNSRNSTPGSSRGNSPARMASGGGETA LALLLLDRLNQLESKVSGKGQQQQGQTVTKKSAAEASKKPRQKRTATKQYNVTQAFGRRGPEQTQGNFGDQDL IRQGTDYKHWPQIAQFAPSASAFFGMSRIGMEVTPSGTWLTYHGAIKLDDKDPQFKDNVILLNKHIDAYKTFP PTEPKKDKKKKTDEAQPLPQRQKKQPTVTLLPAADMDDFSRQLQNSMSGASADST QA
…
12
Blast结果给出的信息
Blast结果会列出跟查询序列相似性比较 高,符合限定要求的序列结果,根据这些 结果可以获取以下一些信息。 1.查询序列可能具有某种功能 2.查询序列可能是来源于某个物种 3.查询序列可能是某种功能基因的同源基因 … 这些信息都可以应用到后续分析中。
NCBI_blast_使用教程 PPT课件

生物序列的同源性
同源性(homology): 指从一些数据中推断出的两个基因或蛋
白质序列具而共同祖先的结论,属于质的 判断。就是说A和B的关系上,只有是同 源序列,或者非同源序列两种关系。而说 A和B的同源性为80%都是不科学的。
5
相似性和同源性关系
序列的相似性和序列的同源性有一定的关系,一 般来说序列间的相似性越高的话,它们是同源序 列的可能性就更高,所以经常可以通过序列的相 似性来推测序列是否同源。
蛋白质序列和核酸数据库中的核酸序列6 框翻译后的蛋白质序列逐一比对。
核酸序列6框翻译成蛋白质序列,再和核 酸数据库中的核酸序列6框翻译成的蛋 白质序列逐一进行比对。
10
Blast相关的问题
怎么获得blast服务,怎么使用的问题?
为什么使用blast,可以获得什么样的信息?
其他问题:实际使用时选择哪种方式(网 络,本地化),参数的选择,结果的解 释…
内容提要
1.基本概念 相似性,同源性
2.Blast介绍 Blast资源和相关问题
3.Blast的应用 网络版,单机版
4.深入了解Blast(改进程序,算法基础) 5.其他的序列相似性搜索工具(fasta)
3
生物序列的相似性
相似性(similarity): 是指一种很直接的数量关系,比如部
分相同或相似的百分比或其它一些合适 的度量。比如说,A序列和B序列的相似 性是80%,或者4/5。这是个量化的关 系。当然可进行自身局部比较。
生物序列的相似性搜索
-blast简介及其应用
2005年3月
生物信息学常见的应用与软件
序列数据的保存格式与相关数据库资源 在数据库中进行序列相似性搜索 多序列比对 进化树构建与分子进化分析 Motif的寻找与序列的模式识别 RNA二级结构,蛋白质二、三级结构的预测 基因芯片的数据分析
第四章 序列相似性搜索工具blast

“The central idea of the BLAST algorithm is to confine attention to segment pairs that contain a word pair of length w with a score of at least T.”
Altschul et al. (1990)
KENFDKARFSGTWYAMAKKDPEG 50 RBP (query) MKGLDIQKVAGTWYSLAMAASD. 44 lactoglobulin (hit)
extend
Hit!
extend
How a BLAST search works: 3 phases
Phase 3: In the original (1990) implementation of BLAST, hits were extended in either direction.
一、 BLAST 简介
BLAST程序是目前最常用的基于局部相似性的数据库 搜索程序,它们都基于查找完全匹配的短小序列片段, 并将它们延伸得到较长的相似性匹配。它们的优势在 于可以在普通的计算机系统上运行,而不必依赖计算 机硬件系统而解决运行速度问题。
BLAST数据库搜索策略
BLAST仅通过部分而不是全 部序列计算最适联配值 ——赢得搜索速度
5’ GTG GGT 5’ TGG GTA 5’ GGG TAG
TWO ASPECTS OF BLAST
BLAST ALGORITHM
Word Hit Heuristic
BLAST STATISTCS
Karlin-Altschul statistics: a general theory of alignment statistics Applicability goes well beyond BLAST
使用 NCBI 查找DNA引物设计BLAST序列比对
最近看到很多战友在论坛上询问如何查询基因序列、如何进行引物设计、如何使用BLAST 进行序列比对……,这些问题在 NCBI 上都可以方便的找到答案。
现在我就结合我自己使用 NCBI的一些经历(经验)跟大家交流一下 BCBI 的使用。
希望大家都能发表自己的使用心得,让我们共同进步!我分以下几个部分说一下 NCBI 的使用:Part one 如何查找基因序列、mRNA、PromoterPart two 如何查找连续的 mRNA、cDNA、蛋白序列Part three 运用 STS 查找已经公布的引物序列Part four 如何运用 BLAST 进行序列比对、检验引物特异性特别感谢本版版主,将这个帖子置顶!从发帖到现在,很多战友对该帖给与了积极的关注,在此向给我投票的(以及想给我投票却暂时不能投票的)各位战友表示真诚的感谢,谢谢各位战友!请大家对以下我发表的内容提出自己的意见。
关于NCBI 其他方面的使用也请水平较高的战友给予补充First of all,还是让我们从查找基因序列开始。
第一部分利用Map viewer 查找基因序列、mRNA 序列、启动子(Promoter)下面以人的 IL6(白细胞介素 6)为例讲述一下具体的操作步骤1.打开Map viewer 页面,网址为:/mapview/index.html 在 search 的下拉菜单里选择物种,for 后面填写你的目的基因。
操作完毕如图所示:2.点击“GO”出现如下页面:3.在步骤二图示的右下角有一个Quick Filter,下面是让你选择的几个复选框,在Gene 前面的小方框里打勾,然后点击Filter. 出现下图:说明一下:1、染色体的红色区域即为你的目的基因所处位置。
2、下面参考序列给出了三个,是不同的部门做出来的,经我验证,序列有微小的差异,但总体来说基本相同。
尽管你分别点击后,序列代码、序列代码等有所差异,但碱基基本一致,不影响大家研究分析序列。
Blast使用技巧解析

PHI-BLAST
模式识别BLAST(Pattern hit intiated BLAST) PHI-BLAST能找到与查询序列相似的 符合某种模式(pattern)的蛋白质 序列
36
Blast的算法基础
基本思想是:通过产生数量更少的但质量 更好的增强点来提高速度。 BALST算法是建立在严格的统计学的基础 之上的。它集中于发现具有较高的相似性 的局部比对,且局部比对中不能含有空位 (blast2.0引入了允许插入gap的算法)。 由于局部比对的限制条件,在大多数情况 下比对会被分解为若干个明显的HSP(Highscore Sequence Pairs)。
设置结果输出显示格式选择需要显示的选项以及显示的文件格式显示数目alignment的显示方式筛选结果e值范围其他一些显示格式参数点击开始搜索16提交任务返回查询号requestid可以修改显示结果格式修改完显示格式后点击进入结果界面17结果页面一图形示意结果18结果页面二目标序列描述部分带有genbank的链接点击可以进入相应的genbank序列匹配情况分值e值19结果页面三详细的比对上的序列的排列情况查询序列和目标序列之间的字母表示两个氨基酸相同加号表示两个氨基酸相似
3
相似性和同源性关系
序列的相似性和序列的同源性有一定的关系,一 般来说序列间的相似性越高的话,它们是同源序 列的可能性就更高,所以经常可以通过序列的相 似性来推测序列是否同源。 正因为存在这样的关系,很多时候对序列的 相似性和同源性就没有做很明显的区分,造成经 常等价混用两个名词。所以有出现A序列和B序 列的同源性为80%一说。
8
Program
blastn
Input
DNA
Database 1 1 6
Blast:大神教你轻松搞定序列比对

Blast:大神教你轻松搞定序列比对Blast (Basic Local Alignment Search T ool) 作为一种序列相似性比对工具,被认为是生物信息分析必须掌握的一款软件。
不管你是做两序列相似性的简单比对,还是引物特异性、基因组成环等个性化分析。
因此,许多看似高大上的基因分析,都可归类于序列间的比较,因此Blast是生信分析中基础性的工具。
今天小编要放大招了,重中之重,送给还在捶胸顿足被一堆数据吓哭的你。
本地Blast本地Blast是该款软件的本地模式,用户可在离线状态下完成目标序列的相似性比对分析。
此种模式不仅可以避免在线提交序列的繁琐和不稳定性,更重要的是能够为用户提供个性化的服务。
若用户需要指定特殊数据库或大量序列的比对,本地Blast则是最优选择。
那么,如何进行本地Blast呢?接下来小编为您献上做本地Blast的基本原则,若您能掌握以下要点,不管对快速应用本地blast还是未来拓展个性化都有很大帮助。
1掌握三个基本要素分别是数据库(database)、待比对序列(query)和目标序列(subject)。
基于这三个基本元素,本地Blast运行方式即是用户选定目标序列(subject)并将其构建成数据库,然后用待比对序列(query)在数据库中搜索,待比对序列遍历数据库中的每一条目标序列后得到最终比对结果。
本地Blast概述:本地Blast是一款集成软件,其中包括blastp、blastx和blastn等模块,通过调用不同的比对模块,blast 实现了五种可能的序列比方式:blastp:蛋白序列与蛋白库作比对,直接比对蛋白序列的同源性。
blastx:核酸序列与蛋白库作比对,将核酸序列先翻译成蛋白序列,再将其与蛋白库作比对。
blastn:核酸序列与核酸库的比对,直接比对核酸序列的同源性。
tblastn:蛋白序列对核算库的比对,现将核酸库翻译成蛋白库,再将蛋白序列与翻译后的蛋白库进行比对。
新手上路,一文秒懂Blast结果图(附序列比对网址)

新手上路,一文秒懂Blast结果图(附序列比对网址)转载请注明:解螺旋·临床医生科研成长平台众所周知,同源性是预测基因和蛋白质功能的主要线索,而序列同源性的判断则离不开两个或多个序列之间相似性的检测。
一般来说,序列间的相似度越高,它们是同源序列的可能性就越高。
其中,序列比对无疑是评估序列相似性的最简单方法。
显然,Blast就是序列比对检测的中坚力量。
Blast自1990年首次亮相以来,凭借从各大数据库(EST、PDB数据库等)获取信息的能力,迅速成为序列比对界的领头羊。
老实说,Blast的界面非常友好,点击相应模块后,大家只需在序列框中丢上自己的靶序列,勾选好物种基因组,点击搜索即可!可看着结果界面涌现出的几十个、数百个甚至数千个候选匹配序列,不少选择困难症的童鞋表示头疼不已:结果辣么多,究竟哪个才是最优解?本文以NM_001206932为例,分解BLAST结果页面,让大家迅速摆脱Blast新手身份。
Blast结果解析首先会看到一个表头,即本次比对的基本信息,如比对类型、序列长度、所选的数据库等等。
如果所选的数据库不合适,请及时迷途知返哦。
接下来就是Blast的结果显示图(Graphic Summary):颜色比例尺,其中相似度从高到低排列分别为:红、紫、绿、蓝、黑,红色区域越多则表示有较好的比对结果。
而在Blast结果的描述区域,两个衡量标准最为重要:Max Score 和E值(E value),前者匹配片段越长,相似性越高则Score值越大;后者是得到上述Score值的概率的大小。
E值越小表示随机情况下得到该Score值的可能性越低。
而点击相应注释名称,又或者在结果显示图(Graphic Summary)中点击对应的线条,均可以查看比对结果的详细信息。
其中,Expect(E值)、Identities(一致性)、Gaps(缺失或插入)三项是评价blast结果的标准。
E值接近零或者为零时,具体上就是完全匹配了;一致性:匹配上的碱基数占总序列长的百分数。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
1、进入网页:/BLAST/
2、点击Search for short, nearly exact matches
3、在search栏中输入引物系列:
注:文献报道ABCG2的引物为5’-CTGAGATCCTGAGCCTTTGG-3’;
5’-TGCCCATCACAACATCATCT-3’
(1)输入方法可先输入上游引物,进行blast程序,同样方法在进行下游引物的blast程序。
这种方法叫繁琐,而且在结果分析特异性时要看能与上游引物的匹配的系列,还要看与下游引物匹配的系列——之后看两者的交叉。
(2)简便的做法是同时输入上下游引物:有以下两种方法。
输入上下游引物系列都从5’——3’。
A、输入上游引物空格输入下游引物
B、输入上游引物回车输入下游引物
4、在options for advanced blasting中:
select from 栏通过菜单选择Homo sapiens
Expect后面的数字改为10
5、在format中:
select from 栏通过菜单选择Homo sapiens Expect后面的数字填上0 10
6、点击网页中最下面的“BLAST!”
7、出现新的网页,点击Format!
8、等待若干秒之后,出现results of BLAST的网页。
该网页用三种形式来显示blast的结果。
(1)图形格式:
图中①代表这些序列与上游引物匹配、并与下游引物互补的得分值都位于40~50分
图中②代表这些序列与上游引物匹配的得分值位于40~50分,而与下游引物不互补
图中③代表这些序列与下游引物互补的得分值小于40分,而与上游引物不匹配
通过点击相应的bar可以得到匹配情况的详细信息。
(2)结果信息概要:
从左到右分别为:
A、数据库系列的身份证:点击之后可以获得该序列的信息
B、系列的简单描述
C、高比值片段对(high-scoring segment pairs, HSP)的字符得分。
按照得分的高低由大到小排列。
得分的计算公式=匹配的碱基×2+0.1。
举例:如果有20个碱基匹配,则其得分为40.1。
D、E值:代表被比对的两个序列不相关的可能性。
E值最低的最有意义,也就是说序列的相似性最大。
设定的E值是我们限定的上限,E值太高的就不显示了
E、最后一栏有的有UEG的字样,其中:
U代表:Unigene数据库
E代表:GEO profiles数据库
G代表:Gene数据库
(3)结果详细信息:
①圈出来的部分代表序列的信息
②第一个大括号代表上游引物与该序列的正链的匹配情况:
共有21个碱基匹配,得分42.1分,E值为0.020
上游引物与序列的2143~2163位点匹配
③第二个大括号代表下游引物与该序列的负链的匹配情况:
共有20个碱基匹配,得分40.1分,E值为0.077。
下游引物与该序列的29360~29379位点互补
注意点:
①上游引物为20个碱基,为什么会变成21个碱基呢?这是因为下游引物的第一个碱基为T,刚好与系列的2163位点的T匹配,因此下游引物的开头的第一个碱基被当成了上游引物了。
同理,上游引物的最后一个碱基为G,被当成了下游引物了。
通过寻找有没有与1~20位点、20~40位点完全匹配的序列,就可以避免这个因素的干扰了。
②为什么与上下游引物匹配的ABCG2序列有多种?
A、为同一个基因来源的不同的mRNA片段
B、为该基因的DNA系列
C、为同一个基因来源的不同的cDNA片段。
结果判断:
①验证文献报道的引物是否正确:如果你可以在所显示的结果中找出你的目的基因,一般说明你的引物正确性没问题。
如果你blast后没有发现你的目的基因,或者分值很低,该引物就可能不适合用
②检测该对引物是否可与其它序列匹配,引起PCR的非特异性扩增。
如果找到了你的目的基因名称,而且找到了一大批同物种的不同基因,(上下游引物分别搜索到相同的基因),而且分数也较高。
这时表明你的引物设计的特异性不高,极有可能在你的扩增产物中出现非特异性产物。