缺血性脑卒中发病机制研究新进展
药物治疗缺血性脑卒中的研究进展

药物治疗缺血性脑卒中的研究进展摘要】脑动脉管腔阻塞或者狭窄能够导致脑局部的血流量突然中断或者减少,从而出现缺血性中风,其是一种脑供氧及供血均减少的病理情况,并能够导致自主神经功能障碍及继发性血管内皮损伤等情况出现。
随着医疗技术发展和人们对对中医药认识的加深,采用中药及针灸对脑卒中患者进行治疗的效果逐渐得到广大临床工作者及患者的认可。
因此,加速中医药现代化的研究与临床应用,开发合理、高效的中医药针剂及中成药,并配合康复及针灸疗法对脑卒中患者进行治疗,既迫在眉睫又任重道远。
【关键词】缺血性脑卒中;发病机制;发病诱因;药物治疗【中图分类号】R543.3 【文献标识码】A 【文章编号】2095-1752(2020)11-0015-02Study on the mechanism of drug therapy for ischemic strokeCheng XinwangChongqing Xiushan Tujia and Miao Autonomous County People's Hospital,Xiushan,Chongqing 409900,China【Abstract】The occlusion or stenosis of the cerebral artery can lead to the sudden interruption or decrease of the blood flow in the brain,which leads to ischemic stroke.This condition is a pathological condition in which the brain oxygen supply and blood supply are reduced,and it can also lead to the autonomic nerve dysfunction and secondary vascular endothelial injury.With the development of medical technology and the deepening of people's understanding of traditional Chinese medicine,the therapeutic effect of traditional Chinese medicine and acupuncture on stroke patients has gradually been recognized by the majority of clinical workers and patients.Therefore,it is urgent and a long way to accelerate the research and clinical application of the modernization of traditional Chinese medicine,develop reasonable and efficient traditional Chinese medicine injection and proprietary Chinese medicine,and treat stroke patients with rehabilitation and acupuncture therapy.【Key words】Ischemic stroke;Pathogenesis;Predisposing factors;Drug therapy1.缺血性脑卒中病发机制和发病诱因1.1 缺血性脑卒中病发机制缺血性脑卒中的发病原理主要为由于机体脑动脉管腔堵塞或者狭窄,进而导致脑局部的血流量突然中断或者减少,造成脑组织供血、供氧和供能减少而引起一系列器官受损的病理状态,该情况能够导致自主神经功能障碍及继发性血管内皮损伤;若持续一定时间,则将导致缺血区的脑组织细胞死亡,即脑梗死。
缺血性脑卒中炎症反应机制探讨

·297·缺血性脑卒中炎症反应机制探讨郑丽娟 张丹峰 河南推拿职业学院 河南洛阳 471000摘 要:缺血性脑卒中后的炎症反应是一个复杂的细胞和分子反应,炎症细胞的活化、炎症介质介导以及炎症信号通路激活是引起缺血性脑卒中级联反应的基础。
炎症过程主要有两部分组成:一是无细胞成分,指的是脑细胞、外周白细胞分泌各种炎性细胞因子、化学趋化因子和类花生酸类物质;二是细胞成分,指的是外周炎性细胞浸润至损伤的脑组织内。
只有充分理解脑梗死的炎症反应和炎症因子时空特点,才能有效的确立治疗时间和治疗的方法。
关键词:炎症反应 炎症细胞 炎症因子脑卒中是目前导致人类死亡的第二位原因。
卒中的高发病率、高死亡率和高致残率给社会、家庭和患者带了沉重的负担[1]。
脑缺血后脑神经细胞内的各种神经因子发生瀑布式的级联反应,启动了对神经细胞的损害过程。
缺血性脑卒中的病理生理进程是一个动态、复杂的过程,受到诸多细胞内外理化因素的影响。
研究表明、炎症反应、兴奋性氨基酸的释放、自由基的生成增加,钙超载、相关凋亡基因的表达等在缺血性脑损伤中发挥了重要的作用[2]。
本文将以炎症细胞与炎症因子为基点分析炎症反应对脑卒中的损害机制。
1炎症反应炎症反应是一种复杂的细胞和分子反应,主要由炎症细胞和炎症介质介导。
炎症细胞浸润,炎症信号通路激活及大量炎症介质产生引发炎症级联反应是缺血性脑卒中炎症反应的基础[3]。
脑缺血后的白细胞浸润所致的炎症反应在缺血性脑损害的发生、发展中所起的重要作用已被相关的研究所证实。
受损的脑细胞产生大量的白介素-1(IL-β)、肿瘤坏死因子-α(TNF-α)等炎症因子,这些炎症因子诱导内皮细胞表达细胞粘附因子,这些粘附因子使中性粒细胞与内皮细胞粘附,中性粒细胞得以穿过血管壁进入脑实质。
5~7天后巨噬细胞和单核细胞进入脑组织。
血液中的炎症细胞也会向靶目标移动。
炎症反应所致的缺血性脑损害机制可能与①中性粒细胞浸润所致的微血管堵塞使缺血进一步加深;②激活的炎性细胞和受损的神经元产生大量调节因子加重了损害;③浸润的中性粒细胞产生诱导型NOS;④缺血的神经元表达COX2;⑤缺血的神经元产生TNF;⑥小胶质细胞也可以产生神经毒素人NO、活性氧及前列腺素等[4]。
急性缺血性脑卒中发病机制研究

急性缺血性脑卒中发病机制研究1. 引言1.1 急性缺血性脑卒中发病机制研究急性缺血性脑卒中是一种常见的脑血管疾病,严重威胁人类的健康和生命。
研究表明,急性脑卒中的发病机制极为复杂,涉及多个方面的因素。
了解脑卒中的发病机制对于预防和治疗脑卒中至关重要。
脑卒中的发病机制包括缺血性和出血性两种类型,其中缺血性脑卒中是最常见的类型。
缺血性脑卒中的发病机制主要是由于脑血管的阻塞或狭窄导致脑组织缺氧、缺血和继而坏死。
在缺血性脑卒中发病过程中,脑缺血引起的神经细胞损伤是一个关键的环节。
神经细胞的损伤会引发一系列生物化学变化,最终导致脑组织的破坏和功能障碍。
免疫炎症在脑卒中发病过程中也发挥着重要的作用。
炎症反应会加剧脑组织的损伤,增加脑卒中的严重程度。
血小板激活与栓子形成也在脑卒中中起着至关重要的作用,它会加重脑部缺血和缺氧的程度,加速神经细胞的死亡。
脑卒中患者的血管再灌注损伤也是一个重要的研究方向,了解再灌注损伤的机制有助于更好地治疗脑卒中患者。
了解急性缺血性脑卒中发病机制对于预防和治疗脑卒中至关重要。
通过深入研究脑卒中的发病机制,可以为未来脑卒中的防治提供重要依据,并有望为寻找新的治疗方法提供突破口。
2. 正文2.1 缺血性脑卒中的病因分析缺血性脑卒中是指由于脑血管狭窄或堵塞引起的脑组织血液供应不足而造成的脑部损伤。
其病因分析主要包括动脉粥样硬化、脑动脉狭窄、脑动脉瘤和栓子形成等多种因素。
动脉粥样硬化是造成缺血性脑卒中最常见的病因之一,其主要特征是血管内动脉壁的斑块形成,导致脑血管狭窄或堵塞。
脑动脉狭窄也是引发脑卒中的重要原因,当脑血管壁发生狭窄时,会导致脑部血液供应不足,容易引发脑卒中。
脑动脉瘤也是导致脑卒中的潜在危险因素之一,当脑动脉瘤破裂时,会导致脑内出血,从而引发急性缺血性脑卒中。
而栓子形成则是由于血液中的血小板在狭窄的脑血管内聚集形成血栓,阻碍了正常的血液流动,造成脑部缺血缺氧。
缺血性脑卒中的病因分析涉及多个方面,包括动脉粥样硬化、脑动脉狭窄、脑动脉瘤和栓子形成等多种因素,对这些病因进行深入的研究有助于更好地预防和治疗脑卒中的发生。
缺血性脑卒中发病机制的研究进展

12 ~ h内 , 即所谓 的第一 阶段 ; 其次 , 大量谷氨 酸与 N A受体结 MD
合, 使细胞 c 通道 反复开放 , a 导致 大量 c 2内流 , a 十 此种结合 可
从 中 毒后 2 h 长 到 5 7 , 4延 ~ d 即所 谓 的第 二 阶段 。 两 阶 段 变 化 因 此 非 N A 和 N A 分 布 重 叠 , 应 交 错 , 上 代 谢 型 谷 氨 酸 MD R MD R 反 加 激 活 , 细 胞 内钙 池 大 量 释 放 c n 导致 致 死 性 C2超 载 。 使 a, a ' 细胞 内 c 超 载 可 致 胞 浆 内或 溶 酶 体 内 C 依 赖 性 酶 类 和 磷 脂 酶 类 大 a a
近年来 , 随着 人们生活水平 的提高及饮食结 构的变化 , 缺血 性脑卒 中的发 生率 呈现逐年增加 的趋势 ,严重影 响了患者 的生
活 质 量 , 卒 中 已经 成 为 威 胁 人 类 健 康 的重 要 杀 手 。因此 探 讨 有 脑 效 的治 疗 手 段 已经 成 为 所 有 医 务 工 作 者 迫 在 眉 睫 的要 求 ,近 年
细胞引起其 功能紊乱 , 造成 内皮 素等缩血管物质分 泌增 加 , 坏 破
1 谷氨 酸的 兴奋 毒性及 钙 超载 与缺 血性 脑 损伤
谷氨酸巾毒 以两种形式存在 :首先谷氨酸与 突触后非 N 一甲
基 一 一天 门 冬 氨 酸 盐 ( MD 受 体 结 合 , 起 N 道 开 放 , D N A) 引 a通 大 量 N a 向细 胞 内流 动 , 时 出 现 继 发 性 c 同 l和 水 份 内 流 , 致 神 导 经 细 胞 肿 胀 、 解 , 至 变 性 坏 死 , 种 情 况 多 发 生 于 中 毒 后 的 溶 甚 此
脑卒中防治的新进展血管内治疗技术

脑卒中防治的新进展血管内治疗技术脑卒中是一种常见且危险的神经系统疾病,由于其高发病率、致残率和死亡率,一直备受关注。
随着医疗技术的不断进步,血管内治疗技术作为脑卒中防治的新进展,已经取得了显著的成效。
本文将重点介绍脑卒中的血管内治疗技术及其新进展。
一、脑卒中概述脑卒中是指由于脑部血液供应中断而引起的脑功能损害,是导致死亡和残疾的重要原因之一。
根据病因不同,脑卒中可分为缺血性脑卒中和出血性脑卒中。
缺血性脑卒中又可分为大脑动脉供血不足和脑血管阻塞。
二、血管内治疗技术血管内治疗技术是指通过导管经血管进入脑血管系统进行治疗。
其优点在于创伤小、恢复快、疗效确切。
下面将介绍几种常用的血管内治疗技术:1. 血管内溶栓血管内溶栓是脑卒中治疗的一项重要技术。
该技术通过导管将溶栓药物直接送达梗塞部位,溶解血栓,恢复脑血流,从而缓解脑缺血症状。
目前常用的溶栓药物有组织型纤溶酶原激活剂(t-PA)等。
血管内溶栓技术适用于缺血性脑卒中,并且在治疗窗口内能够取得良好的效果,减少脑卒中后的残疾发生率和死亡率。
2. 血管内取栓当脑血管阻塞导致脑卒中时,血管内取栓技术能够迅速清除血栓,恢复血流。
这一技术通常通过导管将取栓器具送达梗塞部位,将血栓困住并撤出体外。
血管内取栓技术对于大脑血栓和颈内动脉病变导致的脑梯形征疗效显著,能够改善脑梯形征症状和脑梯形区域的血流灌注。
3. 血管内支架在一些病例中,脑卒中是由于脑动脉狭窄或闭塞造成的。
血管内支架技术通过导管将支架送达病变部位,扩张脑动脉,恢复血液供应。
这一技术适用于脑动脉狭窄或闭塞性脑卒中患者,在改善脑血液循环方面有很好的效果。
三、脑卒中防治的新进展随着技术的不断创新,脑卒中防治的新进展血管内治疗技术取得了突破性的进展。
下面将介绍几项主要的新技术:1. 血管内神经保护剂血管内神经保护剂可通过导管送达脑血管系统,发挥神经保护作用。
这些神经保护剂包括神经生长因子和细胞因子等。
神经保护剂能够减轻脑部缺血缺氧损伤,促进神经元再生,从而恢复脑功能。
《2024年缺血性脑卒中患者脑小血管病总负荷与颈动脉内—中膜厚度的相关性研究》范文

《缺血性脑卒中患者脑小血管病总负荷与颈动脉内—中膜厚度的相关性研究》篇一缺血性脑卒中患者脑小血管病总负荷与颈动脉内-中膜厚度的相关性研究一、引言缺血性脑卒中是一种常见的脑血管疾病,其发病机制复杂,涉及多种因素。
近年来,越来越多的研究开始关注脑小血管病(CSVD)与缺血性脑卒中的关系。
CSVD总负荷作为评估脑小血管病变严重程度的重要指标,与颈动脉内-中膜厚度(IMT)之间是否存在相关性,是本文关注的重点。
本文旨在通过相关性研究,探讨缺血性脑卒中患者CSVD总负荷与IMT的关系,为临床诊断和治疗提供参考依据。
二、方法本研究采用回顾性分析方法,收集了某医院神经内科收治的缺血性脑卒中患者的临床资料。
纳入标准为:年龄≥45岁,经影像学检查确诊为缺血性脑卒中患者。
排除标准为:患有严重心、肝、肾等重要脏器疾病的患者。
所有患者均接受了脑部MRI检查和颈动脉超声检查。
三、数据收集与处理1. 收集患者的年龄、性别、高血压病史、糖尿病病史、吸烟史等基本信息。
2. 通过MRI检查评估CSVD总负荷,包括脑白质高信号(WMH)、微出血(CMB)等病变程度。
3. 通过颈动脉超声检查测量IMT值,并记录双侧颈动脉的IMT值。
4. 根据CSVD总负荷和IMT值进行分类,分为高负荷组和低负荷组,进行组间比较分析。
四、结果1. 缺血性脑卒中患者中,CSVD总负荷较高者占比较高,表明CSVD在缺血性脑卒中患者中普遍存在。
2. 高负荷组患者的IMT值显著高于低负荷组患者,表明CSVD总负荷与颈动脉IMT之间存在正相关关系。
3. 通过相关性分析发现,CSVD总负荷与颈动脉IMT之间存在显著的正相关关系(r=0.682,P<0.01),提示两者在发病机制上可能存在共同的病理生理基础。
4. 进一步分析发现,高血压病史、糖尿病病史和吸烟史等因素对CSVD总负荷和IMT均有影响,可能是导致两者之间正相关关系的重要因素。
五、讨论本研究表明,缺血性脑卒中患者的CSVD总负荷与颈动脉IMT之间存在显著的正相关关系。
缺血性脑卒中后脑水肿的发病机制及治疗进展

缺血性脑卒中后脑水肿的发病机制及治疗进展
李佳楠;李立宏
【期刊名称】《临床医学进展》
【年(卷),期】2024(14)4
【摘要】缺血性脑卒中已成为全球死亡和残疾的主要原因。
脑水肿是缺血性脑卒中的严重并发症,会引起颅内压升高,神经系统症状迅速恶化,形成脑疝,是脑卒中后不良结局的重要危险因素。
迄今为止,脑卒中后脑水肿的详细机制尚不清楚。
这限制了预防和治疗策略以及药物开发的进展。
本文就脑水肿的分类、病理特点、缺血性脑卒中后脑水肿的发生机制与水通道蛋白4、SUR1-TRPM4通道、基质金属蛋白酶9、microRNA、脑静脉回流、炎症反应的关系进行综述。
综述了脑卒中后脑水肿治疗新药的研究进展。
因此,本文综述为进一步研究和临床治疗缺血性脑卒中后脑水肿提供参考。
【总页数】10页(P2769-2778)
【作者】李佳楠;李立宏
【作者单位】西安医学院研究生院西安;空军军医大学唐都医院急诊科西安
【正文语种】中文
【中图分类】R74
【相关文献】
1.蛇毒降纤酶治疗脑卒中后脑水肿的机制研究新进展
2.水通道蛋白在急性缺血性脑卒中后脑水肿发生机制中的研究进展
3.醒后脑卒中的发病机制、危险因素及治疗
研究进展4.铁死亡参与缺血性脑卒中后神经损伤发病机制及其治疗进展5.特鲁索综合征相关缺血性脑卒中发病机制及治疗研究进展
因版权原因,仅展示原文概要,查看原文内容请购买。
脂联素与缺血性脑卒中的研究新进展

阑尾炎切口感染是一种常见并发症 ,为混合感染,应与上述 三个环节失误有关。 应注意: 严格无菌操作, 1 污染器械不可复用,
更不可接触切 口; 2充分止血,结扎仔细;3通畅引流, 采用硅橡
胶管另戳 口引出;注意侧 口不宜过高。4少用或不用电刀、防止 脂肪坏死:另外,缝合j 深或留有死腔,也是导致感染的原因。 我们采用紫外线照射切 口可促进局部血运 , 使炎性和致病 介质清 除加速 ; 使细菌 D A失去功 能;同时增加抗生素抗茵能 N
危险因素、预 防动脉 粥样硬化病变 的发生 、进 展就是预防血
栓 形 成 。炎 症 的血 清 标 志物 水 平 随 动 脉 粥样 硬 化 进 展 的不 同
心病 的独立预测指标 ,还 与众所周知的心血管疾病危险 因素
— —
时期和阶段而动态 变化 ,脂联素是新近研 究的热点之一 ,国 内外研究表 明它参与动脉粥样硬化发 生、发展的各个环 节 , 可能是连 接血 管炎症和 动脉 粥样硬化 的桥梁 ,起着抗 炎、抗 动脉粥样硬化 的作用 。因此 通过监测血清 中脂 联素动态变化 水平 ,有利于对颈动脉 粥样 硬化、脑卒中等各种危 险因子 的 评估 ,尽可能早期应用药物 干预 ,改善颈动脉 粥样 硬化斑块
发生 、发 展 及 预 后 ,稳 定 和 逆 转 斑 块 、 中 断 和 阻止 脑 卒 中 的
发生 。
男性 、2 型糖尿病、血脂异常、高血压、肥胖 、胰岛素抵
抗密切相关 。 脂联素可沉积在受损的动脉壁 , 抑制 内皮炎症反
应 , 外 在 血 浆 和脂 肪 组 织 中脂 联 素与 C P水 平 存 在 显著 的负 此 R
2 具 体 方 法 21 手术 操 作过 程 .
缺血性脑卒中与NO、SOD关系研究进展
目前 , 大 多 数 工 业 化 国 家 , 中 已是 继 心 脏 病 和 癌 症 之 在 卒 后 的 成 人 第 三 大 常见 死亡 原 因 , 且 是 导 致 长 期 致 残 的第 一 位 而 原 因 。 随 着 人 口老 龄 化 E益 加 深 , 年 人 在 人 口中 所 占 比 例 相 t 老 对 升 高 , 使 得 卒 中 的 发 病 率 也 E趋 上 升 。尽 管 国 际 社 会 E益 这 t t 重 视 开 展 脑 血 管 病 的 一 、 级 预 防 尤其 是 二 级 预 防 来 尽 可 能 减 二 少 卒 中发 病 危 险 因素 ( 高 血 压 、 尿 病 、 脂 血 症 等 )但 到 目 如 糖 高 , 前为止 , 于已发卒 中患者 , 了在神 经 系统功 能受 损症状 出 对 除
管平滑 肌而言 , NO 是 有 效 的 血 管 扩 张 剂 , 够 调 节 局部 血 流 , 能
它 还 具 有 抗 血 栓 形 成 、 炎 和 抗 增 殖 作 用 。但 它 的保 护 作 用 持 抗
维普资讯
脑与神经疾病杂志 20 0 7年 第 1 5卷 第 5期
39 1
缺 血性 脑卒 中与 NO、 O S D关 系 研究 进 展
王春 娟 薛连 璧
中 图 分 类 号 :R 4 . 2 7 3 3 文献标识码 : A 文 章 编 号 :1 0 —3 1 2 0 ) 5 3 1 4 0 6 5 X( 0 7 0 —0 9 —0
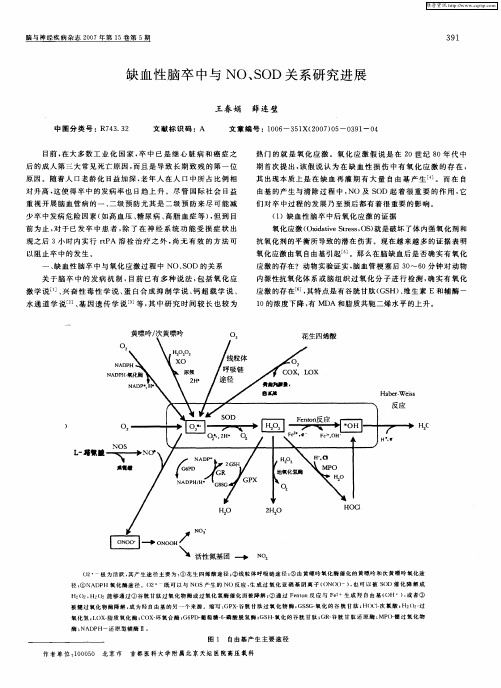
H。 , O2能 够通 过 ① 谷 胱 甘 肽过 氧 化 物 酶 或 过 氧 化 氢 酶 催 化 而 被 降 解 } 通 过 Fe n反 应 与 Fe 生 成 羟 自 由 基 ( O2 H2 ② mo OH ) 或 者 ③ ,
被 髓 过氧 化 物 酶 降解 , 为 羟 自由 基 的 另 一 个 来 源 。缩 写 : X 谷 胱 甘 肽 过 氧 化 物 酶 ; S  ̄ 化 的 谷 胱 甘 肽 ; 成 GP 一 GS G- HOCI 氯 酸 { (2过 一 次 H2)一
缺血性卒中炎症反应的研究新进展

缺血性卒中炎症反应的研究新进展摘要:脑梗死,也就是所谓的缺血性卒中,是世界上造成残疾主要因素同时也是第三大死因。
我国每年大约有一千五百万人患有卒中,其中三分之一的人死亡,三分之一的人残疾,对世界造成了沉重的社会和经济负担。
在这些因素中,80%~85%的卒中是由于脑动脉栓塞或栓塞造成的。
目前已有的研究显示,在脑卒中过程中,免疫炎性反应扮演着重要角色。
脑缺血后,由于炎症反应的发生、脑组织遭受损害、炎性细胞的激活和侵袭,都会引起脑组织的炎症反应,进而导致脑组织的损害。
本文以缺血性卒中炎症反应的研究新进展进行了讨论。
关键词:缺血性卒中;炎症反应;研究进展在中枢神经系统病变中,有许多机理参与,而炎症反应是一个重要的过程。
炎症反应是由于神经系统萎缩,产生胶质细胞,进而诱发循环免疫细胞侵袭大脑,是脑卒中脑损伤的主要原因。
卒中后,脑神经细胞广泛坏死、病变,释放出大量的炎症介质,并释放出与损伤有关的分子模型(DAMP),从而促进了局部的炎症反应。
脑缺血、发炎等疾病的加重,使神经细胞的凋亡速度加快。
因此,缺血后的炎性反应不仅参与了脑梗死的病理生理过程,还与其预后有很大关系。
脑卒中后炎症消退和神经损伤修复是极为重要的过程。
解析出炎症发生的原因,对于脑卒中药物的研究开发有重大意义。
最近,诺华公司发起的CANTOS临床研究也表明了这一点。
他们发现,卡那奴单抗能显著地降低IL-1β和超敏C反应蛋白的浓度,而不会使LDL和胆固醇含量下降,并且显著减少再发的心脑血管事件。
大量的实验表明,IL-1β是导致卒中性炎性损害的重要因素,CANTOS实验首次从临床上证明了炎性反应可直接参与组织缺血损害,因此,抗炎反应控制是减少脑缺血损害的一种有效措施。
1.炎性DAMPs与炎症反应2004年,国外学者研究证实基于内源性疏水性的生物分子能够刺激人体的免疫功能,提出了DAMP的概念[1-4]。
脑缺血后,脑细胞会产生如缺氧、营养缺失、内质网等各种代谢应激,会引起大脑的核心区域缺血导致脑细胞死亡,从而引起缺血性卒中炎症反应。
缺血性脑卒中规范化诊治及新进展答案-2024年华医网继续教育神经内科学答案

缺血性脑卒中规范化诊治及新进展答2024年华医网继续教育神经内科学答案高分辨血管壁MR指导下慢性颈动脉闭塞开通1.慢性颈动脉闭塞最常见病因()A.动脉粥样硬化B.结缔组织疾病C.先天发育异常D.病毒感染E.血管炎正确答案:A2.关于脑动脉血管壁正性重构特征,哪一项表述正确()A.斑块造成的管壁向扩张B.加重管腔狭窄C.斑块面积<40%血管截面积时,动脉管壁表现为正性重构D.DSA检查可见管腔明显狭窄E.斑块面积>40%血管截面积时,动脉管壁表现为正性重构正确答案:C3.高分辨血管壁MR指导下颈动脉闭塞开通的优点是什么()A.建立血管壁完整图像B.减少脑梗死并发症C.明确血肿或溶栓疗法的治疗时态D.对内中膜撕裂如实显示E.评价颅内动脉分支的情况正确答案:A4.慢性颈动脉闭塞临床特异性表现()A.晕厥B.记忆力下降C.偏头痛D.脑卒中E.颈部搏动明显减弱或消失正确答案:D5.颅内动脉易损斑块在HR-VWI扫描特征表现()A.T1高信号B.T1等信号C.T1低信号D.T1增强斑块无明显强化E.T2加权成像正确答案:A卒中相关非运动症状多学科管理专家共识解读1.卒中后认知障碍的治疗目的()A.提高认知水平B.延缓认知障碍的进一步下降C.改善精神行为症状D.提高日常生活能力E.以上均正确正确答案:E2.以下哪项是卒中后患者可能出现的常见精神症状?A.头痛B.咳嗽C.情绪低落D.视力模糊E.肌肉疼痛正确答案:C3.卒中后认知障碍发病相关危险因素中,以下说法错误的是()A.PSCI发生的主要原因是脑小血管病,其中包括动脉硬化和脑淀粉样血管病B.微梗死和脑白质高信号是与PSCI关联最强的表现,其在疾病发展中具有重要作用C.研究发现梗死灶的位置和数目也会影响PSCI的发展,特定部位和传导束被认为是独立的危险因素D.糖尿病与PSCI的风险增加无关E.在卒中前存在认知损害,或者卒中发生后出现并发症,也可能增加发展为PSCI的风险正确答案:D4.卒中后抑郁在卒中幸存者中的累积发病率为55%A.10%B.15%C.35%D.45%E.55%正确答案:E5.急性卒中患者应在发病()内开始胃肠道内营养需管饲肠内营养支持治疗时,应首先选择鼻胃管或口胃管进行肠内营养支持治疗A.7天B.10天C.12天D.15天E.18天正确答案:A卒中后认知障碍干预的新策略1.以下哪项不是PSCI诊断评估中通常考虑的方面?A.症状表现B.影像学检查C.精神心理状态D.血液生化指标E.认知功能测试正确答案:D2.以下关于PSCI对患者病死率影响的研究,以下哪个说法是正确的?A.PSCI对患者病死率没有显著影响B.PSCI与缺血性卒中、其他类型的卒中以及心血管死亡事件均无关C.PICASSO研究中,共有1240名患者参与,且研究时间不足一年D.PSCI会显著增加患者病死率,并与多种不良事件有关E.PSCI在研究中主要关注的是认知功能的改善,而非病死率正确答案:D3.以下关于脑卒中对认知功能影响的说法,哪一项是正确的?A.脑卒中患者几乎不会经历任何认知功能的下降B.在脑卒中后的六个月内,大多数患者会出现至少一个认知领域的损伤C.脑卒中仅影响患者的记忆功能,对其他认知功能无影响D.脑卒中后的认知功能损伤通常在短时间内可以完全恢复E.脑卒中后,只有少数患者会出现全局认知的下降正确答案:B4.关于艾地在治疗神经损伤中的相关指南推荐,以下哪个说法是正确的?A.艾地主要用于治疗严重认知障碍患者B.艾地的每日推荐剂量为60mg,一次口服C.艾地的主要作用不包括激活线粒体功能D.艾地具有较强的抗氧化和清除自由基的作用E.艾地在《中毒迟发型脑病诊断与治疗中国专家共识》中未被提及正确答案:D5.以下哪种药物是能透过血脑屏障的线粒体靶向治疗药物A.伊立替康B.阿托伐他汀C.艾地片D.苯磺酸氨氯地平E.氢氯噻嗪正确答案:C卒中恢复期管理的进展与实践1.以下哪一项不属于对患者及早采取综合的干预措施来提高卒中患者康复管理质量?A.预防B.药物治疗C.精神行为症状治疗D.康复治疗E.立即进行手术治疗正确答案:E2.在急性卒中事件发生后的住院期间对患者进行早期的认知筛查,其后每()进行神经心理评估随访;必要时进行多维度的全套认知功能评估,以明确PSCI的发生及演变A.1周B.1个月C.3个月D.6个月E.1年正确答案:C3.下列关于卒中恢复期的时间划分正确的说法是()A.只有国外有明确是时间划分来界定卒中的恢复期,一般来说为24-48hB.国内外有明确是时间划分来界定卒中的恢复期,一般来说为1年以上C.目前国内外尚没有一个明确的时间划分来界定卒中的恢复期D.国内外没有明确是时间划分来界定卒中的恢复期,一般来说为1年以上E.卒中恢复期的时间划分为整一年正确答案:C4.关于MMSE和MoCA量表在认知功能障碍筛查中的应用,以下哪项描述是正确的?A.MMSE量表主要用于识别轻度认知障碍,而MoCA量表则主要用于痴呆症的诊断B.MMSE量表简单易行,适用于大规模筛查,但无法评估执行功能C.MoCA量表对于文盲和低教育水平的老人具有良好的适用性D.MoCA量表在识别MCI和痴呆方面的敏感性和特异性均较低E.推荐单独使用MMSE或MoCA量表进行认知功能障碍筛查B5.流行病学调查显示:我国不同程度的卒中后认知障碍(PSCI)患病率高达()A.50.97%B.60.97%C.70.97%D.80.97%E.90.97%正确答案:D中国缺血性卒中和短暂性脑缺血发作二级预防指南2022解读1.CISS分型不包括()A.穿支动脉病B.心源性C.大动脉粥样硬化型D.其他原因E.隐源性卒中正确答案:E2.最新脑血管病流行病学权威数据显示,中国每年有多少新发卒中患者()A.100万B.240万C.360万D.480万E.600万正确答案:B3.关于卒中,以下错误的是()A.睡眠呼吸暂停会增加卒中、死亡和心血管疾病(如心脏病、高血压和心房颤动)的风险B.呼吸暂停-低通气指数通常用于评价睡眠呼吸暂停的程度C.高同型半胱氨酸症与卒中及其他血管性疾病的发生风险增高有关D.缺血性卒中或TIA患者膳食种类应单一精细化E.缺血性卒中或TIA患者应注意低盐低脂的均衡膳食正确答案:D4.心源性栓塞最常见的危险因素()A.心房颤动B.高血压C.心室颤动D.心房扑动E.高胆固醇血症正确答案:A5.对发病在24h内、非心源性轻型缺血性卒中(NIHSS评分≤3分)或高风险TIA(ABCD2评分≥4分)患者推荐()A.如无药物禁忌,推荐给予氯吡格雷(75mg)联合阿司匹林(75-100mg)双联抗血小板治疗21d(首次剂量给予氯吡格雷负荷剂量300mg和阿司匹林75-300mg),后改为单药抗血小板治疗B.如无药物禁忌,推荐给予氯吡格雷(75mg)联合阿司匹林(75-100mg)双联抗血小板治疗90d(首次剂量给予氯吡格雷负荷剂量300mg和阿司匹林75-300mg),后改为单药抗血小板治疗C.如无药物禁忌,推荐给予氯吡格雷(75mg)联合阿司匹林(75-100mg)双联抗血小板治疗100d(首次剂量给予氯吡格雷负荷剂量300mg和阿司匹林75-300mg),后改为单药抗血小板治疗D.如无药物禁忌,推荐给予氯吡格雷(75mg)联合阿司匹林(75-100mg)双联抗血小板治疗24d(首次剂量给予氯吡格雷负荷剂量300mg和阿司匹林75-300mg),后改为单药抗血小板治疗E.如无药物禁忌,推荐给予氯吡格雷(75mg)联合阿司匹林(75-100mg)双联抗血小板治疗30d(首次剂量给予氯吡格雷负荷剂量300mg和阿司匹林75-300mg),后改为单药抗血小板治疗正确答案:A急性缺血性卒中的精准抗血小板治疗1.阿司匹林抑制血小板聚集的机制是()A.抑制血小板环氧化酶(COX-1)B.抑制血小板环氧化酶(COX-2)C.P2Y12抑制剂D.非肽类血小板糖蛋白IIb/IIIa拮抗剂E.抑制血小板生成正确答案:A2.对于轻型AIS及高危TIA患者,在发病24h内启动双联抗血小板治疗阿司匹林100mg/d,联合氯吡格雷75mg/d(首日负荷剂量为300mg),并持续()A.14天B.21天C.24天D.60天E.90天正确答案:B3.属于P2Y12抑制剂是()A.阿司匹林B.西洛他唑C.氯吡格雷D.替罗非班E.阿法韦林正确答案:C4.阿司匹林抵抗的诊断标准是()A.即用15μmol/LADP作诱导剂,其平均血小板聚集率≥60%,以及用0.5mg/mLAA作为诱导剂,其平均血小板聚集率≥20%B.即用10μmol/LADP作诱导剂,其平均血小板聚集率≥70%,以及用0.5mg/mLAA作为诱导剂,其平均血小板聚集率≥20%C.即用10μmol/LADP作诱导剂,其平均血小板聚集率≥70%,以及用0.5mg/mLAA作为诱导剂,其平均血小板聚集率≥30%D.即用10μmol/LADP作诱导剂,其平均血小板聚集率≥70%,以及用0.5mg/mLAA作为诱导剂,其平均血小板聚集率≥15%E.即用10μmol/LADP作诱导剂,其平均血小板聚集率≥60%,以及用0.5mg/mLAA作为诱导剂,其平均血小板聚集率≥20%正确答案:B5.中国人群中存在的2种导致CYP2C19酶缺陷的主要等位基因()A.CYP2C19*2B.CYP2C19*6C.CYP2C19*9D.CYP2C19*4E.CYP2C19*3正确答案:A房性心脏病-不明原因栓塞性卒中(ESUS)——一个潜在的重要病因1.ESUS的治疗方式包括以下哪种()A.抗血小板治疗B.抗凝治疗C.血管扩张药物治疗D.手术治疗E.雾化吸入治疗正确答案:B2.STAF评分几分提示房颤所致卒中()A.3B.4C.5D.6E.7正确答案:C3.改良STAF新增的变量包括()A.多血管区多发病灶赋值3分B.NT-proBNP≥431pg/ml赋值3分C.年龄>62岁2分D.左心房扩大2分E.单发病灶的赋值标准变更为梗死灶直径大于15mm 正确答案:B4.CRYSTSLAF试验发现40岁以上,皮下埋藏植入式心脏监测器连续监测36月,房颤发现率为()A.0.1B.0.2C.0.3D.0.4E.0.5正确答案:C5.下面不属于房性心脏病的是()A.心腔扩大B.纤维化内皮细胞功能障碍C.肌细胞功能受损D.卵圆孔未闭E.高血糖引起的视网膜损伤正确答案:D急性缺血性脑卒中的影像学评估1.下列脑卒中的危险因素中属于不可干预因素的是()A.吸烟B.高血脂C.肥胖D.年龄E.饮食习惯正确答案:D2.有关缺血半暗带,下列说法错误的是()A.缺血半暗带与核心梗死区血供不同B.缺血半暗带指存在功能障碍但尚未死亡,可挽救的脑组织C.及时挽救缺血半暗带是AIS治疗的关键D.可通过CT/MR等多种方式进行评估,如CTP、DWI-PWI不匹配、DWI-FLAIR不匹配等E.缺血半暗带区域一定会导致脑功能障碍A3.下列哪种成像技术可帮助识别急性缺血性脑卒中患者可行的血管重建治疗方法()A.传统CTB.MRIC.CTAD.PETE.X线平片正确答案:C4.下列不属于缺血性卒中TOAST分型的有()A.穿支动脉病B.大动脉粥样硬化C.小动脉闭塞D.心源性栓塞E.原因不明型正确答案:A5.下列急性期脑梗死的CT征象中不包括()A.岛带征B.豆状核模糊征C.黑洞征D.大脑中动脉高密度征E.脑血肿征正确答案:CCAS并发症处理和围手术期管理1.为预防脑高灌注综合征,以下那一条措施不合适()A.急性脑梗死症状稳定者2周后手术B.双侧严重狭窄,先行狭窄严重侧,3月后再行对侧C.严重的颈动脉狭窄并脑血流量显著降低者,分期血管成形术D.对于血压难以控制者可加用镇静药物,使血压平稳下降E.急性脑梗死立即手术正确答案:E2.以下哪一项不是CAS并发症()A.心率、血压下降B.TIA和脑栓塞C.高灌注综合征(CHS)D.支架内血栓形成E.颅内感染正确答案:E3.术后并发症是CAS中一大难题,在低血压、心动过缓、心律失常等不良情况下,以下哪个措施是CAS患者急诊医生常用的围手术期管理方法()A.严格静脉注射药物,过早地恢复患者自主呼吸B.常规监测降压药物的浓度和剂量,及时调整药物用量C.给予大剂量的利尿剂,加强尿液排泄量的监测D.维持足够的液体输入,避免低钠低血容量的情况发生E.施行机械通气和有创性呼吸治疗正确答案:B4.假性动脉瘤处理中哪一项不合适()A.加压包扎B.超声引导下瘤体内注射凝血酶C.覆膜支架D.弹簧圈栓塞或外科治疗E.穿刺引流正确答案:E5.诊断脑高灌注综合征的必要条件是()A.术中或术后MCA平均血流速度比术前增加100%以上B.术中或术后MCA平均血流速度比术前增加50%以上C.血压升高D.意识障碍E.癫痫正确答案:A急诊取栓困难路径建立——经桡动脉穿刺1.常见的与桡动脉穿刺相关并发症不包括()A.桡动脉痉挛B.闭塞和前臂血肿C.假性动脉瘤D.动静脉瘘E.前臂坏死正确答案:E2.桡动脉穿刺方法()A.钢针B.套管针C.两者皆可D.其他正确答案:C3.中国女性桡动脉的粗细()A.2.60±0.99mmB.2.70±0.99mmC.2.60±1.1mmD.2.70±1.0mmE.2.50±0.99mm正确答案:A4.中国男性桡动脉的粗细()A.2.55±1.1mmB.2.58±1.1mmC.2.60±0.99mmD.2.55±0.99mmE.2.50±0.99mm正确答案:B5.经桡动脉穿刺是取栓手术中常用的途径之一以下哪种情况不适宜经桡动脉穿刺进行取栓手术()A.股动脉无法穿刺B.患者有过敏体质C.患者无法配合手术D.手臂有畸形或者其他障碍影响穿刺E.经桡动脉穿刺均可适用于进行取栓手术正确答案:E卒中后癫痫的诊断和治疗1.下列哪种类型的癫痫发作会引起抽搐()A.部分性发作B.大发作C.强直-阵挛发作D.空白发作E.癫痫性痴呆发作正确答案:C2.下面关于卒中后癫痫(PSE)早期发作正确的是()A.卒中后1天内发生的痫性发作B.卒中后1周内发生的痫性发作C.卒中后1月内发生的痫性发作D.卒中后1季度内发生的痫性发作E.卒中后1年内发生的痫性发作正确答案:B3.下面关于卒中后癫痫不正确的是()A.卒中后一定时间内出现的癫痫发作B.卒中前有癫痫病史C.卒中前无癫痫病史D.排除脑部及全身系统性疾病E.脑电监测到痫性放电与卒中病变部位一致正确答案:B4.下面关于卒中后癫痫用药疗程正确的是()A.1月B.3月C.半年D.1年E.至少2年正确答案:E5.下面关于PSE预后错误的是()A.导致卒中患者致残率和病死率明显增加B.伴有PSE的青年卒中患者肢体功能恢复较差C.但与其他症状性癫痫相比,PSE药物难治率相对较高D.总体预后较好E.可能会出现认知和情绪障碍,影响生活质量正确答案:C急性缺血性卒中的静脉溶栓治疗进展1.根据指南,对于发病4.5-9小时的AIS患者,如果什么条件下推荐阿替普酶静脉溶栓治疗()A.患者年龄大于80岁B.存在核心灌注不匹配C.患者有轻型致残性卒中D.患者合并多发病和虚弱E.患者年龄大于90岁正确答案:B2.什么情况下不适宜使用静脉溶栓治疗急性缺血性卒中()A.血压高于185/110mmHgB.伴有颅内出血的影像学表现C.近期出血性疾病史D.服用口服抗凝药物治疗的患者E.没有该情况,静脉溶栓治疗可以被广泛使用正确答案:D3.卒中终生风险的国家是哪个()A.美国B.印度C.中国D.巴西E.俄罗斯正确答案:C4.根据指南,对于发病6小时内的AIS患者,如果不适合阿替普酶静脉溶栓治疗,可以考虑使用哪种药物()A.尿激酶B.替奈普酶C.阿司匹林D.去氨普酶E.重组组织型纤维蛋白溶解酶原激活剂(rt-PA)正确答案:A5.根据《中国脑血管病临床管理指南(第二版)》,以下哪个时间段内发病的急性缺血性卒中患者应进行阿替普酶静脉溶栓治疗()A.发病0-1小时内B.发病1-3小时内C.发病3-4小时内D.发病4-4.5小时内E.发病4.5-9小时内正确答案:B缺血性脑卒中的脑保护治疗1.我国脑卒中不具有以下特点()A.高发病率B.高死亡率C.高致残率D.低救治率E.低卒中复发率正确答案:D2.关于缺血性脑卒中脑保护药物描述错误的有()A.存在基础-临床转化困难B.新药应针对缺血级联反应的多个环节C.得到了各国指南的普遍推荐D.可考虑将既往尝试失败的药物作为溶栓或取栓的辅助治疗E.脑保护药物的临床应用往往需要进行大规模的多中心随机对照试验,以确证其疗效和安全性,并确认相应的适应证和有效剂量等问题正确答案:C3.脑核心梗死区描述错误的是()A.CBF<10ml/100g/minB.扩散异常C.可逆性缺血D.细胞毒性水肿E.梗死区细胞坏死后,可通过再灌注进行部分修复正确答案:B4.脑卒中最常见的类型是()A.急性缺血性脑卒中B.出血性脑卒中C.短暂性脑缺血发作D.硬膜下血肿E.暴发性脑血管病毒感染正确答案:A5.下列哪项措施不是缺血性脑卒中的脑保护治疗常用措施()A.气管插管B.血糖控制C.调整D.营养支持E.血压控制正确答案:A针刺治疗缺血性卒中研究进展1.针刺治疗缺血性卒中的作用机制不包括()A.可促进神经元增殖分化B.可有效改善脑缺血再灌注损伤C.可增加CBF,缓解卒中后血脑屏障功能障碍D.可抑制细胞凋亡和自噬的机制E.预处理可诱导小胶质细胞活化正确答案:E2.所有卒中发病病例中,缺血性卒中占比()A.50-60%B.60-70%C.70-80%D.40-50%E.30-40%正确答案:B3.下面关于全球卒中负担错误的是()A.全球估计共有1220万卒中发病病例B.卒中患病人数估计共有1.01亿C.卒中导致655万人死亡D.所有卒中发病病例中,有90%为缺血性卒中E.缺血性卒中比例在老年人中更高正确答案:D4.针刺治疗缺血性卒中的研究现状表明,下列哪个方面是针刺的主要作用机制()A.促进血管生成和神经再生B.降低血压和血脂C.抑制炎症反应和氧化应激D.增强免疫功能和体液循环E.促进胶质细胞代谢和脑组织重建正确答案:C5.针刺治疗早期缺血性卒中的安全性与有效性研究表明,下列哪种方法是最适合的针刺方式()A.激光针刺B.穴位贴压C.电针刺D.指压按摩E.经皮穿刺正确答案:E急性缺血性卒中的抗凝策略与挑战1.在抗凝治疗中,如何避免出血风险()A.尽可能使用更高效且口服便捷的抗凝药物B.在开始治疗前对患者的出血风险做评估C.提高药物剂量以取得更好的抗凝效果D.避免与其他药物同时使用以避免不良互作用E.口服抗凝药物开始治疗后,可忽略继续监测正确答案:B2.房颤患者的死亡风险是无房颤患者的()倍A.1B.1.6C.2D.3E.4正确答案:B3.抗凝治疗出血可纠正危险因素()A.年龄>65岁B.既往大出血史C.严重肾功能不全(透析或肾移植)D.严重肝功能不全(肝硬化)E.血小板计数减低正确答案:E4.下面关于CHA2DS2-VASc-60评分错误的是()A.高血压1分B.充血性心衰1分C.卒中2分D.年龄>75岁2分E.女性1分正确答案:D5.目前国际上多采用原则实施房颤相关卒中的抗凝治疗,下面正确的是()A.TIA患者,发病第3d启动抗凝治疗B.轻型卒中患者(NIHSS<6分),发病第3d启动抗凝治疗C.对于中度卒中患者(NIHSS8-15分),发病第6d如无出血转化则启动抗凝治疗D.对于重度卒中(NIHSS>20分),发病第12d除外出血转化后启动抗凝治疗E.对于中度卒中患者(NIHSS8-15分),发病第3d如无出血转化则启动抗凝治疗。
后循环缺血的研究新进展

后循环缺血的研究新进展摘要】缺血性脑卒中中约有20%是由后循环缺血引起的。
近年来,随着神经影像技术和相关临床研究的发展,人们对后循环缺血有了更深入的了解。
本文对后循环缺血的病因、发病机制、临床表现、诊断、治疗和预后等方面的进展作一综述;尤其其治疗方面的共识及循证证据,借以提高医生对后循环缺血的再认识。
【关键词】后循环缺血溶栓神经保护介入治疗【中图分类号】R743 【文献标识码】A 【文章编号】2095-1752(2012)14-0094-02后循环指椎-基底动脉系统,由椎动脉、基底动脉及分支组成,主要供血区域为脑干、小脑、丘脑、枕叶、部分颞叶、上段脊髓;椎-基底动脉系统狭窄或闭塞、血流动力学异常、栓子等常引起后循环缺血(PCI)。
1 认识及定义[1-2]1.1 认识19世纪50年代至20世纪70年代,因沿用“颈内动脉供血不足”且由于对PCI认识的滞后,“椎基底动脉供血不足”(VBI)概念被广泛使用,并产生一些错误认识。
随着神经影像学和相关临床研究的进展,80年代以后,形成了如下认识: ①动脉粥样硬化(AS)是主要病因,颈椎骨质增生仅是罕见的情况; ②栓塞是最主要机制; ③无论是临床或影像学检查都无法可靠地界定既非正常又非缺血的状态; ④虽然头晕/眩晕是常见症状,但此症状的最常见病因却是良性发作性位置性眩晕(BPPV)。
基于以上认识,90年代,PCI代替了VBI。
1.2 定义是指椎基底动脉系统缺血引起的各种一过性或持续性的症状,根据缺血的程度及持续时间可分为TIA和IS。
鉴于DWI发现约50%的后循环TIA有明确的梗死改变致使TIA 与IS的界限越来越模糊。
2 发病机制和危险因素2.1 发病机制[1,2,3]PCI主要原因是AS;最常见的动脉病变在椎动脉颈部及颅内部分,不在基底动脉内;最常见机制是栓塞,其次依次为大动脉粥样硬化、小穿支动脉疾病和动脉夹层,偏头痛、血管肌纤维发育不良、高凝状态、药物滥用等相对少见。
缺血性脑卒中的病理机制及药物研究进展

缺血性脑卒中的病理机制及药物研究进展作者:马聪吉杜晓华杨为民来源:《医学信息》2014年第11期摘要:缺血性脑卒中已成为威胁人类健康的重要因素,其发病率和死亡率呈不断上升趋势,因此探讨缺血性脑卒中的发病机理及其药物研究,对于预防和治疗该病具有重要的意义。
本文阐述了缺血性脑卒中的发病机制及治疗药物的研究进展。
关键词:缺血性脑卒中;发病机制;治疗药物脑卒中,又称中风,是一种突然起病的脑血液循环障碍性疾病。
脑卒中分为缺血性脑卒中和出血性脑卒中,其中缺血性脑卒中大约占所有脑卒中的80%,是指局部脑组织区域血液供应障碍,导致脑组织缺血缺氧而发生病变坏死。
缺血性脑卒中具有高发病率、高致残率和高死亡率的特点,已成为人类健康的一大威胁。
通过研究缺血性脑卒中的发病机制,可为临床预防和治疗该病提供有效的理论依据。
1病理机制缺血性脑卒中发生后,由于大脑血流供应中断,引起能量代谢障碍和兴奋性神经递质的释放。
能量代谢障碍①诱导诱导氧自由基的产生和线粒体功能损伤,从而导致细胞膜的完整性遭到破坏;②则导致离子泵功能障碍,使大量的Ca2+、Na+等离子内流,诱导了大量酶及炎症因子的产生,导致DNA断裂和细胞骨架的破坏。
大量的兴奋性神经递质丛神经轴突末端释放后与相应的受体作用而产生兴奋性毒性。
能量耗竭、Ca2+内流、兴奋性毒性以及炎症反应等机制共同导致了细胞凋亡。
1.1能量耗竭和酸中毒脑组织在缺血、缺氧状态下,细胞的能量代谢转为无氧酵解,使细胞出现能量耗竭。
无氧酵解引起脑组织缺血性乳酸酸中毒,细胞Na+-K+泵功能损伤,K+大量外溢,同时Na+、Cl-及Ca2+大量流人细胞内引起细胞损伤;缺血区乳酸堆积还可引起神经胶质和内皮细胞的水肿和坏死,加重缺血性损害。
1.2细胞内Ca2+超载细胞Ca2+超载可通过下述机制导致细胞死亡:①大量Ca2+沉积于线粒体,干扰氧化磷酸化,使能量产生障碍;②细胞内Ca2+依赖性酶类过度激活可使神经细胞骨架破坏;③激活磷脂酶,使膜磷脂降解,?訩通过生成大量自由基加重细胞损害;?訪可激活血小板,促进微血栓形成,在缺血区增加梗死范围;④脑缺血时,脑血管平滑肌和内皮细胞均有明显的Ca2+超载。
急性缺血性脑卒中发病机制研究

急性缺血性脑卒中发病机制研究急性缺血性脑卒中(ischemic stroke)是指脑动脉或脑血管突然发生血液供应不足的情况,导致脑部局部缺氧及神经功能障碍。
这种病因复杂,病变过程迅速,给患者的生命和健康带来严重威胁。
为了有效预防和治疗急性缺血性脑卒中,科学家们对其发病机制进行了深入研究。
1. 动脉病变:大脑动脉粥样硬化是最常见的引发急性缺血性脑卒中的病理基础。
随着年龄的增长,血管壁不断受到各种损伤因素的影响,如高血压、高血糖、高血脂等,血管壁内膜逐渐增厚、钙化,形成斑块。
斑块破裂或者形成血栓,堵塞脑血管,导致脑部缺血。
2. 血栓形成:急性缺血性脑卒中的另一个重要机制是血栓形成。
血液中的凝血系统平衡异常会引起血栓的形成。
局部缺血状态下,受损的内皮细胞会释放一系列活性物质,诱导血小板聚集和激活,促进血栓的形成。
3. 溶栓系统紊乱:溶栓系统是人体一个重要的保护机制,能溶解血管内生成的血栓。
肾上腺素激素和其他应激物质会抑制溶栓系统的正常功能。
在急性缺血性脑卒中中,患者可能伴随应激状态,促使溶栓系统受到抑制,而造成血栓无法迅速溶解,进而加重缺血程度。
4. 炎症反应:炎症过程也参与了急性缺血性脑卒中的发病机制。
血液循环不足引起的缺氧及损伤会导致炎症反应的发生。
炎症细胞释放的细胞因子会进一步加重血管损伤、促进斑块破裂和血栓形成。
急性缺血性脑卒中的发病机制是多种因素共同作用的结果。
动脉病变、血栓形成、溶栓系统紊乱和炎症反应都是导致脑部缺血性损伤的重要原因。
进一步的研究可以帮助我们更好地理解这些机制,从而为急性缺血性脑卒中的防治提供更有效的策略。
中国急性缺血性脑卒中诊治指南急救与脑血管再灌注治疗的新进展

中国急性缺血性脑卒中诊治指南急救与脑血管再灌注治疗的新进展脑卒中是一种常见且严重的疾病,严重威胁着人们的生命和健康。
据统计,全球每年有超过1300万人因脑卒中而死亡或残疾。
而其中,急性缺血性脑卒中占据了脑卒中的绝大多数,因此,对于急性缺血性脑卒中的诊治具有重要的意义。
为了指导临床诊治工作,中国脑卒中学会制定了《中国急性缺血性脑卒中诊治指南》,该指南基于大量的临床研究和专家经验,着眼于急性缺血性脑卒中的早期诊断、治疗和护理,为医生提供了规范化操作的指导。
首先,对于急性缺血性脑卒中的急救措施,《指南》明确了急救人员在到达现场后的首要任务是迅速评估患者的意识和生命体征。
如果患者陷入昏迷状态,要保持气道通畅,并尽快实施静脉溶栓治疗。
对于意识清醒的患者,及时进行血压监测和控制,并尽快转运到具备脑卒中治疗条件的医院。
其次,诊断是治疗的基础。
《指南》指出,急性缺血性脑卒中的诊断主要依据临床表现和影像学检查。
临床上,患者常常出现突发性的头痛、意识障碍、肢体活动不协调等症状。
而在影像学检查上,脑血管造影和颅脑CT等技术则有助于明确脑卒中的类型和病因。
针对急性缺血性脑卒中的治疗,《指南》提出了脑血管再灌注治疗的新进展。
脑血管再灌注治疗是通过介入手术等方法,恢复患者脑血流灌注和神经功能,以减少脑卒中后遗症的目的。
该治疗方法在近年来取得了显著的进展,已成为急性缺血性脑卒中治疗的重要手段之一。
在脑血管再灌注治疗中,机械取栓和溶栓是常见的方法。
机械取栓是指通过血管内介入手术,将栓塞物从血管中取出,恢复血流通畅。
而溶栓则是通过静脉注射溶栓药物,使栓子溶解,恢复血流。
这两种方法各有优劣,医生需要根据患者的具体情况进行选择。
此外,《指南》还强调了抗血小板治疗的重要性。
抗血小板药物可以减少血小板的聚集,防止血栓形成,从而降低再次发生脑卒中的风险。
常用的抗血小板药物有阿司匹林、氯吡格雷等,医生可以根据患者的具体情况进行选择。
在日常护理中,及时评估和处理并发症是非常重要的。
医学文档急性缺血性脑卒中

提要一、概述二、病因与发病机制三、临床表现与诊断四、治疗进展五、问题与展望一、概述1概念缺血性脑卒中(cerebral ischemic stroke)又称脑梗死(cerebral infarction),是各种原因导致脑动脉血流中断,局部脑组织发生缺血缺氧性坏死,而出现相应的神经功能缺陷。
2脑血管病的分类1995年10月第四届全国脑血管病学术会议将脑血管疾病分为十类:无症状性脑血管病,短暂性脑缺血发作,脑卒中,脑血管性痴呆,高血压脑病,颅内动脉瘤,颅内血管畸形,脑动脉炎,其他动脉疾病和颅内静脉病,静脉窦及脑部静脉血栓形成。
3中西病名的界定:脑卒中又称脑血管病,中医称为中风;由于该病起病急骤、变化迅速、症状多样,就象自然界变幻莫测的风,故取类比象曰中风; 中风临床四大症突然昏仆、不省人事;半身不遂;口眼歪斜;言语不清或失语; 中风是目前危害人类健康的常见病和多发病,具有四高一低的特点。
4流行病学;我国现有脑卒中患者约500万人,每年新发病例高达150万人,其致残率约70%,而重度致残者占40%以上。
美国每年由该病所造成的直接经济损失可达500亿美元。
脑卒中现已成为使老年人致残的三大疾病之一二.病因与发病机制病因1.血管壁的病变 : 动脉粥样硬化和高血压引起的脑小动脉硬化,及在此基础上发生的血栓形成是脑梗塞常见原因。
其他如先天性动脉瘤、脑血管畸形及各种原因引起的动脉炎、颅静脉病变等也可引起脑梗死。
动脉内膜损伤(包括动脉营养血管)破裂后,胆固醇沉积于内膜下层,引起血管壁脂肪透明变性,进一步使纤维组织增生,管壁增厚,动脉弹性回缩力降低,血小板及血液中其他有形成分、纤维素等附着于受损粗糙的内膜上,易于造成动脉管壁的血栓形成。
在一些诱发因素的影响下,若有局部或全身血压降低,血液灌流减少,血流缓慢,血液粘度增加,血管痉挛等,使血栓逐渐扩大,最终使动脉完全闭塞。
脑动脉硬化好发于颅底大血管、大脑中动脉及其分支、基底动脉及颈动脉的颈内、外动脉分叉处。
《2024年MHR、NPAR、SII对进展性缺血性脑卒中发生预测价值的研究》范文

《MHR、NPAR、SII对进展性缺血性脑卒中发生预测价值的研究》篇一一、引言进展性缺血性脑卒中是一种常见的神经系统疾病,其发病机制复杂,病程进展迅速,给患者带来极大的健康风险。
因此,准确预测其发生对于制定有效的治疗方案和改善患者预后具有重要意义。
近年来,随着医学研究的深入,MHR(Mean Hematocrit Ratio,平均血细胞比容比)、NPAR(Neurological Parameters of Acute Stroke,急性卒中神经学参数)和SII(Systemic Immune-Inflammation Index,全身免疫炎症指数)等指标在缺血性脑卒中的预测和评估中逐渐受到关注。
本文旨在探讨MHR、NPAR、SII 对进展性缺血性脑卒中发生的预测价值,以期为临床诊断和治疗提供参考依据。
二、研究方法本研究采用回顾性分析方法,收集了某医院近三年内收治的缺血性脑卒中患者的临床数据。
纳入标准为年龄≥18岁、诊断为缺血性脑卒中、有完整的MHR、NPAR和SII等指标数据。
排除标准为患有其他严重神经系统疾病或恶性肿瘤等影响研究结果的患者。
根据患者病情发展情况,将患者分为进展性缺血性脑卒中组和非进展性组。
采用统计学方法比较两组患者在MHR、NPAR、SII等指标上的差异,并分析这些指标对进展性缺血性脑卒中发生的预测价值。
三、研究结果1. 患者基本情况本研究共纳入200例缺血性脑卒中患者,其中进展性组80例,非进展性组120例。
患者年龄、性别、基础疾病等方面无明显差异。
2. MHR与进展性缺血性脑卒中的关系研究结果显示,进展性组患者的MHR值明显高于非进展性组(P<0.05)。
进一步分析表明,MHR值越高,患者发生进展性缺血性脑卒中的风险越大。
3. NPAR与进展性缺血性脑卒中的关系NPAR指标在进展性组和非进展性组之间也存在显著差异(P<0.05)。
与MHR类似,NPAR值越高,患者发生进展性缺血性脑卒中的风险也越大。
缺血性脑卒中的发病机制及其临床治疗方法

探究缺血性脑卒中的发病机制及其临床治疗方法摘要:目的:探究缺血性脑卒中的发病机制及其临床治疗方法。
方法:选取76例缺血性脑卒中患者作为治疗组,采用抗凝治疗、神经细胞保护及溶栓治疗,选择69例正常人作为对照组,对比治疗前后患者的凝血酶原时间(pt)、活化部分凝血时间(aptt)、纤维蛋白原(fib)和血浆凝血酶时间(tt)及脑卒中神经功能缺损评分。
结果:治疗前两组各项凝血指标和脑卒中神经功能缺损评分具有差异性,p0.05,差异无统计学意义,p<0.05,差异具有统计学意义。
2结果治疗前治疗组患者与对照组正常人神经功能评分、凝血酶原时间(pt)、活化部分凝血时间(aptt)、纤维蛋白原(fib)及血浆凝血酶时间(tt)组间比较p<0.05,差异具有统计学意义;治疗组治疗后与治疗前相比神经功能评分、凝血酶原时间(pt)、活化部分凝血时间(aptt)、纤维蛋白原(fib)及血浆凝血酶时间(tt)等评价指标均得到显著改善,p<0.05,差异具有统计学意义。
通过研究报道看出,大多数重症加强治疗病房(icu)中,假丝酵母菌中的白假丝酵母菌为最常见的致病菌,在大多数的icu中,临床分离的治病真菌中占大约60%左右。
通过我们的研究发现,本院icu发生下呼吸道真菌感染的32例患者中,致病真菌主要以白色念珠菌和热带假丝酵母菌为主,分别为11株和9株,合计占到了下呼吸道真菌感染的62.4%。
这些结果证明了本院icu病房造成真菌感染的致病菌与大部分临床报道的结果相一致,白色念珠菌仍然是占有重要地位的致病菌株。
通过学者的研究表明真菌感染主要以下呼吸道真菌感染为主,其感染部位大都在下呼吸道,约占全部真菌感染的50%以上。
真菌感染的症状并不是很明显,易与其他疾病症状造成混淆,并且其临床表现形式也是多种多样,会给诊断造成极大的困难。
所以,很多医院对其早期的治疗无从下手,使得目前多数医院的真菌感染病例数居高不下。
多肽类药物防治缺血性脑卒中的研究进展

多肽类药物防治缺血性脑卒中的研究进展【摘要】静脉溶栓治疗是临床治疗缺血性脑卒中最经常使用的方式,但因为医治时间较短,其临床医学运用受明显限制。
除此之外,再灌注还会进一步造成神经细胞损害,其病理学全过程和发病机制繁杂,包含氧化损伤、发炎、细胞凋亡、兴奋性毒性等繁杂机制。
近些年,越来越多研究表明,多肽类药对缺血性脑卒中(IS)具备较好的防治和疗效。
依据其作用机制,本文综述了Hsp27仿真模拟肽、NBD多肽、扇贝多肽、神经肽Humanin(HN)等多肽药品预防IS的研究成果。
为进一步科学研究多肽药品预防缺血性脑卒中给予参照,为其临床医学开发设计运用给予新的基本思路。
【关键词】缺血性脑卒中;多肽类药物;炎症;氧化损伤;凋亡缺血性脑卒中广义上来讲是指血液供应不足,难以满足脑组织代谢需求,从而产生一系列症状的综合征。
严重的脑缺血会造成脑功能不可逆的损伤,甚至是死亡。
缺血性脑卒中实际上是一个比较宽泛的名词,医学上缺血性脑卒中的相关疾病包括且不限于短暂性脑缺血发作(TIA)、缺血性卒中(脑梗死)、慢性脑供血不足等等。
缺血性脑卒中具备高复发性和高死亡率的特性,其防治的任务紧急而严峻。
近期的科学研究加强了多肽类药物和缺血性中风相互关系。
从天然提取到人工合成,多肽作为一种新药,在临床应用和生产制备方式上均显示出独特的优越性。
在临床上,多肽药物与重组蛋白药物、单抗药物类似,具有特异性强、疗效好等优势;在生产制备方式上,多肽药物接近小分子化合物,具有纯度高、质量可控且结构容易确定等特点,所以目前多肽治疗已被认为是具有高选择性、有效且相对安全的潜在疗法[1]。
近年来,全球多肽药物市场快速发展,多肽药物也成为国内外新药研发的重要方向。
大型跨国制药企业,均通过收购或并购形式加大了对多肽药物研发的投入,并相继收获了不少上市药物。
相比发达国家和地区,我国多肽药物领域仍然存在较大差距,产品结构也有差异,其中免疫增强类产品居多,而针对肿瘤、糖尿病、罕见病等的产品占比较小,市场还未成熟,也远未饱和。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
缺血性脑卒中发病机制研究新进展大连市中心医院神经外科(116023)陈东综述大连医科大学附属二院神经外科(116023)赵贵德审校大连市中心医院神经外科(116023)考宏盛审校摘要:短暂或持久的局灶性脑缺血可引起一系列病理生理变化导致脑损害,且随时间和缺血程度增加而加重。
本文就中枢神经系统中兴奋性氨基酸(谷氨酸)三种受体:NMDA、AMPA、metabotropic(促代谢)受体,神经元及神经胶质细胞的去极复极化,缺血后的炎症反应及细胞凋亡几方面在缺血性脑卒中中的作用机制作一综述。
关键词:脑卒中;脑梗塞;发病机制缺血性脑卒中是由于脑主要动脉的血流短暂或持久减少引起。
大部分病例中血流减少是由于脑动脉被栓子堵塞或血栓形成所致。
工业化国家每10万人中有250~400人发生脑缺血,死亡率大约30%,仍为第三大死因。
脑缺血病理生理学方面研究的突破,使人们认识了脑缺血早期症状。
在这些努力下,对脑缺血临床治疗取得成功。
而与此同时,大量药物治疗却不能令人满意,说明脑缺血病人神经保护治疗并不象在动物实验中那样简单、明确。
1能量衰竭和兴奋性氨基酸细胞毒性作用脑组织对氧和葡萄糖的代谢需求远较其他组织为高,而且几乎只依赖氧化磷酸化过程来产能。
脑局灶血流减少使物质转化(尤其是氧和葡萄糖)发生障碍。
使维持离子梯度所必需的能量生成障碍[1]。
因为能量缺乏,膜电位消失,神经元和神经胶质细胞去极化,并有谷氨酸从突触前神经末梢释放,胶质细胞和神经元对神经递质的再摄取一般均需耗能,结果兴奋性氨基酸被突触前膜再摄取受阻,导致细胞间隙谷氨酸累积,NMDA受体和metabotropic谷氨酸受体激活导致钙超载,通过PLC (磷脂酶C)和Ins(1、4、5)P3传递信号。
这样Na+、Cl-通过一价离子通道进入神经元内,Na+、Cl-内流较K+外流多,因此水被动流入导致细胞水肿。
水肿可影响梗塞灶周边区域的灌注,同时使颅内压增高、脑血管受压。
卒中后数小时内病人C T和MRI的脑水肿影像对估计预后有重要意义。
线粒体:自由基引发的线粒体膜脂质过氧化反应(LPO)或细胞内形成的脂质过氧化物作用于线粒体膜,使膜的流动性及液态改变,从而导致线粒体功能障碍。
高能磷酸化物质产生减少,自由基产生增多,线粒体肿胀,嵴断裂,细胞色素C释放,导致细胞凋亡。
血液动力学、能量代谢、离子变化对缺血范围的影响并不一致,在灌注缺损区中心,脑血流量只减少20%[2],而缺氧性去极化却发展非常迅速,脂肪和蛋白质分解、生物能衰竭后的微管解聚和体内离子平衡的破坏使细胞快速死亡[3]。
在这些致命区和正常区之间存在半暗带,此区勉强保持血流以进行新陈代谢[4]。
如果长时间不给予治疗,兴奋性细胞毒作用不断发展或继发性损害过程,如快速的去极化、缺血后炎症反应、细胞凋亡使该区迅速发展成梗塞灶。
因此神经保护的根本目的是挽救半暗带。
虽然有充足的证据证明人类卒中病人存在这个半暗带,但其范围和时间变化规律仍不十分清楚,其范围可能比较小,存在时间也比较短[5]。
2谷氨酸受体:兴奋性氨基酸细胞毒作用途径以上说明谷氨酸受体激活后离子平衡的破坏、收稿日期:2000-03-6;修回日期:2000-12-28作者简介:陈东,1970出生,男,主治医师,硕士,主要从事脑血管病方面的研究。
Foreign Medical Sciences Section on Neurology&Neurosurgery2001,28(1)细胞内钙离子浓度增加,是梗塞灶内细胞死亡的主要原因。
NMDA受体控制Ca2+、Na+、K+通道。
在MC A阻塞而形成的永久或暂时梗塞的动物模型中应用NMDA受体拮抗剂发现有明显的神经保护作用。
但受体拮抗剂的治疗时间窗非常有限,大约在动脉阻塞后1-2小时内。
AMPA(突触后受体)控制Na+、K+通道,AMPA受体或kaina te受体介导Na+内流使膜静息电位去极化间接介导Ca2+内流;在脑局灶缺血梗塞区又发现新的谷氨酸受体metabotropic受体,该受体激活时可促进细胞内结合钙的释放。
晚近研究发现metabotropic受体II和III 亚型激活后具有神经保护作用[6]。
因此拮抗metabotropic受体不但能发挥神经保护作用,同时也产生一些严重的后果如精神病、呼吸抑制、心血管功能障碍。
这大概可以解释某些临床应用谷氨酸受体治疗失败的原因。
因此应用超选择性(针对受体亚型如I型)受体拮抗剂治疗缺血性卒中临床才更安全。
兴奋性氨基酸细胞毒作用可以直接导致急性细胞死亡,也可通过其他途径导致细胞凋亡。
3梗塞灶周边(半暗带)去极化在大鼠和猫的中风模型中发现缺血的神经元和神经胶质细胞由于能量缺乏、K+释放和谷氨酸在细胞外积聚而去极化。
缺血中心区的细胞只去极化而不复极[7]。
而半暗带区的细胞却可复极,但以能量消耗为代价,如果细胞外的K+和谷氨酸增加,这些细胞也只去极化,随着去极化细胞数量的增大,梗塞灶范围也在不断扩大[8]。
NMDA和AMPA 受体拮抗剂的神经保护作用也可以用此机制解释。
但梗塞灶周边(半暗带)去极化至今在人体中仍未被神经电生理仪探测到,因此其对人类脑缺血的病理生理学方面的影响至今仍不十分清楚。
4炎症反应细胞内第二信使系统的激活、氧自由基的增加和缺氧本身均可通过诱导转录因子(核因子-J B[9]、干扰素调节因子1[10]、STAT3[11])的合成来触发大量前炎症介质基因的表达。
这样,受损的脑细胞产生大量血小板激活因子、肿瘤坏死因子A、白介素1B等炎症介质[12]。
结果,诱导内皮细胞表面粘附分子(ICAM-1、P-selectins、E-selectins)表达,粘附分子与中性粒细胞表面的补体受体反应,中性粒细胞与内皮细胞粘附,穿过血管壁进入脑实质,5~7天后巨嗜细胞和单核细胞也到达缺血的脑组织[13]。
化学因子如IL-8、单核细胞化学粘附蛋白1由受损的脑细胞产生并引导血液中的炎性细胞向靶目标迁移。
脑胶质细胞也参与炎症反应,缺血后4~6小时,星形胶质细胞肥大,而小胶质细胞的突起卷缩如阿米巴样。
越来越多的证据表明缺血后的炎症导致缺血性脑损伤。
以下四种措施可以减少缺血性脑损伤:¹诱导系统中性白细胞减少;º粘附分子或其受体被其抗体所封闭;»关键性炎性因子如IL-1被封闭;¼编码干扰素调节因子1的基因缺失。
有很多机制参与缺血后炎症反应导致的缺血性损害:¹中性白细胞浸润所致的微血管阻塞使缺血程度加重;º激活的炎性细胞和受损神经元产生大量调节因子产生严重后果;»浸润的中性粒细胞产生iNOS,生成大量有毒的NO。
¼缺血的神经元表达COX2(一种通过产生超氧化物和前列腺素来调节缺血损害的酶);½缺血的神经元产生肿瘤坏死因子A,加重缺血性损害;¾激活的小胶质细胞也可产生神经毒素如NO、活性氧和前列腺素。
5细胞凋亡过量的谷氨酸受体的激活、钙超载、氧自由基、线粒体和DNA的损害等因素均可导致脑细胞的坏死和凋亡。
这部分取决于刺激的性质和强度、受损细胞的种类及在生命周期中所处的阶段[14]。
急性、持续的血管阻塞引起坏死,而轻微受损尤其在半暗带引起细胞凋亡。
对线虫遗传学研究表明,细胞凋亡的调节必须有ced-3、ced-9和ced-4三个基因的参与。
ced -3基因激活可促进凋亡,而ced-9基因活化则抑制凋亡,二者间作用的平衡由ced-4基因参与调节而起/分子开关0样作用[15]。
在哺乳动物细胞内的白介素1B转化酶(ICE)蛋白酶家族与ced-3基因产物CED-3高度同源,活化该家族蛋白可促进细胞凋亡。
因该家族蛋白酶具有均为半胱氨酸蛋白酶类和特异酶切Asp氨基位点两大特点,故又称为Caspase蛋白酶家族,其中Caspase-3与线虫CED-3极为相似,激活后可导致多种细胞死亡,故又称为/死亡蛋白酶0。
Caspase如何促使缺血细胞死亡的?Bcl-2,cyt-C(细胞色素C)和Cas-pase蛋白酶为细胞凋亡所需的三个效应器。
Bcl-2抑制细胞凋亡,细胞在各种促凋亡刺激作用下释放cyt-C,可激活凋亡所需的Caspase蛋白酶。
运用TUNEL(原位末端标记)法,大鼠大脑中动脉阻塞30分钟再灌注,分别于再灌注后24、48、72、168国外医学神经病学神经外科学分册2001年第28卷第1期小时做梗塞区大脑切片,TUNEL阳性细胞在72小时切片中最多,168小时切片最少[16]。
因为局灶缺血后cyt-C从线粒体膜释放,激活Caspase蛋白酶。
动物实验发现急性心肌梗塞中细胞凋亡发生的同时伴有Bcl-2表达的降低,推测此现象可能与凋亡的发生有关。
Caspase蛋白酶阻滞剂不仅可以缩小局灶缺血组织死亡的面积,也可减轻神经功能损害。
与NMDA受体拮抗剂不同,Caspase蛋白酶阻滞剂在缺血后几小时注射也可发挥明显的效果。
有人在30分钟可逆脑缺血模型[17]后9小时脑室内注射zDE VD1F MK(一种高选择性Caspase-3阻滞剂),细胞保护作用至少可以维持22天。
综上所述,脑局灶缺血损害是一个过程,神经功能障碍随时间延长而加重,缺血边缘的神经元可以存活几小时至几天,呈迟发性损害,如果侧枝循环建立,半暗区的脑神经功能仍可恢复。
以上五种机制可以引起脑局灶缺血区的神经元死亡,是坏死抑或凋亡,由不同因素决定,据此可以选择不同的治疗手段,使缺血性脑卒中的临床治疗更加有效。
参考文献01Martin,R.L,Lloyd HG,Cowan AI.The early events of oxygen and glu-cos e deprivati on:setti ng the scene for neuronal death?Trends Neurosci, 1994;17:251-25702Hoss mann,KA.Viability thresholds and the penumbra of focal ischemia.Ann Neurol,1994;36:557-56503SiesjoBK,Elmer E,J anelidze S,et al.Role and mechani sms of sec ondary mitochondrial failure.Acta Neuroc hir Suppl(Wien),1999;73:7-1304Obrenovitch,TP.The ische mic penumbra:twenty years on Cerebrovasc Brain Metab.Rev.1995;7(4):297-32305Kaufmann AM,Firlik AD,Fukui MB,et al.Ische mic core and penrmbrain human stroke.Stroke,1999;30(1):93-9906Bond A,O.Neill MJ,Hicks CA,et al.Neuroprotecti ve effects of a s ys-te micall y active group II metabotropic glutamate receptor agonis t LY35470in a gerbil model of global i schaemi a.Neuroreport,1998;9: 1191-119307Hoss mann,KA.Periin farct depolariz ati ons.Cerebrovasc.Brain Metab.Rev,1996;8:195-20808M ies G,Iiji ma T,Hoss mann KA.Correlation bet ween peri-infarc t DC s hifts and ischae mic neuronal damage in rat.Neuroreport,1993;4:709 -71109O.Neill LA,Kaltschmidt C.NF-kappa B:a c rucial transcription fac tor for glial and neuronal cell functi on.Trends Neurosci,1997;20(6):252 -25810Iadecola C,Salkowski CA,Zhang F,et al.The transcription factor inter-feron re gul atory fac tor1is express ed after cerebral i schemia and con-tri butes to i schemic brain injury.J Exp Med,1999;189(4):719-727 11Planas AM,Soriano MA,Berruez o M,et al.Inducti on of s tat3,a signal transducer and teanscreption factor in reactive microglia following tran-s ient focal cerebral ischae mia.Eur J Neuros ci,1996;8(12):2612-261812Rothwell NJ,Hopki ns SJ.Cytoki nes and the nervous s ys tem II:Ac tions and mechanis ms of action.Trends Neurosci,1995;18:130-13613Iadecola C.Bright and dark s ides of nitric oxi de i n ischemic brain in-jury.Trends Neurosci,1997;20:132-13914Leist M,Nicotera P.Apoptosi s,excitotoxici ty,and neuropathology.Exp Cell Res,1998;239(2):183-20115Chinnai yan A M,O.Rourke K,Lane BR,et al.Interacti on of CED-4 w i th CED-3and CED-9:a molecular framework for cell death.Sc-i ence,1997;275(5303):1122-112616Dirnagl U,Iadecola C,Mos kowi tz MA.Pathobiol ogy of ischae micstroke: aninte gratedvie w.Trends Neurosci,1999;22(9):391-39717Fi nk K,Zhu J,Na mura S,et al.Prologed therapeutic w i ndo w for is-chemic brain damage caus ed by delayed caspase activati on.J Cereb Blood Flo w Metab,1998;18:1071-1076Foreign Medical Sciences Section on Neurology&Neurosurgery2001,28(1)。