第4章-第五节遗传性代谢缺陷与分子病
分子病与先天性代谢缺陷病

C.密码子插入或缺失:珠蛋白基因序列中插入或缺失1个或数个密 码子,发生读码顺序移动或使珠蛋白肽链增加或减少相应的1个或数个 氨基酸。
D.融合基因:同源染色体的不等交换,导致这些异常的血红蛋白链 由两种不同珠蛋白部分肽链连接而成。
控制珠蛋白肽链的α基因簇在16p13.2,人类二 倍体细胞中共有4个α基因;类β基因簇位于11p1 5.4。
(一) 血红蛋白病
2.异常血红蛋白病 (1)异常血红蛋白病的珠蛋白基因突变类型: A.单个碱基的取代:①错义突变 如(镰型细胞贫血症),系血红蛋
白β链第6位密码子由6(谷氨酸)突变为6(缬氨酸)。②无义突变 如 , 系血红蛋白β链第145位密码子由(酪氨酸)突变为终止密码子。③终止 密码子突变 如 ,由血红蛋白α链的终止密码子突变为(谷氨酰胺), 使α链非正常延长至172个氨基酸才终止。
(一) 血红蛋白病
(2)β 地中海贫血:①重型β 地中海贫血:患者正常β 链缺乏或合 成量很少,血红细胞中无或含量很少,和2含量增高,α 珠蛋白链过剩 而沉积到红细胞膜上,改变了红细胞膜的性能,引发严重的溶血性贫 血。主要临床症状表现为患儿出生几月后溶血反应,并伴有腹泻、发 热、生长发育迟缓、身材矮小、骨髓增生,可出现鼻塌眼肿、上颌前 突、头大额隆等特殊的“地中海贫血面容”。②中间型β 地中海贫血: 临床表现介于轻型和重型之间,中度贫血,脾脏轻或中度肿大,黄疸 可有可无,骨骼改变较轻。③轻型β 地中海贫血:带有一个正常的β 珠蛋白基因,一般无临床症状或有轻微贫血和脾脏肿大。
(四) 膜转运载体蛋白病
肝豆状核变性的遗传又称病,由编码P型铜转运酶的基因(7)突变引起, 基因定位于13q14.3,为常染色体隐性遗传,7基因突变多为错义突变 或无义突变。临床表现以神经系统和肝脏受损为主,晚期表现为痴呆。 患者可通过服期 治疗,因此本病的早期诊断对于临床治疗很有意义。
医学遗传学习题集名词解释

医学遗传学习题集名词解释第一章绪论1 遗传性疾病(genetic disease):是指其发生需要有一定的遗传基础,通过这种遗传基础、并按一定的方式传于后代发育形成的疾病。
2 先天性疾病(congenital disease):一般是指婴儿出生时就已表现出来的疾病。
3 家族性疾病(familial disease):是指一个家族中多个成员都表现出来的同一种病,即某一种疾病有家族史。
第二章人类基因1卫星DNA(satellite DNA):以5bp、10bp或20bp、200bp为一个重复单位,经过多次重复串联,长度可达105bp,约占整个基因组10%~15%的简单序列DNA。
2 基因组(genome):一个物种所有遗传信息的总称。
通常表述为一个单倍体细胞中全部的基因或遗传物质。
3 结构基因(structural gene):是决定合成某一种蛋白质或RNA分子结构相应的一段DNA。
结构基因的功能是把携带的遗传信息转录给mRNA,再以mRNA为模板合成具有特定氨基酸序列的蛋白质或RNA。
4 断裂基因(split gene):是真核生物的结构基因,编码序列往往被非编码序列所分割,呈现断裂状的结构。
5 基因家族(gene family):是一组来源相同、结构相似、功能相关的基因。
它们在基因组中的拷贝只有微小的差别,并行使相关的功能。
6 单一基因(solitary gene):指在基因组中只有单个或极少数拷贝的基因。
7 外显子和内含子(exon and intron):外显子(exon)是指编码氨基酸的序列,内含子(intron)是指位于外显子之间的非编码序列。
8 调节基因(regulator gene):是指能控制结构基因转录起始和产物合成速率并能影响其他基因活性的一类基因。
9 串联重复基因(tandemly repeated gene):连续或不连续的首尾串联重复排列的多拷贝基因。
10 拟基因(假基因,pseudogene):在多基因家族中,某些成员不产生有功能的基因产物的基因。
医学遗传学:分子病与先天性代谢缺陷

Hereditary persistence of fetal hemoglobin (HPFH)
6
人类正常血红蛋白的组成
血红蛋白
血红素
血红蛋白单体
7
8
地中海贫血
疾病简介:临床体征、地域分布
分子和细胞病理学机制:致病基因,常见突变, 发病机制
临床处置:诊断、治疗和预防
主要内容:
血红蛋白病 血友病 胶原蛋白病 膜通道蛋白病 结构蛋白病 角蛋白病
氨基酸代谢异常 糖代谢异常 核苷酸代谢异常 脂代谢异常 溶酶体储积症
5
血红蛋白病
• 异常血红蛋白
Abnormal hemoglobin
• 珠蛋白生成障碍性贫血(地中海贫血 ,地贫)
Thalassemia
50%~55 %
1%~3%
发病时间较晚,无自发性出血,在拔牙或外科手术 后异常出血 只有大手术后才发生出血
30
血友病A的致病基因
31
血友病A的常见突变
突变类型 倒位突变 无义突变 插入/缺失突变
中国南方不同高发地
区人群携带率 1 % ~ 23%
地贫的分类:按合成速率降低的珠蛋白类型
14
地中海贫血的致病基因:珠蛋白基因
α-珠蛋白基因突变
缺失型突变 基因簇发生大片段缺失 80%~85%
非缺失型突变 突变导致生成无功能的mRNA 突变导致mRNA加工障碍 突变导致生成不稳定的珠蛋白链
α-珠蛋白基因缺失突变
地贫临床表型差异与相关基因型的关系
2 1
2 1 2
2 1 2 1
2 1 2 1
2 1 2 1
正常 (/ )
静止型 (-/ )
分子病的定义

分子病的定义一、分子病的分类分子病是指由于基因突变导致蛋白质或酶的结构异常,从而引起一系列的病理生理变化。
根据其病因和临床表现,分子病可以分为以下几类:1.血红蛋白病:由于血红蛋白的珠蛋白β链发生了突变,导致血红蛋白的结构异常,从而引起贫血、黄疸、脾肿大等症状。
常见的血红蛋白病包括镰状细胞贫血、地中海贫血等。
2.囊性纤维化:囊性纤维化是由于囊性纤维化基因(CFTR基因)突变导致的遗传性疾病,主要累及肺部、肠道和胰腺等器官,引起呼吸困难、腹泻、胰腺炎等症状。
3.苯丙酮尿症:苯丙酮尿症是由于苯丙氨酸羟化酶基因突变导致苯丙氨酸代谢异常,从而引起智力低下、鼠尿臭味等症状。
4.免疫缺陷病:免疫缺陷病是由于免疫系统基因突变导致免疫功能缺陷,从而引起反复感染、自身免疫性疾病等症状。
常见的免疫缺陷病包括先天性无丙种球蛋白血症、共济失调性毛细血管扩张症等。
5.其他分子病:除上述几种常见的分子病外,还有许多其他类型的分子病,如神经性疾病、代谢性疾病等。
二、分子病的诊断与治疗对于分子病的诊断,主要依赖于基因检测和生化检测等方法。
基因检测可以直接检测基因突变位点,从而确定病因;生化检测可以检测相关代谢产物和酶活性等指标,从而辅助诊断。
对于分子病的治疗,应根据不同类型采取不同的治疗方法。
对于血红蛋白病和免疫缺陷病等,可以通过输血、免疫球蛋白替代等方法进行治疗;对于囊性纤维化等,可以使用CFTR调节剂等药物进行治疗;对于苯丙酮尿症等,应限制苯丙氨酸的摄入,并使用特殊配方奶粉进行治疗。
三、展望随着基因组学和蛋白质组学等生物技术的不断发展,对分子病的认识将越来越深入。
未来,针对不同类型的分子病,将会有更加精准的诊断和治疗方法。
同时,随着基因治疗技术的发展,对一些严重的遗传性疾病,如囊性纤维化等,有望实现根治。
然而,基因治疗技术仍存在一些挑战和风险,需要进一步研究和探索。
总之,分子病是一类严重的遗传性疾病,其诊断和治疗需要多学科合作和综合治疗。
分子病

分子病与先天性 代谢缺陷病
川北医学院
分子病与代谢缺陷病
基因突变或基因缺陷可导致其编码蛋 白质的结构或合成量异常,引起机体 功能障碍而导致疾病。
根据突变基因编码蛋白质的功能及对 机体所产生的影响不同,这类疾病分 为分子病和先天性代谢缺陷病。
川北医学院
分子病与代谢缺陷病
第一节 分子病
川北医学院
川北医学院
58组→酪) HbM (α Boston 分子病与代谢缺陷病
HbMMilwaukee(β63组→谷) HbMHydepark(β92组→酪) HbMSakatoon(β58组→酪) HbMIwate(α87组→酪)
川北医学院
分子病与代谢缺陷病
4.血红蛋白C病 发病机制:血红蛋白分子β6谷→赖 遗传方式:AR
全世界已发现681种异常血红蛋白。
国内发现67种,20种世界首次报道。
仅约40%的异常血红蛋白对人体有不同程
度的功能障碍。
1.镰形细胞贫血症
发病机制: 血红蛋白β6谷氨酸→缬氨酸, 导致正常HbA变成异常HbS 。
遗传方式:AR
川北医学院
分子病与代谢缺陷病
HbS HbS
HbA HbS
川北医学院
蛋白质构象病。
川北医学院
分子病与代谢缺陷病
什么是镰刀形贫血? 引起的原因是什么?
川北医学院
分子病与代谢缺陷病
一、血红蛋白病
血红蛋白病:由于珠蛋白基因变异 或缺陷引起血红蛋白分子结构异常 或合成速率降低所导致的遗传性血 液病。
(一)血红蛋白的分子结构及遗传控制
1.血红蛋白的分子组成
α β
ε
δ
分子病与先天性代谢缺陷病
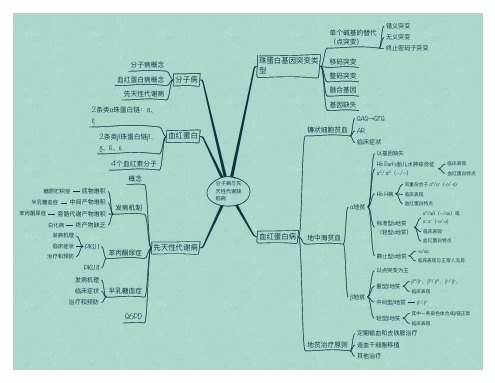
β⁰/β⁺、 β⁰/ β⁰、 β⁺/ β⁺、 临床表现
中间型β地贫 β⁺/ β⁺
轻型β地贫
其中一一条染色色体合成β链正常 临床表现
定期输血血和去铁胺治疗
地贫治疗原则 造血血干干细胞移植
其他治疗
临床表现 血血红蛋白白特点
α地贫
地中海海贫血血
双重杂合子子 α⁰/α⁺(-α/-α)
Hb H病 临床表现
血血红蛋白白特点
标准型α地贫 (轻型α地贫)
α⁰/αA(--/αα)或 α⁺ α⁺(-α/-α) 临床表现 血血红蛋白白特点
静止止型α地贫 以点突变为主
-α/αα 临床表现与正常人人无无异
β地贫
重型β地贫
PKU II 发病机理理 临床症状 治疗和预防
半乳糖血血症
先天性代谢病
G6PD
分子子病与先 天性代谢缺 陷病
珠蛋白白基因突变类 型
单个碱基的替代 (点突变)
移码突变
错义突变 无无义突变 终止止密码子子突变
整码突变
融合基因
基因缺失
血血红蛋白白病
GAG→GTG
镰状细胞贫血血 AR
临床症状
以基因缺失
Hb Bart's胎儿儿水水肿综合征 α⁰/ α⁰(--/--)
分子子病概念 血血红蛋白白病概念
先天白白链:α、 ξ
2条类β珠蛋白白链β、 γ、δ、ε
血血红蛋白白
4个血血红素分子子
概念
糖原贮积症 底物堆积
半乳糖血血症 中间产物堆积 苯丙酮尿尿症 旁路路代谢产物堆积
发病机制
白白化病 终产物缺乏
发病机理理
临床症状 治疗和预防
PKU I
苯丙酮尿尿症
遗传性代谢疾病

遗传性代谢疾病遗传性代谢疾病是由基因突变引起的一类疾病,其特点是机体无法正常代谢某些物质,导致代谢产物堆积在体内或缺乏某种代谢产物,从而引发一系列临床症状。
本文将介绍遗传性代谢疾病的基本概念、常见类型、诊断和治疗等方面内容,旨在加深对遗传性代谢疾病的了解。
一、基本概念遗传性代谢疾病是由遗传突变引起的一类疾病,主要表现为机体无法正常代谢某些物质,常见的包括糖代谢异常、脂代谢异常和氨基酸代谢异常等。
这些突变可遗传于下一代,并且影响人体正常生理功能。
二、常见类型1. 糖代谢异常:糖代谢异常是一类由于某些酶的缺陷而导致的代谢障碍。
常见的糖代谢异常疾病有糖尿病、葡萄糖酮症和乳酸酮症等。
2. 脂代谢异常:脂代谢异常是指机体对脂类物质的代谢异常。
常见的脂代谢异常疾病有高脂血症、脂肪酸氧化障碍和糖脂代谢紊乱等。
3. 氨基酸代谢异常:氨基酸代谢异常是指机体对氨基酸的代谢异常。
常见的氨基酸代谢异常疾病有苯丙酮尿症、甲基丙二酮尿症和酪氨酸尿症等。
三、诊断遗传性代谢疾病的诊断通常需要结合临床表现、家族史以及实验室检查等综合考虑。
临床症状可以包括生长迟缓、智力发育迟滞、肝脾肿大等。
此外,实验室检查可以通过检测相关酶活性、代谢产物浓度以及基因突变等方法进行确诊。
四、治疗遗传性代谢疾病的治疗通常包括对症治疗、饮食控制和基因治疗等。
对症治疗可以通过补充缺乏的酶活性或代谢产物来缓解症状。
饮食控制通常是限制某些物质摄入,以减少代谢产物的积累。
基因治疗是一种新兴的治疗方法,具体包括基因替代、基因修复和基因编辑等。
总结遗传性代谢疾病是由基因突变引起的一类疾病,其特点是机体无法正常代谢某些物质,导致代谢产物堆积或缺乏。
常见类型包括糖代谢异常、脂代谢异常和氨基酸代谢异常。
该疾病的诊断需要综合考虑临床表现、家族史和实验室检查等。
治疗方面可采用对症治疗、饮食控制和基因治疗等手段。
随着基因治疗的发展,相信对于遗传性代谢疾病的治疗将有更好的前景。
遗传性代谢病演稿PPT课件

其他治疗方法
肝移植
对于某些严重遗传性代谢病, 如尿素循环障碍等,肝移植是 一种有效的治疗方法。
基因治疗
随着基因技术的不断发展,基 因治疗逐渐成为遗传性代谢病 治疗的新方向,通过纠正基因 缺陷来根治疾病。
支持性护理
对于某些无法根治的遗传性代 谢病,提供支持性护理措施, 如心理疏导、康复训练等,以 提高患者的生活质量。
加强遗传性代谢病的早期筛查 和诊断,提高患者的早期诊断 率,减少漏诊和误诊。
针对不同类型的遗传性代谢病 ,开展更加深入的临床研究, 为患者提供更加个性化的治疗 方案。
THANK YOU
感谢聆听
80%
限制天然有害物质摄入
对于某些遗传性代谢病,限制天 然有害物质的摄入,如苯丙酮尿 症患者的苯丙氨酸摄入。
100%
特殊配方食品
针对不同疾病制定特殊配方食品 ,以满足患者营养需求,同时降 低代谢负担。
80%
补充营养素
对于某些代谢异常,适当补充特 定的营养素,如维生素、矿物质 等,以维持正常的生理功能。
保持积极心态
家庭成员应鼓励患者保持积极心态,增强战 胜疾病的信心和勇气。
05
遗传性代谢病的研究进展与展望
研究现状
遗传性代谢病是遗传基因突变导致的代谢异常性疾 病,具有较高的致残率和致死率。
目前,遗传性代谢病的研究主要集中在病因、诊断 、治疗等方面,取得了一定的进展。
遗传性代谢病的诊断方法包括生化检测、基因检测 和影像学检查等,治疗手段包括饮食控制、药物治 疗和手术治疗等。
动态监测
对于已经确诊的患者,需要定期进行复查和监测 ,了解疾病进展和治疗效果。
综合分析
结合临床观察、实验室检查、基因检测等多种手 段进行综合分析,提高诊断准确性。
遗传代谢性疾病

遗传代谢性疾病
contents
目录
• 遗传代谢性疾病概述 • 常见遗传代谢性疾病 • 遗传代谢性疾病的诊断与治疗 • 遗传代谢性疾病的科研进展 • 社会对遗传代谢性疾病的关注与支持
01 遗传代谢性疾病概述
定义与分类
定义
遗传代谢性疾病是由于基因突变 导致酶缺陷或细胞内代谢途径异 常而引起的疾病。
丙酸血症
总结词
丙酸血症是一种罕见的有机酸代谢障碍疾病,由于丙酸分解代谢异常导致。
详细描述
患者可能出现呕吐、喂养困难、肌张力低下、惊厥等症状,目前尚无特效治疗方法,需要加强遗传咨 询和早期干预。
03 遗传代谢性疾病的诊断与 治疗
诊断方法
01
03
04
临床观察
观察患者的症状和体征,如生 长发育迟缓、智力障碍、肌无
遗传代谢性疾病的流行病学
流行病学特征
遗传代谢性疾病的流行病学特征因疾病类型而异,但总体来 说,这些疾病在人群中的发病率较高,且存在一定的地区和 种族差异。
预防与控制
针对遗传代谢性疾病的预防与控制,主要采取早期筛查、诊 断和治疗措施,以提高患者的生活质量和生存率。同时,加 强遗传咨询和产前诊断也是预防遗传代谢性疾病的重要手段 。
详细描述
患者可能出现语言障碍、视力减退、 听力丧失、肌无力等症状,目前尚无 特效治疗方法,需要加强遗传咨询和 早期干预。
枫糖尿症
总结词
枫糖尿症是一种罕见的遗传性疾病,由于支链氨基酸代谢异常导致。
详细描述
患者可能出现智力低下、肌张力低下、惊厥等症状,目前尚无特效治疗方法,需要加强遗传咨询和早期干预。
鼓励志愿者参与遗传代谢性疾病的宣传、教 育、关爱和救助活动,为患者提供心理和社
(遗传与优生)第四章第四节 多基因遗传病
四、脂类代谢病
• 由于某些脂质在代谢中酶遗传性缺陷, 导致相应代谢物在体内沉积过多。 • 从广义上来说,脂类代谢异常包括血浆 脂质(脂蛋白)代谢异常、固醇类激素 代谢异常、神经鞘脂类贮积和粘脂类贮 积。 • 经鞘脂贮积病是由于细胞膜的组成成分---神经鞘脂类化合物代谢障碍所致。
Bye—Bye
!
二、血浆蛋白病
血浆蛋白:血液中含量高、种类多、功能
重要的一类蛋白质,在体内起着物质运输、
凝血和免疫防御等作用。
基因缺陷导致血浆中某种蛋白质缺陷引起
血浆蛋白病。
血友病
血友病:凝血因子遗传性缺乏引起严重凝
血功能障碍的出血性疾病。
甲型(A型,凝血因子VIII缺乏),最高 乙型(B型,凝血因子IX缺乏) 丙型(C型,凝血因子XI缺乏)
三、
常见基因遗传病
• 多基因遗传病: 由多对基因控制的且受环境因素影响 的遗传病。
例:先天畸形、精神分裂病、哮喘、消 化性溃疡、先天性心脏病。
三、
常见基因遗传病
(一)易患性和发病阈值
易患性:多基因遗传病中一个个体在遗传
基础和环境因素共同作用下患某种多基因 遗传病的风险称易患性。
易感性:多基因遗传病中由遗传基础决定
1.白化病
• 是一种常见的先天性皮肤及其附属器官 黑色素缺乏所引起的疾病。
临床表现:皮肤白皙、毛发银白色或淡黄色、畏光 遗传病因:酪氨酸酶基因缺陷。
基因定位:全身性:AR;11q14—q21。 局部性:AD;15p 治疗:避免日晒,用5%对氨基甲酸护肤液涂皮肤暴 露部分,外出戴有色眼镜。
2.苯丙酮尿症
二、糖代谢病
(一)半乳糖血症(galactosemia):AR (案例9-5)
遗传性代谢缺陷与分子病课件

第二节 分子病
分子病是指由于结构基因突变而造成 蛋白质分子结构或合成量异常所引起的疾病。
•遗传性代谢缺陷与分子病
一、血红蛋白病
由血红蛋白分子合成异常引起的疾病称为血红蛋白病。
(a)血红蛋白亚单位 (b) 血红蛋白四聚体 人类正常血红蛋白的结构
•遗传性代谢缺陷与分子病
正常人体血红蛋白
白化病患者
•遗传性代谢缺陷与分子病
尿黑酸尿症
1
发病率
1/250000
2
遗传方式 AR
3
基因定位
3q21-q23
•遗传性代谢缺陷与分子病
4、尿黑酸尿症的发病原理
苯丙氨酸
酪氨酸
尿黑酸(堆积) 尿黑酸氧化酶(缺乏)
乙酰乙酸
CO₂+H₂O
•遗传性代谢缺陷与分子病
5、尿黑酸尿症的临床特征
新生儿期即可由尿排出大量尿黑酸, 新鲜尿的颜色正常,放置空气中则变为棕 色或黑色。成年以后因被氧化的尿黑酸长 期沉积于结缔组织中,致使中廓、巩膜、 鼻、颊等变为褐色或蓝黑色而出现褐黄病, 晚期可累及关节,进展为褐黄病性关节炎。
2
遗传方式 AR
3
基因定位 12q24.1
•遗传性代谢缺陷与分子病
4、苯丙酮尿症的发病原理
苯丙氨酸羟化酶(缺乏)
苯丙氨酸 (堆积)
酪氨酸
酪氨酸酶(活性降低)
苯丙酮酸 (堆积)
黑色素(减少)
苯乳酸(堆积) 苯乳酸(堆积)
•遗传性代谢缺陷与分子病
5、苯丙酮尿症的临床特征
①神经系统症状:如兴奋不安、多 动或嗜睡、萎靡、肌张力增高、腱 反射亢进、惊厥、智能发育落后, 80%有脑电图异常。
医学遗传学名词解释

第一章绪论无第二章遗传的细胞学基础1.常染色质:间期核内纤维折叠盘曲程度小、分散度大、能活跃地进行转录的染色质。
2.异染色质:间期核内纤维折叠盘曲紧密、呈凝聚状态,一般无转录活性的染色质,又分为结构异染色质和兼性异染色质两大类。
3.兼性异染色质:是在特定细胞的某一发育阶段由原来的常染色质失去转录活性,转变成凝缩状态的异染色质,二者的转化可能与基因的表达调控有关。
4. Lyon假说:(1)雌性哺乳动物体细胞内仅有一条X染色体有活性,其他的X染色体在间期细胞核中螺旋化而呈异固缩状态的X染色质,在遗传上失去活性。
(2)失活发生在胚胎发育的早期(人胚第16天);在此之前所有体细胞中的X染色体都具有活性。
(3)X染色体的失活是随机的,但是是恒定的。
5.剂量补偿:由于正常女性体细胞中的1条X染色体发生了异固缩,失去了转录活性,这样就保证了男女性个体X染色体上的基因产物在数量上基本一致,这称为X染色体的剂量补偿。
第三章遗传的分子基础1.外显子和内含子:真核生物的基因为断裂基因,即结构基因是不连续排列的,中间被不编码的插入序列隔开,编码序列称为外显子,编码序列中间的插入序列称为内含子。
2.单一序列和高度重复序列:单一序列是在一个基因组中只出现一次或少数几次,大多数编码蛋白质和酶类的基因即结构基因为单一序列。
重复序列是指在基因组中有很多拷贝的DNA序列,有些重复序列与染色体的结构有关。
3.基因突变:是指基因在结构上发生碱基对组成或排列顺序的改变。
4.转换和颠换:转换是指一个嘌呤被另一个嘌呤所取代,或是一个嘧啶被另一个嘧啶所取代。
颠换指嘌呤取代嘧啶,或嘧啶取代嘌呤。
5.同义突变:是指碱基替换使某一密码子发生改变,但改变前后的密码子都编码同一氨基酸,实质上并不发生突变效应。
6.错义突变:是指碱基替换导致改变后的密码子编码另一种氨基酸,结果使多肽链氨基酸种类和顺序发生改变,产生异常的蛋白质分子。
7.无义突变:是指碱基替换使原来为某一个氨基酸编码的密码子变成终止密码子,导致多肽链合成提前终止。
分子病

分子病分子病(molecular disease)由于遗传上的原因而造成的蛋白质分子结构或合成量的异常所引起的疾病。
蛋白质分子是由基因编码的,即由脱氧核糖核酸(DNA)分子上的碱基顺序决定的。
如果DNA分子的碱基种类或顺序发生变化,那么由它所编码的蛋白质分子的结构就发生相应的变化,严重的蛋白质分子异常可导致疾病的发生。
实际上任何由遗传原因引起的蛋白质功能异常所带来的疾病都是分子病,但习惯上把酶蛋白分子催化功能异常引起的疾病归属于先天性代谢缺陷而把除了酶蛋白以外的其他蛋白质异常引起的疾病称为分子病。
分子病这一名词是1949年美国化学家L.C.波林在研究镰形细胞贫血症时提出的,他发现患者的异常血红蛋白β链N端的第6位的谷氨酸被缬氨酸所替代并把它称为血红蛋白S(HbS)。
迄今已发现的血红蛋白异常达300多种,包括由于血红蛋白分子结构异常导致的异常血红蛋白病和血红蛋白肽链合成速率异常导致的血红蛋白病如地中海贫血。
分子病除了血红蛋白病以外,还有各种血浆白蛋白异常、球蛋白异常、脂蛋白异常、铜蓝蛋白异常、转铁蛋白异常、补体异常、受体蛋白异常等。
目前已能应用遗传工程的方法作血红蛋白病等分子病的产前诊断(见重组DNA 技术)。
例如α-地中海贫血(巴特氏胎儿水肿综合征)是由4个α结构基因全部缺失引起的。
通过分析羊水中胎儿脱屑细胞的DNA分子是否存在α珠蛋白基因即可诊断本病。
分析时先提取人类α珠蛋白信使核糖核酸(mRNA),用反向转录酶制备互补DNA(cDNA),再将cDNA用32P标记,然后与从羊水细胞中分离获得的DNA 进行分子杂交,再用放射自显影的吸印法来检查,即可判定是否有珠蛋白基因存在。
在某些情况下,限制性核酸内切酶的方法更为优越。
由于基因突变可以造成某种限制酶切点的丧失或新切点的出现。
在这种情况下,用同一种限制酶处理正常的和发生突变的基因就会出现长短不相同的DNA片段。
例如用限制酶HpaI切割正常人的D NA,切点是在距β珠蛋白基因3′端5 000个核苷酸处,切下的β基因包含在一个7个碱基对(7.6Kb)的DNA片段中。
分子病名词解释

分子病名词解释分子病是一种由基因突变或染色体异常引起的遗传病。
基因是生命的基本单位,它包含着控制身体内部生物化学反应和生长发育的遗传信息。
基因突变会导致生物体基因表达的异常,从而引发各种疾病。
分子病包括单基因遗传病和多基因遗传病两类。
单基因遗传病通常由单个基因的突变引起,如先天性免疫缺陷综合症、囊性纤维化、血友病等。
多基因遗传病则是由多个基因的异常相互作用引发的,如某些心脏病、精神病等。
分子病的发生与个体的基因组有直接关系,其中有些分子病表现为遗传性疾病,意即父母携带有该基因突变并将其遗传给子代,而有些分子病则是在个体本身的细胞遗传物质发生突变后引起。
分子病在临床上的表现十分复杂,其病因、发病机制和临床症状有很大的异质性。
一些分子病表现为疾病的早发型,患者在出生后不久即表现出明显的症状,如营养代谢障碍症、遗传代谢病等。
而一些分子病则表现为疾病的晚发型,症状出现的晚,如乳腺癌、结直肠癌等。
此外,分子病还可以表现为不同种类的疾病,如遗传性高胆固醇血症可引起心血管疾病、家族性结节性甲状腺癌可引起甲状腺癌等。
诊断分子病一般需要进行基因检测,通过寻找基因突变以及检测其影响的蛋白质的功能来确定疾病的基因突变和相关产物。
分子病的治疗方法主要有三个方面,一是基因治疗,即通过基因修补或替代等手段来纠正基因突变;二是蛋白质治疗,即通过补充或调节相关蛋白质来缓解疾病;三是细胞治疗,即通过移植或修复患者受损细胞来缓解疾病。
总的来说,分子病是由基因突变或染色体异常引起的遗传病,其病因和临床表现差异很大。
目前的诊断和治疗方法主要集中在基因检测、基因治疗和蛋白质治疗等领域,但仍面临许多挑战。
未来随着技术和研究的不断发展,预防和治疗分子病的方法将会更加多样化和精准化。
遗传性代谢缺陷与分子病课件

疾病预防与遗传咨询
遗传风险评估
01
通过家族史、基因检测等手段,评估个体遗传代谢缺陷的风险
,提供针对性的预防建议。
遗传咨询
02
为携带遗传代谢缺陷基因的家庭提供遗传咨询,解释疾病风险
、传递方式等问题,指导家庭生育决策。
新生儿筛查
03
通过大规模新生儿筛查,早期发现潜在的遗传代谢缺陷患者,
进行早期干预和治疗,降低疾病对患儿的影响。
治疗措施
探讨针对该遗传性代谢缺陷的 治疗措施,如饮食控制、酶替
代疗法、基因治疗等。
案例二:一种分子病的综合诊断与治疗
01
02
03
04
案例介绍
详细介绍某种分子病的病例, 包括患者的临床表现、疾病的
遗传基础等。
分子病的发病机制
阐述该分子病的发病机制,包 括相关基因、突变位点、蛋白
质功能异常等。
诊断流程
遗传性代谢缺陷的发病机制涉及基因突变,这些突变可能导致酶活性的降低或丧失,代谢 通路受阻,以及代谢产物在体内的异常累积。这些异常代谢产物可能对机体产生毒性作用 ,引起不同组织和器官的损害。
遗传性代谢缺陷的分类
单基因遗传代谢缺陷
由单一基因突变引起的代谢缺陷,如苯丙酮尿症、囊性纤维化等 。
多基因遗传代谢缺陷
05
典型案例分析
案例一:某种遗传性代谢缺陷的分析
案例介绍
详细介绍某种遗传性代谢缺陷 的病例,包括患者的临床表现
、家族史、遗传方式等。
诊断方法
介绍对该遗传性代谢缺陷的诊 断方法,包括生化检测、基因 检测等手段。
代谢缺陷的生化机制
阐述该遗传性代谢缺陷的生化 机制,如涉及的代谢途径、关 键酶、代谢产物的变化等。
分子病与先天性代谢缺陷病PPT课件

(3)血红蛋白M病:珠蛋白基因突变使血红蛋白和亚铁离子结合的 正常氨基酸被另一个不同氨基酸取代,导致亚铁离子变为铁离子,使 部分铁原子变为稳定的高铁状态,影响了血红素和氧的结合能力,导 致组织缺氧。本病为常染色体显性遗传。
(一) 血红蛋白病
3.地中海贫血 地中海贫血为珠蛋白基因突变导致蛋白肽链合成量异 常而引起的贫血症。 (1)α 地中海贫血:①Hb Bart 胎儿水肿综合征:4个α 珠蛋白基 因全部缺失或缺陷,完全不能合成α 珠蛋白链。胎儿严重缺氧,发育 到8~10个月全身水肿而死亡。②Hb H病:3个α 珠蛋白基因缺失或缺陷, 表现为轻度或中度贫血,患者肝脾大,有轻度或间歇发生黄疸,可发 生继发性感染。③轻型(标准型)α 地中海贫血:2个α 珠蛋白基因缺 失或缺陷。临床轻度溶血性贫血或无症状。④静止型α 地中海贫血:1 个α 珠蛋白基因缺失或缺陷,一般无症状。
(一) 血红蛋白病
(2)β 地中海贫血:①重型β 地中海贫血:患者正常β 链缺乏或合 成量很少,血红细胞中无HbA或HbA含量很少,HbF和HbA2含量增高,α 珠蛋白链过剩而沉积到红细胞膜上,改变了红细胞膜的性能,引发严 重的溶血性贫血。主要临床症状表现为患儿出生几月后溶血反应,并 伴有腹泻、发热、生长发育迟缓、身材矮小、骨髓增生,可出现鼻塌 眼肿、上颌前突、头大额隆等特殊的“地中海贫血面容”。②中间型 β 地中海贫血:临床表现介于轻型和重型之间,中度贫血,脾脏轻或 中度肿大,黄疸可有可无,骨骼改变较轻。③轻型β 地中海贫血:带 有一个正常的β 珠蛋白基因,一般无临床症状或有轻微贫血和脾脏肿 大。
(四) 膜转运载体蛋白病
肝豆状核变性的遗传又称Wilson病,由编码P型铜转运ATP 酶的基因(ATP7Base)突变引起,基因定位于13q14.3,为常 染色体隐性遗传,ATP7Base基因突变多为错义突变或无义 突变。临床表现以神经系统和肝脏受损为主,晚期表现为 痴呆。患者可通过服用青霉胺或锌剂加速清除体内贮积的 铜离子而进行早期治疗,因此本病的早期诊断对于临床治 疗很有意义。
第4章-第五节遗传性代谢缺陷与分子病

人民卫生出版社
(三)苯丙酮尿症
❖ 临床上表现为精神发育迟缓,皮肤、毛发和虹膜 色素减退,头发呈赤褐色,癫痫,湿疹,特殊的 鼠样臭味尿。
❖ 婴儿出生后立即进行PKU的筛查,一经确诊,立即 给患儿停乳,喂给低苯丙氨酸水解蛋白,禁荤食 、乳类、豆类和豆 制品。可多吃蔬菜和水果。
人民卫生出版社
苯丙氨酸的代谢途径 1.苯丙酮尿症;2.尿黑酸尿症;3.白化病
人民卫生出版社
二、分子病
❖分子病(molecular disease) 是由遗传性基因突变或获得性基因突变使蛋白
质的分子结构或合成的量异常,从而引起机体功能 障碍的一类疾病。
人民卫生出版社
二、分子病
❖分子病(molecular disease) (一)血红蛋白病 血红蛋白分子合成异常引起的 疾病称血红蛋白病(hemoglobinopathy),习惯上 分为异常血红蛋白和地中海贫血两类。分子遗传学 研究表明,它们的分子基础都是珠蛋白基因的突变 或缺陷所致。
镰状细胞贫血的发病机制
人民卫生出版社
(2)地中海贫血 ❖ 地中海贫血(tha1assemia)是由于珠蛋白基因缺
失或突变导致相应珠蛋白链合成障碍降低,造成一 些肽链缺乏,另一些肽链相对过多,出现肽链数量 的不平衡,导致溶血性贫血。是一种常染色体隐性 遗传病。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
(三)苯丙酮尿症
❖ 临床上表现为精神发育迟缓,皮肤、毛发和虹膜 色素减退,头发呈赤褐色,癫痫,湿疹,特殊的 鼠样臭味尿。
❖ 婴儿出生后立即进行PKU的筛查,一经确诊,立即 给患儿停乳,喂给低苯丙氨酸水解蛋白,禁荤食 、乳类、豆类和豆 制品。可多吃蔬菜和水果。
人民卫生出版社
苯丙氨酸的代谢途径 1.苯丙酮尿症;2.尿黑酸尿症;3.白化病
人民卫生出版社
学习目标
1.掌握遗传性代谢缺陷病和分子病的概念。 2.掌握遗传性代谢缺陷病的特征。 3.熟悉遗传性代谢缺陷病的分子机制。 4.熟悉主要的分子病的分子机制。 5.了解主要的遗传性代谢缺陷病和分子病的临床
症状 。
人民卫生出版社
一、遗传性代谢缺陷
❖遗传性代谢缺陷(inborn errors of metabolism) 也称先天性代谢缺陷或遗传性酶病,指由于遗传上 的原因(通常是基因突变)而造成的酶蛋白质分子 结构或数量的异常,从而导致代谢紊乱的机体功能 障碍性疾病。
人民卫生出版社
(四)白化病
❖ 白化病(albinism)也是一 种氨基酸代谢异常的遗传病 。
❖ 正常人黑色素是由黑色素细 胞中的黑色素体合成,它含 有的酪氨酸酶可将酪氨酸转 变,最终生成黑色素。
❖ 白化病人由于基酪氨酸酶因 突变,该基因定位于11q14q21,使该酶缺乏,导致黑色 素生物合成缺陷 。
基
酶结构异常→功能性缺乏 酶活性↓
因
突
酶稳定性↓ → 酶降解速率↑
变
改变酶与辅助因子结合的部位
调控基因变异 → 酶合成数量↓
人民卫生出版社
(一)半乳糖血症
❖主要表现:
患儿对乳糖不耐受, 婴儿哺乳后呕吐、腹泻 ,继而出现白内障、肝 硬化、黄疸、腹水、智 力发育不全等。
发病率约为1/50000。 ❖半乳糖血症属于常染 色体隐性遗传,致病基 半乳糖的体内代谢途径 因定位于9p13。
镰状细胞贫血的发病机制
人民卫生出版社
(2)地中海贫血 ❖ 地中海贫血(tha1assemia)是由于珠蛋白基因缺
失或突变导致相应珠蛋白链合成障碍降低,造成一 些肽链缺乏,另一些肽链相对过多,出现肽链数量 的不平衡,导致溶血性贫血。是一种常染色体隐性 遗传病。
人民卫生出版社
1)α地中海贫血(α-地贫)
人民传性代谢缺陷两种原因: 一是由于编码酶蛋白的结构基因发生突变,引
起酶蛋白结构异常或缺失; 二是基因的调控系统发生异常,使之合成过少
或过多的酶,引起代谢紊乱。
人民卫生出版社
基因、mRNA、酶和代谢过程的相互关系
人民卫生出版社
酶活性降低或缺乏的原因
结 构
酶完全不能合成——酶缺乏
人民卫生出版社
预防和治疗: ➢ 新生儿筛查 ➢ 患儿戒奶,吃不含乳糖和半乳糖的代乳品
( 出生3个月开始 )。
人民卫生出版社
(二)葡糖-6-磷酸脱氢酶缺乏症
❖ 由于葡糖-6-磷酸脱氢酶(G6PD)缺乏而引起的葡 糖-6-磷酸脱氢酶缺乏症(G6PD)是一种常见的X连 锁不完全显性遗传病。
❖ 临床主要表现为一组溶血性疾病,包括:“蚕豆病 ”、药物性溶血、新生儿黄疸、某些感染性溶血和 慢性非球形细胞溶血性贫血。
人民卫生出版社
导学案例
“黄种”父母为何会生出“白种”小孩 一天在遗传咨询门诊来了一对夫妻,带着一个 全身皮肤雪白、就连眉毛、毛发等全为白色或浅 黄色,眼睛虹膜和瞳孔呈现淡红色。丈夫说“我 们夫妻俩都是正宗的黄种人,也经常做体检,没 有发现什么问题,可是不知怎么回事,我们的小 孩却是“白种”的。听说是遗传的,但我们俩家 族都从来没有生出过“小洋人”,“为什么他会 出现全身发白的现象,能治疗吗?”
①Hb Bart’s胎儿水肿综合征 发
病于胎儿期,基因型为α0地中海
贫血基因纯合子(--/--),这
是α地中海贫血中最严重者,4
个α珠蛋白基因全部缺失。
图 Hb Bart’s胎儿水 肿
人民卫生出版社
②血红蛋白H病(HbH):患者为α0地中海贫血基因和 α+地中海贫血基因的双重杂合子,基因型为(-/-α)。HbH病患儿在出生时几乎无明显的症状, 只有轻度贫血,至1周岁左右便出现HbH病的临床症 状。
人民卫生出版社
二、分子病
❖分子病(molecular disease) 是由遗传性基因突变或获得性基因突变使蛋白
质的分子结构或合成的量异常,从而引起机体功能 障碍的一类疾病。
人民卫生出版社
二、分子病
❖分子病(molecular disease) (一)血红蛋白病 血红蛋白分子合成异常引起的 疾病称血红蛋白病(hemoglobinopathy),习惯上 分为异常血红蛋白和地中海贫血两类。分子遗传学 研究表明,它们的分子基础都是珠蛋白基因的突变 或缺陷所致。
❖ 在我国多数G6PD缺乏者没有临床症状,但在诱因作 用下发病。
❖ G6PD基因定位于Xq28。男性半合子发病,女性杂合 子具有不同的表现度。
人民卫生出版社
(三)苯丙酮尿症
❖ 苯丙酮尿症(phenylketonuria,PKU)一种严重的 常染色体隐性遗传性氨基酸代谢病。
❖ PKU患者由于肝脏内苯丙氨酸羟化酶(PAH)缺乏, 苯丙氨酸不能转变为酪氨酸,后者转化为苯丙酮酸 和苯乳酸并在体内累积,并导致血液和尿液中苯丙 氨酸及其衍生物排出增多 。
人民卫生出版社
(1)镰状细胞贫血 ❖ 是因β基因缺陷所引起的一种疾病,呈常染色体隐
性遗传。HbS纯合子(HbSHbS)患者β珠蛋白基因 的第6位密码子由正常的GAG突变为GTG(A→T), 使其编码的β珠蛋白N端第6 位氨基酸由正常的谷 氨酸变成了缬氨酸,形成HbS。
人民卫生出版社
(1)镰状细胞贫血
医学遗传学基础
教材主编 林小珊
医学副编 主遗者编 传莫 莉学基础 孙元田 海南医学院 林小珊 广州医科大学卫生职业技术学院 林 文 惠州卫生职业技术学院 莫 莉 广西科技大学医学院 植瑞东 肇庆医学高等专科学校 赖燕燕 广西卫生职业技术学院
人民卫生出版社
第四章 遗传性疾病
第五节 遗传性代谢缺陷与分子病
白化病
人民卫生出版社
❖ 临床表现: ➢ 为全身皮肤、眼睛、毛发等的黑色素缺乏,全身白化
,终身不变。 ➢ 患者眼睛视网膜无色素,虹膜和瞳孔呈现淡红色,羞
明怕光,眼球震颤,常伴有视力异常。 ➢ 患者对阳 光敏感,暴晒可引起皮肤角化增厚,并诱
发皮肤癌。 ➢ 发病率约1/10000~1/20000,呈常染色体隐性遗传。