western blot失败经验总结
Western blot结果异常原因分析及对策(个人总结)

结果异常
原因
对策
1.假阴性,不出现 ⑴一抗、二抗等不匹配
⑴重新订购试剂,认真选取一抗与组织
蛋白条带或者蛋
种属,并设置内参。
白条带很弱
⑵一抗或/和二抗浓度低
⑵增加抗体浓度,延长孵育时间
⑶封闭剂或无关蛋白与一抗或二抗有交叉反应 ⑶封闭用温和去污剂,如TWEEN 20,
⑷SDS-PAGE 电泳上样 前, 煮沸 10
min,以增强蛋白质解聚变性
⑸蛋白样本降解
⑸新鲜制备标本,使用蛋白酶抑制剂
3. 背 景 非 特 异 染 ⑴封闭不充分
⑴延长封闭时间,更换合适的封闭剂
色过强
(脱脂奶粉,BSA,血清等)
⑵一抗浓度过高
⑵增加一抗稀释倍数
⑶抗体孵育温度过高
⑶4℃孵育
⑷二抗非特异性结合或与封闭剂交叉反应
(无关蛋白中含有与待测抗原结构类似多肽(共 或更换封闭剂(脱脂奶、BSA或明胶)
同抗原表位),与特异抗体发生交叉反应)
⑷一抗不识别目的物种的靶蛋白
⑷检查说明书,或做 ClustalW 比对,
设阳性对照
⑸样本中无靶蛋白或靶蛋白含量过低
⑸模索条件,设置阳性对照,增加标本
(抗原无效)
上样量,样本制备时使用蛋白酶抑制剂,
4. 背景出现不均 匀污点、斑点
5. 转膜不充分
⑴转膜时有气泡或抗体分布不均 ⑵抗体与封闭剂结合 ⑶镊子或玻璃平皿污染 ⑷膜湿润不够 ⑸标记膜采用不规范笔 ⑴膜没有完全均匀湿透 ⑵靶蛋白分子量小于 10,000 ⑶靶蛋白等电点等于或接近转移缓冲 液 pH 值 ⑷甲醇浓度过高 ⑸转移时间不够Thick gel
⑷设置二抗对照(不加一抗),降低二抗
wb实验失败总结

wb实验失败总结前言在创作者的道路上,失败是必然存在的一部分。
无论是成功的创作者还是资深的创作者,都会遇到失败。
今天我们要探讨的是”wb实验失败”这个案例。
通过总结这个实验失败的原因和经验教训,我们可以更深入地理解创作过程中的挑战与机遇。
正文1. 实验目标•确定实验目标是十分关键的。
在进行”wb实验失败”之前,我们需要明确我们要实现什么目标,这样才能有一个清晰的方向。
2. 方法选择•在进行创作实验时,我们需要选择一个合适的方法。
在”wb实验失败”中,我们可以反思我们选择的方法是否适合我们的目标。
3. 资源准备•在创作过程中,我们需要合理地分配资源。
是否有足够的时间、人力和经费来支持我们的实验?在”wb实验失败”中,我们可以回顾一下我们是否充分准备了所需的资源。
4. 团队协作•如果创作者有一个团队,良好的团队协作是非常重要的。
在”wb 实验失败”中,我们可以思考我们的团队是否有良好的沟通和合作,是否能够充分发挥每个人的优势。
5. 反馈与调整•创作者需要对实验进行及时的反馈和调整。
在”wb实验失败”中,我们要检验自己是否在实验过程中及时收集反馈,并根据反馈做出相应的调整。
6. 学习与成长•无论实验结果如何,我们都要从失败中学习并不断成长。
在”wb 实验失败”中,我们可以思考我们从这次失败中学到了什么,并如何应用这些经验教训到我们未来的创作中。
结尾尽管”wb实验失败”可能会让我们感到沮丧,但我们不能停止创作的脚步。
通过总结失败的原因和经验教训,我们可以更加明确自己的创作方向,并不断提升自己的创作能力。
只有经历失败,我们才能更好地迎接未来的挑战。
让我们坚持创作,并始终相信自己的能力和潜力!。
western blot实验经验总结

western blot实验经验总结1.抗体的选择对于国内的大多数实验室来讲,做western blot实验选择抗体是个头疼的问题。
原因很简单,买进口抗体捉襟见肘,买国产抗体得需要大无畏的勇气,对于我所在的兰州地区的实验者而言,感触尤深。
在这五年的western blot实验历程里,我先后用过进口抗体,进口抗体国内分装包装,国产抗体,质量良莠不齐。
进口抗体一般不会出现闪失:abcam品种全,质量过硬,但价高(3400元/100微升),而且说是100微升,但至多能吸出来90微升;Sigma的价最贵;比较有性价比的是CST的抗体,现在好像是2200元/100微升,我前后用过二十几种, 1:1000的稀释比下,还没有失过手,100微升通常能完成所有的免疫组化和western blot 实验,还有一个优点是通常量比100微升多出来10微升,唯一的不足是CST的品种实在不多。
Santa的多克隆抗体质量可以,但是选用Santa的单抗还是有风险,估计这也是业界共识了吧。
进口抗体国内分装包装我也用过不少,呵呵,毕竟是穷人(420元/100微升),大概好抗体的比例约为50%,如果能做出来,也存在一个问题,就是抗体大多只能用一次。
我曾经把Santa的原装抗体和分装抗体做过比对(抗体品种,货号等完全一致),在都能做出来的前提下,原装抗体能重复使用的次数要多出许多。
具体原因我已经揣测了好几年,不敢说出来,我一直在想是不是冬天西瓜切成牙和整个卖,也会有所不同。
揣测归揣测,假如只为了发论文毕业走人,可以考虑选用进口分装的多克隆抗体。
国产抗体比较知名的就几家,但质量确实不敢恭维。
western blot能做出来的确实不多,而且杂带多,背景不干净。
我们周边的实验室大多买国产抗体做免疫组化,怎么说呢,应付硕士论文够了。
我也帮别人自制过抗体,再用抗原亲和纯化。
效价非常不错,夸张的时候1:10000都能做出条带,唯一的麻烦是兔子太骚(骚臭),不知道这是不是“兔女郎”名号的来由。
WesternBlot(WB)操作中需要注意事项与结果不理想可能的原因自查

WesternBlot(WB)操作中需要注意事项与结果不理想可能的原因自查Western Blot(WB)操作中需要注意事项与结果不理想可能的原因自查第一部分 WB操作中需要注意事项一、蛋白样品的准备1、样品质量所抽取样品的蛋白含量,蛋白变性是否充分等等,还有,蛋白样品的PH值是否在7~8之间。
这会直接影响到样品浓缩的效果。
此外,蛋白样品是否降解、目的蛋白是否已抽取充分都会对最终结果的可靠性有影响。
2、细胞水平要做WB,多少细胞提的蛋白够做WB一般5×106就足够3、小分子量蛋白10KD需要的注意点1)可选择孔径0.22um的PVDF膜或者NC膜,转膜时间缩短,另外可采用Tricine-SDS-PAGE 体系2)也可以选择PSQ膜,同时缩短转移时间。
也可以将两张膜叠在一起,再转移。
其他按步骤即可4、大分子量蛋白200KD需要的注意点1)做200kd蛋白的WB时要注意,分离胶最好选择>7%的;剥胶时要小心2)转移时间需要相应延长;要做分子量参照(否则出现杂带不知道如何分析)3)转膜液中甲醇含量可适当降低,推荐使用湿装转膜效率更高二、制胶1、凝胶质量不连续SDS-PAGE胶对凝胶的要求较高,分离胶的PH值应在8.8左右,而浓缩胶的PH应在6.8左右,最好不要差过0.5个PH单位,因为这个PH条件是保证电泳液中的甘氨酸不电离的必要条件,也是保证能充分压缩样品的前提。
因此,制备凝胶的时候所用Tris-HCl缓冲液的PH值是否稳定是很重要的。
2、在用ddH2O压分离胶的时候为什么经常存在中间凸的现象1)加水时不能对着胶冲,要沿着玻璃板缓慢流行2)加水满后一定要保证上方水平面是平齐的3)加胶要避免气泡,也要避免加胶太慢而提前凝固等现象4)有其他人的经验是用异丙醇封胶封胶后左右晃一下三、加样1、点样样品尽量不要与电泳液混合,这样能提高浓缩的效果,因为电泳液PH值为8.3,而样品为7.5,混合后会影响到样品的PH值,进而影响浓缩效果。
(完整版)westernblot问题汇总

(完整版)westernblot问题汇总WesternBlot问题解答1.凝胶肿胀或卷曲:注意转膜之前,切割下来的凝胶一定要在转膜缓冲液中浸泡平衡5-10分钟,要含有20%的甲醇。
2.条带歪斜、或漂移因为转膜仪使用时间太长,两块板之间的海绵由于长期使用变得很薄。
当按照阴极-海棉-滤纸-胶-膜-滤纸-海绵依次放好后两块板不能压紧,因而在胶和膜之间可能存在潜在的间隙,电转移时蛋白从胶和膜之间的空隙中流走,形成如图所示的形状。
使用十几张普通的草纸垫在两块海棉之间,条带就可以变得非常的漂亮。
另外,转膜时转膜液一定要浸没凝胶和膜,否则也可能出现上述情况。
3.单个或多个白点:转膜时在膜与胶之间有气泡。
做三明治时一定要逐层压紧,赶尽气泡。
同时转膜液要提前配置好,加好甲醇,保证转膜前夜体内气泡排净。
气泡存在的局部会抵抗蛋白转膜,降低转膜效率。
4、转膜缓冲液过热:缓冲液离子强度太低,或电压及电流过高。
按照上述方式配液,同时转膜过程中注意降温。
我在冰水混合物中转膜,效果很好。
5背景过高!1)封闭不全,我用5%-10%的光明脱脂奶粉,效果还可以,现在做基本上没什么背景。
如果你觉得不够可以加点BSA,1%吧。
2)一抗或者是封闭液与膜蛋白的交叉反应。
这种情况通常可以通过加入TWEEN-20或多次洗膜加以解决。
如果问题不能解决,那么降低一抗浓度,或更换封闭液种类可能解决3)一抗二抗中某些不纯的物质也可以造成背景。
4)抗体浓度太高,或孵育时间太常都可能遇到这种情况。
那么可以适当降低一抗二抗的浓度,或是用提高温度的方式来代替杂交时间的延长;另外,少量多次得洗膜效果要优于多量而长时间得洗膜效果。
5)曝光时间的控制。
通常我们线曝光一分钟试试,条带清晰背景也强,可以缩短曝光时间,或者过一段时间等荧光减弱以后再发光,一般也可以发出比较好的条带。
如果背景强于条带,那么肯定是前面默默个原因的一种,这是可以将膜在TBST中漂洗以下再发光,有时会有比较好的效果(仅限于国外较好的试剂盒,国产的不行,洗一下啥也没了!)这个时候可以洗膜,洗膜后重新发光。
wb实验失败总结(一)

wb实验失败总结(一)前言•对于一个资深的创作者来说,实验失败往往是一种难以避免的经历。
正如冰山只露出海面上一小部分一样,成功背后常常隐藏着无数次的失败。
•在这次WB实验失败后,我要积极总结教训,从中吸取经验,以便下次能够更好地进行创作实践。
正文1. 失败原因的分析•实验前准备不充分:我没有充分研究WB实验的相关背景和先前的类似案例,也没有对实验中可能遇到的问题进行足够的预估。
•实验设计不完善:我在实验设计上存在一些漏洞,导致实验过程中出现了许多意外情况,无法顺利实现预期的目标。
•实验过程中出现的问题没有及时处理:在实验中,我遇到了一些问题,但是没有及时采取措施进行处理,导致问题逐渐扩大,最终导致实验失败。
2. 从失败中学到的教训•充分准备好实验前的背景知识和案例分析,确保自己对实验的理解和预期目标的清晰明确。
•仔细设计实验方案,考虑到可能出现的各种情况,并制定相应的解决方案。
•在实验过程中及时发现问题并进行处理,不要拖延或忽视,以免问题进一步扩大化。
3. 下次实验的改进计划•更加细致的实验准备:充分研究实验背景,做足准备工作,确保对实验内容的全面了解。
•更加完善的实验设计:考虑各种可能出现的情况,对实验过程中可能出现的问题进行预判,并制定相关解决方案。
•更加及时的问题处理:发现问题后,立即采取措施进行处理,以避免问题不断扩大。
结尾•实验失败并不可怕,关键是能够从中吸取经验教训并进行改进。
作为资深的创作者,我深知失败乃成功之母,只有不断总结和反思,才能不断进步。
我相信下次实验一定会更加出色!前言•对于一个资深的创作者来说,实验失败往往是一种难以避免的经历。
正如冰山只露出海面上一小部分一样,成功背后常常隐藏着无数次的失败。
•在这次WB实验失败后,我要积极总结教训,从中吸取经验,以便下次能够更好地进行创作实践。
正文1. 失败原因的分析•实验前准备不充分:我没有充分研究WB实验的相关背景和先前的类似案例,也没有对实验中可能遇到的问题进行足够的预估。
Western Blotting 各种问题及解决办法

可能原因 1.抗体浓度过高 2.封闭液选择不当
3.封闭不完全
背景高且均一
4.抗体与封闭液中其他蛋 白发生交叉反应 5.清洗不充分 6.曝光时间过长
7.膜出问题
8.缓冲液污染或长菌 1.抗体浓度过高 2.封闭液选择不当
3.封闭不完全
背景高,且以斑点形式存 在
4.抗体与封闭液中其他蛋 白发生交叉反应
11.剥膜造成影响
12.膜上抗原降解 13.转好的膜存放时间过长 1. 抗体浓度过高 2.SDS造成非特异性结合 3. 二抗试剂的非特异结合 4.一抗特异性差 1.抗体浓度过高 2.上样量过高 抗体浓度过高
转膜不完全
பைடு நூலகம்
可能采取的措施 降低抗体(一抗/二抗)浓度 比较不同封闭液 根据不同的体系优化封闭液 提高封闭物浓度 优化封闭时间和(或)温度,室温下至少1小时或4℃过夜 在封闭液中添加终浓度0.05%的Tween-20 用含终浓度0.05%的Tween-20的封闭液稀释抗体 更换封闭液 avidin-biotin系统避免使用奶粉,奶粉中含有biotin 检测交叉反应:封闭一张干净的不含蛋白的膜,抗体孵育后检测 增加清洗次数及清洗液体积 如果之前未添加Tween-20,可添加Tween-20至终浓度0.05% 缩短曝光时间 确保转膜前,已根据厂商指导将膜彻底浸润 更换新膜 确保膜始终有足够的液体覆盖,避免干掉 避免徒手操作,应配戴手套,使用镊子 每一步孵育都应确保充分 重新配制缓冲液 降低抗体(一抗/二抗)浓度 比较不同封闭液 根据不同的体系优化封闭液 提高封闭物浓度 优化封闭时间和(或)温度,室温下至少1小时或4℃过夜 在封闭液中添加终浓度0.05%的Tween-20 用含终浓度0.05%的Tween-20的封闭液稀释抗体 更换封闭液 avidin-biotin系统避免使用奶粉,奶粉中含有biotin 检测交叉反应:封闭一张干净的不含蛋白的膜,抗体孵育后检测 确保转膜前,已根据厂商建议将膜彻底浸润 避免徒手操作,应配戴手套,使用镊子 更换新膜 确保膜始终有足够的液体覆盖,避免干掉 每一步孵育都应确保充分 膜过多时,应避免相互堆叠 操作时应小心,对膜造成的损伤会产生非特异性反应 使用新的缓冲液 用前过滤 确保转膜仪、杂交仪等仪器清洁无污染 确保转膜后膜上没有残留的胶 转膜结束后,染胶以确定转膜效率 转膜时胶与膜应充分接触 确保转膜时滤纸与膜装配正确 按厂商指导将膜充分浸润
western 出错原因

成功过的战友讨论下,你曾经WESTERN做不出来是那部分出问题了.看看各部分出问题的几率,为还没有成功,仍然苦苦寻找结果的战友提供点参考(由于本人经验有限,不足之处还请斑竹和各位大侠改正):1、蛋白提取问题,蛋白降解,或者上样量少导致检测失败。
2、蛋白电泳失败。
3、转膜问题:转膜不充分,过转,温度过高导致蛋白结构改变等。
4、抗体问题。
5、显影问题。
6、其它。
请战友提供各部分问题的主要说明和对策,谢谢!祝蛋白版越来越旺!大家来讨论,加分从优!抗体问题排在第一,抗体的特异性,一抗二抗的比例,这些都需要摸索条件。
蛋白电泳第二,胶的配置,由于APS的失效,tris-Hcl PH不准,丙烯酰胺的氧化(以上问题一般都是配置时间较久以后)导致电泳失败或泳道奇形怪状。
我认为western bloting 中蛋白提取是关键,最好用试剂盒提取,尤其目的蛋白是在亚细胞器表达的蛋白;其次是一抗二抗,一抗最好单克隆抗体,二抗国产好厂家的也行;ECL 发光试剂一定要灵敏;制胶,电泳和转膜个实验室都做得比较常规了;显影定影按说明书作做活实验是经验就行。
jiangyuqing123 wrote:大家来讨论,加分从优!偶不懂,特来支持,我做了很久的WB,最大的一个难点感觉在于样品制备上!蛋白的降解有些时候是你想象不到的,可能出奇的糟糕,也可能完全没事~~如果单独做WB的话,感觉以“快速彻底裂解,先变性后保存”的原则比较有效,尽量选择足够强的裂解液,甚至直接给loading buffer 裂解,然后取出部分蛋白定量,其余加入loading buffer100度充分变性,再分装保存。
有些时候比蛋白酶抑制剂更有效。
另一个感觉就是样品中目的蛋白的量,限于WB的检测下限问题,一般目的蛋白量最好别低于1ng(虽然ECL法下限是0.1ng,即100pg),最好在10ng以上为好。
这就要求在做WB之前必须充分考虑目的蛋白的可能丰度,最好充分的准备工作然后再设计细胞量或者组织量,千万别把样品搞稀了,否则就不好办了。
原因分析和解决方法在这里!

原因分析和解决方法在这里!
Western Blot虽是实验室最常用的蛋白分析技术,但是由于它步骤繁琐、操作流程长、影响因素多,所以,导致我们在做实验时会遇到各式各样的问题。
今天我们总结了一些WB实验中经常遇到的各种“奇葩”条带问题,并为你推荐了常用的解决方法,希望对大家有所帮助。
一、目标条带没有信号
二. 图片背景过高,难以分辨条带
三、 膜上出现黑点和黑斑
四、 出现非特异性条带
五、条带中出现整条白色空斑
六、条带中出现白圈
七、 条带拖尾
八、条带变形
九、条带呈哑铃装
十、最边缘条带弯曲
十一、背景非均一性
十二、 条带呈“微笑”或“倒微笑”状
十三、其他问题
以上是我们对WB条带出现各种状况及解决方法的一个总结,希望可以对大家有所帮助。
western blot转膜常见现象及处理方法

1,转膜不充分/过转对于非小分子量尤其是大分子量(>100kd)蛋白而言,一般不会出现过转情况,转膜不充分是主要原因,增加转膜力度无非是加大电压/电流,延长时间。
但对于小分子蛋白(〈30kd就要注意了,<15kd尤其要当心)很可能出现过转,丽春红染膜或观察marker有没有穿膜可以确认,没有丽春红也可以考染转膜后的胶,如果是一片空白,则要小心了,可适当降低转膜力度。
对于低分子量一般转膜液不要加SDS以防过转,《抗体技术实验指南》提供的针对中低分子量的湿转缓与高分子量的相比也是不含SDS的。
2,封闭时间不足这一点似乎多数人都不重视,所以有时候会有些意外.我一般室温2~4h,但4度过夜确实是最好的。
另外在平皿中摇似乎比封口膜更容易掌握并获得较好的效果,对新手似乎更好。
3,抗体与封闭液有交叉。
实际上牛奶对于多数抗体来说还是很好的,但所有二抗也都写着:与牛奶可能有交叉反应。
BSA蛋白单一,可降低因牛奶其他成分引起的背景。
单用TBST也可以,此法虽然减少了交叉,但单就封闭能力而言却也因此而降低。
4,抗原—抗体浓度过高一般10min*3次就足够了,如果不放心可按自己意志加强时间、次数、洗脱液用量。
5,暴光时间过长降低曝光时间,以牺牲敏感度为代价其最低标准是使目的条带能够正常显影,对一切原因有效,但效果局限。
6,抗原—抗体浓度过高降低抗体浓度或蛋白上样量,以牺牲敏感度为代价其最低标准是使目的条带能够正常显影,仅对膜上蛋白和一抗引起的有效。
7,转膜不良(如有气泡在胶—膜之间)主要是气泡在胶—膜之间引起的绝缘区使蛋白无法通过而不能到达膜上,这一步在操作时尤其要小心,我自己犯过,也看到其他人犯过。
我的办法是把胶铺到膜上,先使其一边接触膜的一边,这样就会在胶和膜交界的地方形成一个水相前缘,然后慢慢落下凝胶,这样这个水相前缘就会逐渐推进直至胶膜重合,如是则不会有气泡产生。
把胶铺到膜上而非相反是因为胶是透明的,可以看到水相前缘以及有无气泡产生。
(整理)westernblot失败经验总结.

western blot失败经验总结1我做了很久的WB,最大的一个难点感觉在于样品制备上!蛋白的降解有些时候是你想象不到的,可能出奇的糟糕,也可能完全没事~~如果单独做WB的话,感觉以“快速彻底裂解,先变性后保存”的原则比较有效,尽量选择足够强的裂解液,甚至直接给loading buffer裂解,然后取出部分蛋白定量,其余加入loading buffer100度充分变性,再分装保存。
有些时候比蛋白酶抑制剂更有效。
另一个感觉就是样品中目的蛋白的量,限于WB的检测下限问题,一般目的蛋白量最好别低于1 ng(虽然ECL法下限是0.1ng,即100pg),最好在10ng以上为好。
这就要求在做WB之前必须充分考虑目的蛋白的可能丰度,最好充分的准备工作然后再设计细胞量或者组织量,千万别把样品搞稀了,否则就不好办了。
最后,还是多看文献,在做某个蛋白之前最好看看别人是如何做的,有什么特殊要求,内参如何选等等。
western blot失败经验总结2我就跑胶和转膜谈一点儿自己的体会1、跑胶电泳Buffer重复利用的时候要注意,不能混浊,有时候能用个3-4次,有时候第二次都不行了,事先前预电泳一下,看看气泡的均匀度;Buffer一定要淹过梳子孔,否则就会发现今天的蛋白不会跑;跑的时候先用低电压跑过浓缩胶,再换高电压跑分离胶,不要追求速度,慢慢跑条带会分离的更好,特别是对于高分子量的目的蛋白;2、转膜我用的是湿转,Buffer一定要预冷,最好提前一天配好放4度;滤纸不要大过PVDF膜,防止短路;胶在负极,膜靠近正极,放入Tank前检查一遍会使你免以体会放反的痛心疾首;膜要标记好正反面;转膜过程中,经常过去看看,防止意外,如电泳仪接触不好。
WESTERNBLOTTING心得1.总蛋白提取(胞膜,胞浆,胞核):组织匀浆后,15000g, 4度, 20分钟后,取上清.Buffer:NP40 750ul, 脱氧胆酸钠0.5克, SDS 0.1克, 加1*PBS 至100ml. 再加入PMSF(7.4mg/m l) 0.1ml. (现用现加)7.4mg/ml PMSP异丙醇溶液: 取A mg PMSF 加A/7.4 ml 异丙醇.工厂-20度储存2.组织膜蛋白提取:所在操作冰上进行(1)取组织,用PBS冲洗干净组织上的血液. 加入10ml Buffer A , 用剪刀尽可能剪碎组织,于冰上充分匀浆。
血泪教训,Westernblot最全避雷手册。

⾎泪教训,Westernblot最全避雷⼿册。
谨以此⽂,纪念那些年我做过的Western blot。
—— ⼀位不愿透露姓名的科研民⼯提起Western blot (WB),很多⼈都能哭诉半宿。
这是⼀个基础实验,也是⼀个让⼤家头疼的实验,简称“⽞学”。
话不多说,做过的都懂。
今天给⼤家提供WB最全避雷⼿册,希望⼤家能愉快实验,顺顺利利。
This is the dividing line.倾尽全⼒,长⽂预警1、WB有什么优点?答:灵敏,可达ng级,⽤ECL显⾊法可达pg级。
灵敏,相对便宜且特异性⾼。
2、为什么我的细胞提取液中没有⽬标蛋⽩?答:a) 你的细胞中并不表达这种蛋⽩质,换⼀种细胞或查⽂献弄清楚;b)你的细胞中的蛋⽩质被降解掉了,你必需加⼊PMSF,抑制蛋⽩酶活性;c) 你的抗体不能识别⽬的蛋⽩,检查抗体说明书,看是否有问题。
3、我的细胞提取液有的有沉淀,有的很清亮,为什么呢?答:a)有沉淀可能因为你的蛋⽩没有变性完全,可以适当提⾼SDS的浓度,同时将样品煮沸时间延长,⼀般需96℃以上10-15min左右;b) 也不排除你的抗原浓度过⾼,这时需再加⼊适量上样缓冲液。
4、我做的蛋⽩质分⼦量很⼩(10KD左右),请问怎么做WB?答:尽量选择0.2µm的膜,缩短转移时间;也可将两张膜叠在⼀起再电转。
5、我的⽬的带很弱,怎么加强?答:最主要是加⼤抗原上样量;同时也可以将⼀抗稀释⽐例上调。
6、胶⽚背景很脏,有什么解决⽅法?答:减少抗原上样量,降低⼀抗浓度,改变⼀抗孵育时间和温度(尽量4℃过夜,此孵育条件⽐37℃1h要温和很多),并提⾼封闭液浓度。
7、⽬标带是空⽩,周围有背景,是为什么?答:你的⼀抗浓度较⾼,⼆抗上HRP催化活⼒太强,同时你的显⾊底物处于⼀个临界点,反应时间不长,将周围底物催化完,形成了空⽩即“反亮现象”。
将⼀抗和⼆抗浓度降低,或更换新进⼝的显⾊底物。
8、我的胶⽚是⼀⽚空⽩,是怎么回事?答:如果能够排除下⾯的⼏个问题那么问题多半出现在⼀抗和抗原制备上。
Western blot常见14个问题解决
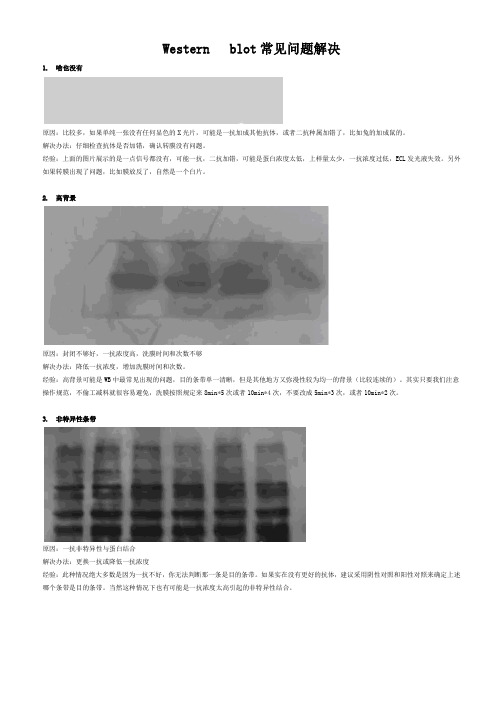
Western blot常见问题解决1. 啥也没有原因:比较多,如果单纯一张没有任何显色的X光片,可能是一抗加成其他抗体,或者二抗种属加错了,比如兔的加成鼠的。
解决办法:仔细检查抗体是否加错,确认转膜没有问题。
经验:上面的图片展示的是一点信号都没有,可能一抗,二抗加错,可能是蛋白浓度太低,上样量太少,一抗浓度过低,ECL发光液失效。
另外如果转膜出现了问题,比如膜放反了,自然是一个白片。
2. 高背景原因:封闭不够好,一抗浓度高,洗膜时间和次数不够解决办法:降低一抗浓度,增加洗膜时间和次数。
经验:高背景可能是WB中最常见出现的问题,目的条带单一清晰,但是其他地方又弥漫性较为均一的背景(比较连续的)。
其实只要我们注意操作规范,不偷工减料就很容易避免,洗膜按照规定来8min*5次或者10min*4次,不要改成5min*3次,或者10min*2次。
3. 非特异性条带原因:一抗非特异性与蛋白结合解决办法:更换一抗或降低一抗浓度经验:此种情况绝大多数是因为一抗不好,你无法判断那一条是目的条带。
如果实在没有更好的抗体,建议采用阴性对照和阳性对照来确定上述哪个条带是目的条带。
当然这种情况下也有可能是一抗浓度太高引起的非特异性结合。
4. 条带中出现边缘规则的白圈原因:电转中膜和胶之间存在气泡。
解决办法:转膜前去掉膜和胶之间的气泡经验:我们常常将电转液倒入一个盘子里,倒入的液体不能太多也不能太少,最好的高度是与放上第一层滤纸齐平,然后往滤纸上浇点转膜液,把电泳胶用清水清洗下,将电泳胶平铺到滤纸上,仔细检查滤纸与胶之间是否有气泡,可以左右前后观察,不同方向观察之后确认无气泡,然后再往胶上面浇点电转液,用两只手的拇指和食指轻轻夹住PVDF膜的两侧,使膜成U型,然后将U型的底部接触胶的中间,慢慢往两边放下膜,这样一般气泡很少。
然后上层滤纸同样用U型的放置方法,用玻璃棒稍微贴实下,然后盖上海绵。
注意不要来回赶气泡,这样反而会带入气泡。
Western Blot常见问题及解决方法

Western Blot(蛋白质印迹)技术是生物医学研究中常用的蛋白质分析技术之一,可以用于检测蛋白质样品中的特定抗原。
然而,在进行Western Blot实验时,可能会遇到一些问题,下面列举了一些常见问题及解决方法:1.电泳条带不清晰或无条带:解决方法:•检查样品质量,确保待检测样品是可用的,无降解或污染。
•调整电泳参数,如电压、电流和时间等,以优化电泳效果。
•更换抗体,确认抗体的特异性,避免非特异性结合。
2.膜上背景过重:解决方法:•在抗体孵育之前,使用封闭剂封闭膜上的非特异性位点,减少抗体非特异性结合。
•减少一抗和二抗的浓度,减轻非特异性结合。
•增加洗涤次数和时间,以清除未结合的抗体和蛋白质。
3.目的蛋白信号弱或无:解决方法:•调整一抗和二抗的浓度,提高特异性结合的信号强度。
•选择更灵敏的显色方法,如化学发光法等。
•在转膜前,优化样品处理,如提取、纯化和浓缩等步骤。
4.非特异性条带:解决方法:•检查抗体特异性,确认抗体的特异性,更换抗体以减少非特异性结合。
•检查样品处理步骤,避免样品中的杂蛋白干扰。
•增加洗涤次数和时间,以清除未结合的抗体和非特异性结合的蛋白质。
5.显影后膜上条带处变为黄色或白色:解决方法:•可能是由于曝光时间过长或显影液浓度过高导致的,可以降低曝光时间和减少显影液浓度来解决问题。
•在膜清洗时使用甲醇或乙醇等有机溶剂,清除膜上的多余抗体和显色剂。
6.条带内无显影的白点:解决方法:•在转膜前检查是否存在气泡,气泡会影响转膜效果和条带形成。
•增加转膜时间和电流,以促进蛋白质的转移。
•检查显影液是否过期或存在质量问题。
7.存在散在小圆斑:解决方法:•这可能是由于封闭液中的杂质颗粒导致的,可以通过过滤封闭液来清除杂质。
•检查一抗和二抗是否含有杂质抗体或存在抗体污染的情况,更换抗体可以解决问题。
•检查显色液是否存在问题,如过期或存在杂质等,更换显色液可以解决问题。
8.条带变形或不均匀:解决方法:•检查电泳胶的质量和制备过程是否存在问题。
western常见问题及解决方法

#
#
细胞和组织的处理
• 问题三.液氮研磨后离心样本不能分层 • 研磨的时候不充分,会形成絮状的液态
物质,如有手动匀浆器可以再进行处理
注意:
• 裂解时抑制剂的添加非常重要,做一般 的蛋白加入PMSF即可,个别蛋白需要 加Aprotinin, Leupeptin ,做磷酸化 的蛋白需要加入磷酸酶抑制剂 (cocktail)
#
电转
• 膜的选择还要考虑到目的蛋白分子 量的大小,如果目的蛋白分子量为 20KD以下,最好用0.22微米的膜, 大于20KD可以选择0.45微米的膜。
#
电转
• 问题二.电转时膜、滤纸、胶大小有何讲究?
如果是用的是半干转,顺序为:阴极-》滤纸 -》胶-》膜-》滤纸。滤纸的长宽分别比胶 小1-2mm,而膜的长宽分别比胶大1-2mm。
#
细胞和组织的处理
• 问题四.提取的蛋白样本如何保存最合适? 解答:温度越低越好 1、液氮 2、-80度冰箱 3、-30度冰箱 4、加入loading buffer煮后保存于-20度。
#
实验流程
蛋白提取 √ SDS凝胶配制
电泳 电转 封闭及一抗 显色
#
SDS凝胶配制
• 问题一.胶凝的效果不好是什么原因? 1、AP时间过长,重新配制AP 2、可以适当多加入TEMED 3、室温低 4、混合不均匀
#
电泳
• 问题二.跑电泳的时候配的胶总是“缩” 是什么原因呢?是有的成分不对吗? 胶里的水分被蒸发了。过夜时拿保鲜膜 包起来,在里面加点水保持湿度就可以 了。 也可能母液(30%聚丙烯酰胺)有问题, 你可以重新配制一份观察
Western blotting 常见问题分析

Western blotting 常见问题分析SDS-PAGE凝胶电泳常见的问题分析Western Blotting实验流程第一步:蛋白样品制备蛋白抽提质量的好坏直接决定着Western结果的好坏,因此,根据标本类型和检测类型选择合适的蛋白制备方法是至关重要的。
作为沃尔森的优势产品,我们为您提供RIPA改良型试剂盒以提取细胞或组织的总蛋白。
RIPA改良型试剂盒(WB0001,WB0002)中提供蛋白酶抑制剂、磷酸酶抑制剂、裂解缓冲液Lysis Buffer、PMSF等,可在非变性条件下从哺乳动物组织和培养细胞中提取总蛋白,获得的总蛋白可用于Western Blotting、免疫共沉淀等后续研究。
第二步:蛋白定量为确保每个蛋白样品的上样量一致,收集的蛋白样品均应测定蛋白浓度。
蛋白浓度的测定方法,应根据组织细胞裂解或匀浆所使用的方法及裂解液的不同选用,因为去垢剂和还原剂等对不同蛋白浓度测定方法的影响差别很大。
如使用沃尔森公司生产的RIPA改良型试剂盒,建议您使用BCA法测定蛋白浓度(WB0003,WB0004)。
第三步:电泳(1) SDS-PAGE凝胶配制沃尔森的SDS-PAGE凝胶配制试剂盒(WB0006)提供了所需的试剂以及配制各种浓度SDS-PAGE的配方,我们也为您提供了制胶所需的各种组分:30% Acr-Bis(29:1)(WB0014); 40% Acr-Bis(39:1)(WB0015);PMSF(100mM)(WB0016);0.5M Tris-HCl,pH6.8(WB0017); 1.5M Tris-HCl,pH8.8(W0018)。
(2) 样品处理每80μL样品加入20μL的5×SDS-PAGE蛋白上样缓冲液(WB0005),混匀。
置于100℃的水浴加热10min,14000g离心20min,取上清液进行电泳。
(3) 上样与电泳第四步:转膜转膜缓冲液可以参考《分子克隆》自行配制。
Westernblot离完美总差一步,可能因这7个小误区

Westernblot离完美总差一步,可能因这7个小误区转载请注明:解螺旋·临床医生科研成长平台遥想当年,小鱼初接触WB时,只顾一味瞎折腾,完美地避开了所有正确操作,以至于每次做完该实验,就觉得自己的血槽已经被耗光了。
好在多次与WB斗智斗勇后,也积攒了不少经验,这就将wb中常见的7大错误分享给大家,与君共勉。
1乌糟糟的蛋白样品“良好的开始是成功的一半”,其实,对于WB这座大厦而言,好的蛋白样品的准备无疑是打下了良好的地基,而坏的样品则奠定了“豆腐渣”工程的基调。
因而,正确的蛋白质提取和样品制备是至关重要的。
尽管每个实验室都有相应的样品准备方法,但做实验的小伙伴在制备蛋白样品时仍需要考虑以下因素:盐离子浓度、缓冲液种类、抑制剂、还原剂和加热时间等。
一个最佳Protocol应该最大程度上提高蛋白质的溶解度和稳定性,有助于WB更好得可视化目的蛋白。
2失败的凝胶制备上样前,要谨记完美的凝胶和好的样品才是最配的,不然只能使之前的种种努力付之东流。
为了避免前功尽弃,在使用SDS-PAGE凝胶前需要仔细检查并警惕以下情况出现:1)凝胶中不仅出现气泡,而且孔道之间并不均一,这会严重影响蛋白质在凝胶中的迁移。
2)当凝胶边缘开始收缩时,说明凝胶已经变得非常干燥,需要重新配制。
3)凝胶凝固时间太短,仍处于可以流动状态。
为了确保凝胶完全凝固,可将胶轻轻倾斜来进行检查。
3转膜中的层层“陷阱”WB中,转膜是对Western最终结果影响最大的一步,转膜质量的好坏直接决定了实验结果的好坏。
那么转膜中又存在哪些容易被人忽视的陷阱存在呢?1)徒手抓蛋白转移膜。
毫无疑问,此时手指上的油脂与蛋白会封闭转移膜,容易产生背景污斑影响实验结果。
因此建议实验全程要用手套及镊子。
2)忽略了分离胶的上下方向以及膜的正反面。
通常使用预染Marker可解决此问题,此外还可以将胶和膜的一角剪掉来进行区分。
3)转移膜和凝胶不幸干涸了。
一般转膜前可将凝胶放在缓冲液里平衡一下防止胶变形,也有助于进一步去掉有碍转膜的杂质。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
western blot失败经验总结1我做了很久的WB,最大的一个难点感觉在于样品制备上!蛋白的降解有些时候是你想象不到的,可能出奇的糟糕,也可能完全没事~~如果单独做WB的话,感觉以“快速彻底裂解,先变性后保存”的原则比较有效,尽量选择足够强的裂解液,甚至直接给loading buffer裂解,然后取出部分蛋白定量,其余加入loading buffer100度充分变性,再分装保存。
有些时候比蛋白酶抑制剂更有效。
另一个感觉就是样品中目的蛋白的量,限于WB的检测下限问题,一般目的蛋白量最好别低于1 ng(虽然ECL法下限是0.1ng,即100pg),最好在10ng以上为好。
这就要求在做WB之前必须充分考虑目的蛋白的可能丰度,最好充分的准备工作然后再设计细胞量或者组织量,千万别把样品搞稀了,否则就不好办了。
最后,还是多看文献,在做某个蛋白之前最好看看别人是如何做的,有什么特殊要求,内参如何选等等。
western blot失败经验总结2我就跑胶和转膜谈一点儿自己的体会1、跑胶电泳Buffer重复利用的时候要注意,不能混浊,有时候能用个3-4次,有时候第二次都不行了,事先前预电泳一下,看看气泡的均匀度;Buffer一定要淹过梳子孔,否则就会发现今天的蛋白不会跑;跑的时候先用低电压跑过浓缩胶,再换高电压跑分离胶,不要追求速度,慢慢跑条带会分离的更好,特别是对于高分子量的目的蛋白;2、转膜我用的是湿转,Buffer一定要预冷,最好提前一天配好放4度;滤纸不要大过PVDF膜,防止短路;胶在负极,膜靠近正极,放入Tank前检查一遍会使你免以体会放反的痛心疾首;膜要标记好正反面;转膜过程中,经常过去看看,防止意外,如电泳仪接触不好。
WESTERNBLOTTING心得1.总蛋白提取(胞膜,胞浆,胞核):组织匀浆后,15000g, 4度, 20分钟后,取上清.Buffer:NP40 750ul, 脱氧胆酸钠0.5克, SDS 0.1克, 加1*PBS 至100ml. 再加入PMSF(7.4mg/m l) 0.1ml. (现用现加)7.4mg/ml PMSP异丙醇溶液: 取A mg PMSF 加A/7.4 ml 异丙醇.工厂-20度储存2.组织膜蛋白提取:所在操作冰上进行(1)取组织,用PBS冲洗干净组织上的血液. 加入10ml Buffer A , 用剪刀尽可能剪碎组织,于冰上充分匀浆。
每次30秒,重复3-5次.(2)离心机1000rpm,4℃离心10min 后,所得上清液转入超速离心管。
(去掉大块组织及结缔组织)(3)100000g,4℃离心1hr, 弃去上清.(此为胞浆蛋白,若只提取胞浆蛋白,可用10000rpm, 4度, 30分钟离心,取上清即可)。
沉淀用适量的Buffer B重悬,4度过夜后,分装至EP管,Epp endorf 台式离心机10000rpm,4℃离心30min。
(4)收集所得上清液即为膜组份。
Buffer A:0.32M 蔗糖,5mM Tris-HCl(PH 7.5),120mM KCl,1mM EDTA, 1mM EGTA, 0.2mM PMSF, 1ug/ml Leupeptin, 1ug/ml Pepstatin A, 1ug/ml Aprotinin。
冰上预冷。
Buffer B 20mM HEPES(PH 7.5),10% 甘油,2% Triton X-100, 1mM EDTA, 1mM EGTA, 0.2mM,PMSF, 1ug/ml Leupeptin, 1ug/ml Pepstatin A, 1ug/ml Aprotinin。
冰上预冷。
所得蛋白分装EP管(每管约50ul),取一管测蛋白浓度,其它放入-70度深冻冰箱保存。
如果要定量的话,最好能立即根据浓度,加入上样缓冲,煮沸,4度保存。
反复冻融蛋白会降解,浓度不准。
考马斯亮兰G250测蛋白浓度:考马斯亮兰G250配方:G250 0.4克, 95%乙醇100ml, 85% 磷酸200ml, 加水至2000ml,过滤后使用。
BSA标准液:0.5mg/ml BSA, 双蒸水配制。
配制标准液(浓度分别为0,10,20,30,40,50ug/ul):1 2 3 4 53 6G250 3ml 3ml 3ml 3ml 3ml 3ml0.5mg/mlBSA ------ 20ul 40ul 60ul 80ul 100ulddH2O 100ul 80ul 60ul 40ul 20ul -----待测蛋白配液:每管3ml G250,2ul样品,98ul ddH2O。
(若加入样品后,液体没有变蓝色,则可再加2ul样品,ddH2O减为96 ul,标准液浓度可适当扩大。
)紫外分光光度计,595nm,用1号管调零,制出标准曲线, 标准方程及R值(R值应大于0.99, 否则操作误差较大),测量待测蛋白OD值,算出蛋白浓度。
(若加入2ul样品,则浓度值需除2,方为真正浓度;若加4ul样品,则除4)聚丙烯酰胺电泳:1.自来水,洗涤剂清洗玻璃板,用ddH2O冲洗干净,立放于盒子内,60度烤箱烘干备用。
2.配胶:凝胶储备液(30% Acr/Bise):丙烯酰胺29克,双体甲叉双丙烯酰胺1克,加水至100ml。
(有说要棕色瓶保存,3个月换新的,30%的丙烯酰胺如有沉淀,最好弃掉)10%APS:0.1克过硫酸铵,加ddH2O至1 ml.(4度保存,一周内使用),本人都是配新鲜的用。
10%SDS: 1克SDS,10 ml ddH2O. (不溶时,可加热50度左右助溶.)TEMED: 原液即可.0.5M Tris-HCl (PH6.8);1.5M Tris-HCl (PH8.8): 9.0855克Tris, 加入ddH2O溶解后,用HCL调PH至8.8,加ddH2O到50 ML配胶的浓度和方法书上都有,不详细说了!本人做的是70KD 和17KD 的分子量,分别配了10%和12%的分离胶,4%的浓缩胶。
摇动溶液充分混合(不要过于剧烈以免引入过多地氧气)灌入2/3的分离胶后(估计距梳子底1CM)应立即用ddH2O封胶。
等到能看清水和胶的分界线,则可配浓缩胶。
用面巾纸吸干水后,注入浓缩胶,至顶。
插入梳子,不要有气泡。
蓝色字体是从别人贴子上看到的这些小孔的孔径随“双丙烯酰胺~丙烯酰胺” 比率的增加而变小,比率接近1:20 时孔径达到最小值。
SDS聚丙烯酰胺凝胶大多按“双丙烯酰胺~丙烯酰胺”为1:29 配制,试验表明它能分离大小相差只有3% 的蛋白质。
分离胶及浓缩胶均可事先配好(除AP及TEMED外),过滤后作为储存液避光存放于4℃,可至少存放1个月,临用前取出室温平衡(否则凝胶过程产生的热量会使低温时溶解于储存液中的气体析出而导致气泡,有条件者可真空抽吸3分钟),加入10%AP(0.7~0.8:100, 分离胶浓度越高AP浓度越低,15%的分离胶可用到0.5:100)及TEMED(分离胶用0.4:1000, 15%的可用到0. 3:1000,浓缩胶用0.8:1000)PAGE配胶的试剂和配方比例对电泳结果的质量当然有决定性的影响,这个配胶的比例,在《分子克隆》上有详细的论述。
容易忽视的问题主要在于过硫酸铵(AP)一定要新鲜——最好用小指管配AP(写日期)保存在-20度,超过2周的AP扔掉算了,或者已经反复打开使用多次的A P都别用。
胶浓度<10%时可用0.1%的SDS封,浓度>10%时用水饱和的异丁醇或异戊醇,也可以用0.1%的SDS),封胶后切记,勿动。
如用醇封胶需用大量清水及ddH2O冲洗干净,然后加少量0.1%的SDS,目的是通过降低张力清除残留水滴。
等浓缩胶凝的时间,就可以配蛋白了。
按照想要的浓度加入LOADING BUFFER配制。
本人按每孔总体积20ul, 终浓度1* RSB,含50ug蛋白混合样品,不足的体积用1* PBS补足。
短暂离心后,沸水中煮5分钟,放室温,用时再短暂离心。
例如. 50ug/20ul,做2.5孔, 即50ul,125ug1 2 3 4蛋白浓度 10.33 ug/ ul 13.91 ug/ ul 14.87 ug/ ul 19.03 ug/ ul所需体积 12.1 ul 9 ul 8.4 ul 6.6 ul5*RSB 10 ul 10 ul 10 ul 10 ul1* PBS 27.9 ul 31 ul 31.6 ul33.4 ul5*RSB: 1.89克Tris-HCL(PH 6.8), 5克SDS, 0.125克溴酚蓝, 25ML 甘油.(很粘稠)Beta-巯基乙醇用时现加,按25%比例.10* PBS: Nacl 4.25克,Na2HPO4. 12H2O 15.4g, NaH2PO4 2H2O 1.4g, 调PH到7.2-7.4,加ddH 2O到500ML约30分钟左右, 取下梳子,将胶板放入电泳槽内,加入1*电泳缓冲液, 冲洗加样孔. 微量进样器逐个加样,空泳道加入1* RSB. 两胶板之间的电泳液要满. 接好正负极, 开始电泳, 可先小电压,约50V ,大约跑过浓缩胶后再改为100V. 观察MARKER的情况, 适时终止电泳.建议说目的带要跑到分离胶的2/3处比较好,但本人一直都在上1/3处,结果也还可以.预染MARKER很重要,开始选了一个国产的,总是跑不好,后来换成NEB的,效果很好,而且也不贵,范围也比较广,把我要的7.,42.17全都包括在内.如果浓缩胶跑的好的话, 几个孔的蛋白会跑成一条直线.注意加样时间要尽量短,以免样品扩散,为避免边缘效应,可在未加样的孔中加入等量的样品缓冲液未聚合的丙烯酰胺具有神经毒性,操作时应该戴手套防护.梳子插入浓缩胶时,应确保没有气泡,可将梳子稍微倾斜插入以减少气泡的产生,梳子拔出来时应该小心,不要破坏加样孔,如有加样孔上的凝胶歪斜可用枕头插入加样孔中纠正但要避免枕头刺入胶内.10* 电泳缓冲液: Tris7.58克, 甘氨酸36克, SDS 2.5克, ddH2O定容到250ML.不好溶,可用搅拌器.可重复使用,每次加一些新的进去.但本人每次都是配新的,也不麻烦.转膜跑完电泳后,取出胶,可以一块做考马思亮蓝R250染色约30分钟, 脱色液脱色, 看看蛋白条带跑的如何,上样量如何.另一块可用做转膜. 放入转移缓冲液中摇30分钟左右(有次着急只晃了10分钟,结果也挺好的)。
开始用的PVDF膜, 0.45um, 用国产的MARKER后,总是染的不清楚, 而且无论时间长短, 最后一块滤纸都会被染色. 后来换了NEB后,PVDF也不容易上色(可能和PVDF的质量有关,我买的虽然也是MILLIPORE,但好像比较便宜20CM*10CM才50块钱). 丽春红染色也很少能看清条带,后来发现,染色后用甲醇洗,比较容易看出来,但是很难洗掉红色,不知道有没有影响。