N2在Pd金属表面的吸附行为
n2 物理吸附-脱附表征

n2 物理吸附-脱附表征
物理吸附-脱附表征是指利用吸附-脱附技术来研究材料的表面
性质和孔隙结构。
这种技术主要用于研究吸附剂、催化剂和多孔材
料等。
在物理吸附-脱附表征中,常用的技术包括氮气吸附法(BET 法)、氩气吸附法、比表面积测定法、孔体积测定法等。
首先,物理吸附-脱附表征可以通过氮气吸附法来评估材料的比
表面积。
氮气吸附法是利用氮气在不同相对压力下吸附到材料表面
的原理来测定材料的比表面积。
通过绘制吸附等温线和脱附等温线,可以计算出材料的比表面积,进而了解材料的表面活性和孔隙结构。
其次,物理吸附-脱附表征也可以通过氩气吸附法来评估材料的
孔体积。
氩气吸附法利用氩气分子在不同相对压力下进入材料孔隙
的原理,来测定材料的孔体积分布。
通过分析吸附等温线和脱附等
温线的形状,可以得到材料的孔体积分布信息,从而了解材料的孔
隙结构特征。
另外,物理吸附-脱附表征还可以结合比表面积测定法和孔体积
测定法来全面评估材料的吸附性能。
比表面积和孔体积是影响材料
吸附性能的重要因素,通过综合分析两者的数据,可以更全面地了
解材料的吸附-脱附特性,为材料的应用提供重要参考。
总的来说,物理吸附-脱附表征是一种重要的材料表征技术,通过测定材料的比表面积和孔体积等参数,可以全面了解材料的表面性质和孔隙结构特征,为材料的研究和应用提供重要的参考依据。
n2吸附-解吸

n2吸附-解吸N2吸附-解吸是指氮气(N2)在固体表面上的吸附和解吸过程。
氮气是地球大气中的主要成分之一,也是许多工业过程中不可或缺的重要气体。
了解N2在固体表面上的吸附-解吸行为对于理解气体的吸附性质、催化反应和材料科学等领域具有重要意义。
N2吸附-解吸的过程可以通过实验或模拟方法进行研究。
实验方法主要包括吸附等温线测量和解吸过程的动力学研究。
吸附等温线测量是通过在不同温度下将氮气暴露在固体表面上,然后测量吸附气体的量来确定吸附等温线。
吸附等温线的形状和斜率可以提供有关吸附过程的信息。
动力学研究则通过跟踪吸附和解吸速率随时间的变化来研究吸附-解吸过程的动力学行为。
N2吸附-解吸性质的研究对于理解气体在材料表面上的相互作用和吸附机制至关重要。
吸附过程的动力学行为可以揭示吸附过程的速率控制步骤,从而帮助我们设计更高效的吸附材料。
此外,N2吸附-解吸性质的研究可以用于评估材料的孔隙结构和比表面积。
通过测量吸附等温线的形状和斜率,可以计算出材料的比表面积和孔隙容积,这对于材料科学和催化反应的研究具有重要意义。
N2吸附-解吸的研究还可以帮助我们理解气体在纳米尺度上的行为。
纳米材料具有较大的比表面积和较高的吸附活性,因此对于纳米材料上N2吸附-解吸过程的研究对于理解纳米材料的性质和应用具有重要意义。
通过研究N2在纳米材料上的吸附和解吸行为,可以揭示纳米材料的表面化学性质和吸附机制,为纳米材料的设计和应用提供指导。
除了对气体吸附性质的研究,N2吸附-解吸还在环境和能源领域具有重要应用。
例如,N2吸附-解吸可以用于气体分离和储存,在石油和天然气开采中起到重要作用。
此外,N2吸附-解吸还可以用于制备催化剂和吸附剂,促进化学反应的进行。
N2吸附-解吸是氮气在固体表面上的吸附和解吸过程。
通过研究N2吸附-解吸的性质,我们可以深入了解气体在材料表面上的相互作用、吸附机制和纳米材料的性质。
此外,N2吸附-解吸还在环境和能源领域具有重要应用。
氧气分子在金纳米颗粒表面的吸附行为研究

氧气分子在金纳米颗粒表面的吸附行为研究金纳米颗粒是一种具有广泛应用前景的纳米材料,其物理和化学特性与单个金原子和金离子有显著的不同。
金纳米颗粒的表面经常被表面活性物质(例如硫化物分子)覆盖,以控制它们的尺寸、形状和活性。
最近的研究发现,氧气分子可以在包含硫化物单层的金纳米颗粒表面上吸附。
本文将介绍这个现象和其可能的应用。
氧气的吸附行为在光催化、被动氧化和其他许多技术中起着至关重要的作用。
氧气在某些金属表面可以与自由电子相互作用,并形成金属氧化物。
这种氧化作用可以将有害气体和有机物转化为无害的物质。
金纳米颗粒在气相反应中也可以具有类似的氧化功能。
研究表明,氧气可以通过物理吸附和化学吸附两种方式在金纳米颗粒上吸附。
物理吸附是相互作用力较弱的过程,吸附分子与固体表面之间只有范德华力和静电力。
化学吸附是一种强化学键形成的过程,分子和固体表面上的原子或离子之间的化学键力是相互作用的核心。
在金纳米颗粒表面上,氧气的化学吸附是难以实现的,因为氧气分子的反键轨道(LUMO)的能级高于金作为电子给体的轨道(例如d轨道)的能级。
因此,氧气对金纳米颗粒表面的亲和力相对较小。
然而,在包含硫化物分子的金纳米颗粒表面上形成了一种特殊结构,其自由电子密度较高,因而可以提高氧气与金纳米颗粒之间的相互作用。
硫化物分子是限制金纳米颗粒氧化的关键因素之一。
硫化物分子将金纳米颗粒表面和气体环境之间隔开,防止气体与金纳米颗粒之间形成凝聚反应。
然而,在氧气存在的条件下,硫化物分子可以发生位移,并使金纳米颗粒表面的自由电子密度增加。
这样,氧气就可以通过物理吸附和氧化反应与金纳米颗粒表面相互作用。
然而,氧气分子与硫化物分子之间也存在相互作用。
例如,氧气中的自由电子可以与硫化物分子进行电子转移,形成硫化物阴离子和超氧根离子。
在此情况下,氧气的化学吸附和金纳米颗粒的氧化反应可以增强,从而影响金纳米颗粒的光学、电学和催化性能。
总的来说,金纳米颗粒表面上氧气分子的吸附行为是一个重要的研究领域,在环境监测、催化合成和其他技术中具有广泛应用前景。
氮吸附原理

动态氮吸附孔径分布测试的原理和方法许多超细粉体材料的表面是不光滑的,甚至专门设计成多孔的,而且孔的尺寸大小、形状、数量与它的某些性质有密切的关系,例如催化剂与吸附剂,因此,测定粉体材料表面的孔容、孔径分布具有重要的意义。
国际上,一般把这些微孔按尺寸大小分为三类:孔径£ 2nm为微孔,孔径=2~50nm为中孔,孔径350nm为大孔,其中中孔具有最普遍的意义。
用氮吸附法测定孔径分布是比较成熟而广泛采用的方法,它是用氮吸附法测定BET比表面的一种延伸,都是利用氮气的等温吸附特性曲线:在液氮温度下,氮气在固体表面的吸附量取决于氮气的相对压力(P/F0), P为氮气分压,P o为液氮温度下氮气的饱和蒸汽压;当P/P0在0.05~0.35范围内时,吸附量与(P/P o)符合BET方程,这是氮吸附法测定粉体材料比表面积的依据;当P/P030.4时,由于产生毛细凝聚现象,即氮气开始在微孔中凝聚,通过实验和理论分析,可以测定孔容、孔径分布。
所谓孔容、孔径分布是指不同孔径孔的容积随孔径尺寸的变化率。
所谓毛细凝聚现象是指,在一个毛细孔中,若能因吸附作用形成一个凹形的液面,与该液面成平衡的蒸汽压力P必小于同一温度下平液面的饱和蒸汽压力P。
,当毛细孔直径越小时,凹液面的曲率半径越小,与其相平衡的蒸汽压力越低,换句话说,当毛细孔直径越小时,可在较低的P/F0压力下,在孔中形成凝聚液,但随着孔尺寸增加,只有在高一些的P/P0 压力下形成凝聚液,显而易见,由于毛细凝聚现象的发生,将使得样品表面的吸附量急剧增加,因为有一部分气体被吸附进入微孔中并成液态,当固体表面全部孔中都被液态吸附质充满时,吸附量达到最大,而且相对压力P/P D也达到最大值1。
相反的过程也是一样的,当吸附量达到最大(饱和)的固体样品,降低其相对压力时,首先大孔中的凝聚液被脱附出来,随着压力的逐渐降低,由大到小的孔中的凝聚液分别被脱附出来。
设定粉体表面的毛细孔是圆柱形管状,把所有微孔按直径大小分为若干孔区,这些孔区按大到小的顺序排列,不同直径的孔产生毛细凝聚的压力条件不同,在脱附过程中相对压力从最高值(P o)向下降低时,先是大孔后再是小孔中的凝聚液逐一脱附出来,显然可以产生凝聚现象或从凝聚态脱附出来的孔的尺寸和吸附质的压力有一定的对应关系,凯尔文方程给出了这个关系:r k = -0.414 log(P/P)................................................ ⑴r K叫凯尔文半径,它完全取决于相对压力P/F0,即在某一P/P o下,开始产生凝聚现象的孔的半径,同时可以理解为当压力低于这一值时,半径为r K 的孔中的凝聚液将气化并脱附出来。
n2 tpd物理化学吸附

n2 tpd物理化学吸附物理化学吸附是指气体分子在固体表面附着的一种吸附现象。
其中,N2 TPD(Temperature Programmed Desorption)是一种常用的实验方法,用于研究气体分子在固体表面的吸附和解吸过程。
N2 TPD实验通常是通过在固体表面吸附N2分子,然后通过升温来观察N2分子的解吸行为。
实验过程中,首先将固体样品置于低温下,使N2分子吸附到固体表面。
然后,通过升温,提高固体样品的温度,使吸附的N2分子逐渐解吸并从固体表面脱附。
解吸的N2分子会通过质谱仪等检测手段进行实时监测,从而得到N2 TPD曲线。
N2 TPD曲线可以提供关于固体表面吸附态和解吸态的重要信息。
曲线上的峰值对应着不同类型的吸附态和解吸态,通过对峰值的位置、峰高和峰形进行分析,可以推断出吸附态和解吸态的性质和特点。
例如,峰值的位置可以用来确定吸附态和解吸态的能量,峰高可以用来估计吸附态和解吸态的覆盖度,峰形可以用来判断吸附态和解吸态的均匀性和分布情况。
N2 TPD实验可以用于研究各种固体材料的表面性质和吸附行为。
例如,通过对催化剂的N2 TPD曲线进行分析,可以了解催化剂表面的活性位点和吸附态,从而优化催化剂的设计和性能。
此外,N2 TPD 还可以用于研究各种气体分子在固体表面的吸附和解吸过程,探索气体分子与固体表面之间的相互作用机制。
然而,需要注意的是,N2 TPD实验只能提供吸附和解吸过程的宏观信息,无法直接观察到分子尺度上的吸附和解吸行为。
因此,在对N2 TPD实验结果进行解释和分析时,需要结合其他表征手段,如X 射线衍射、扫描电子显微镜等,来获取更全面和准确的信息。
N2 TPD物理化学吸附是一种常用的实验方法,用于研究气体分子在固体表面的吸附和解吸行为。
通过对N2 TPD曲线的分析,可以了解固体表面的吸附态和解吸态的性质和特点,从而深入研究固体材料的表面性质和吸附行为。
这为优化催化剂设计和气体分子与固体表面相互作用机制的研究提供了重要的实验手段。
N2吸脱附曲线说明
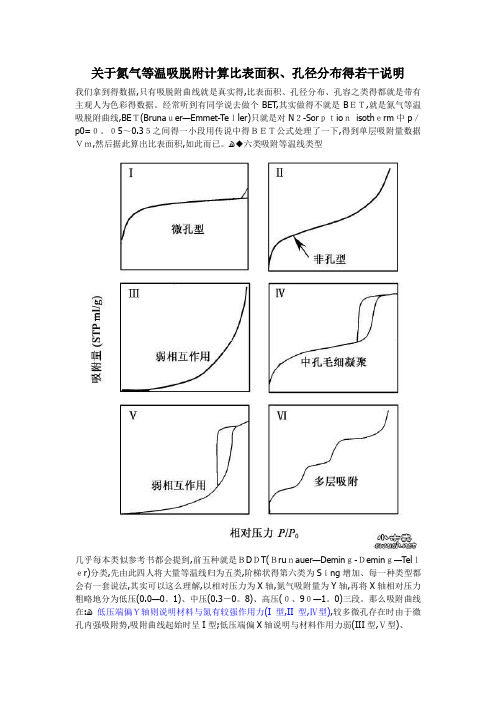
关于氮气等温吸脱附计算比表面积、孔径分布得若干说明我们拿到得数据,只有吸脱附曲线就是真实得,比表面积、孔径分布、孔容之类得都就是带有主观人为色彩得数据。
经常听到有同学说去做个BET,其实做得不就是BET,就是氮气等温吸脱附曲线,BET(Brunauer—Emmet-Teller)只就是对N2-Sorptionisotherm中p/p0=0。
05~0.35之间得一小段用传说中得BET公式处理了一下,得到单层吸附量数据Vm,然后据此算出比表面积,如此而已。
ﻫ◆六类吸附等温线类型几乎每本类似参考书都会提到,前五种就是BDDT(Brunauer—Deming-Deming—Teller)分类,先由此四人将大量等温线归为五类,阶梯状得第六类为Sing增加、每一种类型都会有一套说法,其实可以这么理解,以相对压力为X轴,氮气吸附量为Y轴,再将X轴相对压力粗略地分为低压(0.0—0。
1)、中压(0.3-0。
8)、高压(0、90—1。
0)三段。
那么吸附曲线在:ﻫ低压端偏Y轴则说明材料与氮有较强作用力(І型,ІІ型,Ⅳ型),较多微孔存在时由于微孔内强吸附势,吸附曲线起始时呈І型;低压端偏X轴说明与材料作用力弱(ІІІ型,Ⅴ型)、中压端多为氮气在材料孔道内得冷凝积聚,介孔分析就来源于这段数据,包括样品粒子堆积产生得孔,有序或梯度得介孔范围内孔道。
BJH方法就就是基于这一段得出得孔径数据;ﻫ高压段可粗略地瞧出粒子堆积程度,如І型中如最后上扬,则粒子未必均匀。
平常得到得总孔容通常就是取相对压力为0。
99左右时氮气吸附量得冷凝值。
◆几个常数※液氮温度77K时液氮六方密堆积氮分子横截面积0。
162平方纳米,形成单分子层铺展时认为单分子层厚度为0。
354nmﻫ※标况(STP)下1mL氮气凝聚后(假定凝聚密度不变)体积为0、001547mLﻫ例:如下面吸脱附图中吸附曲线p/p0最大时氮气吸附量约为400 mL,则可知总孔容=400*0、001547=400/654=约0、61mL※STP每mL氮气分子铺成单分子层占用面积4。
n2吸附脱附

氮气等温吸脱附计算比★★注意★★我们拿到的数据,只有吸脱附曲线是真实的,比表面积、孔径分布、孔容之类的都是带有主观人为色彩的数据。
经常听到有同学说去做个BET,其实做的不是BET,是氮气等温吸脱附曲线,BET(Brunauer-Emmet-Teller)只是对N2-Sorption isotherm中p/p0=~之间的一小段用传说中的BET公式处理了一下,得到单层吸附量数据Vm,然后据此算出比表面积,如此而已。
◆六类吸附等温线类型几乎每本类似参考书都会提到,前五种是BDDT(Brunauer-Deming-Deming-Teller)分类,先由此四人将大量等温线归为五类,阶梯状的第六类为Sing增加。
每一种类型都会有一套说法,其实可以这么理解,以相对压力为X轴,氮气吸附量为Y轴,再将X轴相对压力粗略地分为低压()、中压、高压()三段。
那么吸附曲线在:低压端偏Y轴则说明材料与氮有较强作用力(І型,ІІ型,Ⅳ型),较多微孔存在时由于微孔内强吸附势,吸附曲线起始时呈І型;低压端偏X轴说明与材料作用力弱(ІІІ型,Ⅴ型)。
中压端多为氮气在材料孔道内的冷凝积聚,介孔分析就来源于这段数据,包括样品粒子堆积产生的孔,有序或梯度的介孔范围内孔道。
BJH方法就是基于这一段得出的孔径数据;高压段可粗略地看出粒子堆积程度,如І型中如最后上扬,则粒子未必均匀。
平常得到的总孔容通常是取相对压力为左右时氮气吸附量的冷凝值。
◆几个常数※液氮温度77K时液氮六方密堆积氮分子横截面积平方纳米,形成单分子层铺展时认为单分子层厚度为※标况(STP)下1mL氮气凝聚后(假定凝聚密度不变)体积为例:如下面吸脱附图中吸附曲线p/p0最大时氮气吸附量约为400 mL,则可知总孔容=400*=400/654=约※ STP每mL氮气分子铺成单分子层占用面积平方米例:BET方法得到的比表面积则是S/(平方米每克)=*Vm,其中Vm由BET方法处理可知Vm=1/(斜率+截距)◆以SBA-15分子筛的吸附等温线为例加以说明此等温线属IUPAC 分类中的IV型,H1滞后环。
一氧化碳化学吸附法测定金属pd表面积

一氧化碳化学吸附法测定金属Pd表面积一氧化碳化学吸附法是一种常用的测定金属Pd(钯)表面积的方法。
在化学学科中,金属表面积的测定是一个重要的课题,因为表面积的大小直接关系到金属催化剂的活性和选择性。
在本文中,我们将深入探讨一氧化碳化学吸附法测定金属Pd表面积的原理、方法以及实际应用。
一氧化碳(CO)是一种常见的气体,它在金属表面上具有很强的吸附作用。
一氧化碳化学吸附法就是利用一氧化碳在金属Pd表面上的吸附行为来测定金属Pd的表面积。
在此方法中,首先将金属Pd样品暴露于一氧化碳气氛中,一氧化碳会吸附在金属Pd表面上形成一层薄膜。
通过测定一氧化碳吸附前后金属Pd的质量变化,可以计算出金属Pd 的表面积。
在实际测定中,一氧化碳化学吸附法需要精密的实验装置和严格的操作流程。
需要准备高纯度的金属Pd样品,并在实验室中搭建好气体吸附装置。
接下来,将金属Pd样品置于固定的温度和压力下,经过一定时间与一氧化碳气氛接触,再利用高真空系统进行一氧化碳的脱附,最后测定金属Pd样品的质量变化,就可以得到金属Pd的表面积。
一氧化碳化学吸附法测定金属Pd表面积具有一定的优势和局限性。
优势在于这种方法测定简单、可靠性高,且适用于不同形貌和晶面的金属Pd样品。
然而,由于实验条件的要求较高,仪器设备和操作技术的要求也较高,使得这种方法相对来说操作复杂,需要较为专业的技术人员进行操作。
一氧化碳化学吸附法是一种重要的测定金属Pd表面积的方法,它通过测定一氧化碳在金属Pd表面上的吸附行为来计算金属Pd的表面积。
这一方法具有一定的优势和局限性,但无疑对金属Pd表面积的测定提供了一种重要的手段,为金属催化剂的表征和研究提供了重要的支持。
至此,我们对一氧化碳化学吸附法测定金属Pd表面积的原理、方法以及实际应用有了一个全面的了解。
希望本文能够帮助你更深入地理解这一领域的知识,并为你在相关研究和实验中提供一些参考。
我们来探讨一下一氧化碳化学吸附法测定金属Pd表面积的实际应用。
氮气吸附-2

氮气因其易获得性和良好的可逆吸附特性, 成为最常用的吸附质。通过这种方法测定的比 表面积我们称之为“等效”比表面积,所谓 “等效”的概念是指:样品的比表面积是通过 其表面密排包覆(吸附)的氮气分子数量和分 子最大横截面积来表征。实际测定出氮气分子 在样品表面平衡饱和吸附量(V),通过不同 理论模型计算出单层饱和吸附量(Vm),进 而得出分子个数,采用表面密排六方模型计算 出氮气分子等效最大横截面积(Am),即可求出 被测样品的比表面积。
固体表面吸附
固体表面上的原子或分子与液体一样,受力也 是不均匀的,而且不像液体表面分子可以移动,通 常它们是定位的。 固体表面是不均匀的,即使从宏观上看似乎很 光滑,但从原子水平上看是凹凸不平的。 同种晶体由于制备、加工不同,会具有不同的 表面性质,而且实际晶体的晶面是不完整的,会有 晶格缺陷、空位和位错等。 正由于固体表面原子受力不对称和表面结构不 均匀性,它可以吸附气体或液体分子,使表面自由 能下降。而且不同的部位吸附和催化的活性不同。
这公式称为 Langmuir吸附等温式,式中 b称为吸附系数,它的大小代表了固体表面吸 附气体能力的强弱程度。
以q 对p 作图, 得:
1.当p很小,或吸附很弱时,bp<<1,q = bp, q 与 p 成线性关系。 2.当p很大或吸附很 强时,bp>>1,q =1, q 与 p无关,吸附已 铺满单分子层。 3.当压力适中, q pm,m介于0 与1之间。
体积要换算成标准状况STP,101325Pa,273.15K, 1mol标准状况(STP)气体的体积为22.4dm3 (2)单位质量的吸附剂所 吸附气体物质的量。
x a m
物理吸附与化学吸附
具有如下特点的吸附称为物理吸附: 1.吸附力是由固体和气体分子之间的范德华引 力产生的,一般比较弱。 2.吸附热较小,接近于气体的液化热,一般在 几个 kJ/mol以下。 3.吸附无选择性,任何固体可以吸附任何气体, 当然 吸附量会有所不同。
N2吸附脱附测试技术及分析

❖ 由于吸附质与孔壁之间的 强相互作用,吸附开始在 很低的相对压力下。
❖ 由于吸附的分子之间的相 互作用,完全填满孔穴需 要提高相对压力
❖ 在较低的相对压力下( <0.3,氮气吸附)微孔填 充不会观察到毛细管凝聚 现象。(单层吸附具有可 逆性)
2.吸附理论
N2吸附脱附测试技术
1.吸附现象
❖ 当气体或蒸汽与干净的固体接触时,一部分气体 被固体捕获,若气体体积恒定,则压力下降,若 压力恒定,则气体体积减小。从气相中消失的气 体分子或进入固体内部,或附着于固体表面。前 者被称为吸收(absorption),后者被称为吸附( adsorption)
❖多孔固体因毛细凝结(capillary condensation)而 引起的吸着作用也称为吸附作用
孔的毛细效应对 吸附等温线的影响
脱附过程: ➢毛细凝聚后的液面曲率半径小于毛细凝聚前 ➢吸附质在孔壁接触角不为0,前进角大于后退角 氮气的吸附等温线滞后环闭合点 一般在P/P0=0.42~0.50之间。 此时毛细凝聚的张力等于液面的抗拉强度
2.吸附理论
迟滞现象(Hysteresis)
❖ 若吸附-脱附不完全可逆,则吸附-脱附 等温线是不重合的,这一现象称为迟滞 效应,即结果与过程有关,多发生在IV 型吸附平衡等温线。
2.吸附理论
IV型等温线
❖ Ⅴ型等温线很少遇到,而 且难以解释,虽然反映了 吸附质与吸附剂之间作用 微弱的Ⅲ型等温线特点, 但在高压区又表现出有孔 充填(毛细凝聚现象)。
2.吸附理论
毛细管凝结现象
❖ 根据Kelvin公式,凹液面 上的蒸汽压小于平液面上 的饱和蒸汽压,所以在小 于饱和蒸汽压时就有可能 在凹液面上发生蒸汽的凝 结,发生这种蒸汽凝结的 作用总是从小孔向大孔, 随着气体压力的增加,发 生气体凝结的毛细孔越来 越大,这种现象被称为毛 细凝结现象。
有机缓蚀剂的表面活性对金属吸附与腐蚀行为的影响

表面活性是指溶质使溶剂的表面张力降低的性质. 表面 张力指垂直通过液体表面上任一单位长度与液面相切的表 面收缩力 ,是液体的主要物理化学性质之一 [3 ]. 有机缓蚀剂 的表面活性可用表面张力随浓度的变化 ( - dγ/ dc)表示 ,但 常定量表征表面活性的是缓蚀剂的临界胶浓度或 PC20 ( PC20
( 10 )
π = (Γm / n) R T ln ( 1 + kcn )
( 11 )
π = (Γm / n) R T ln ( 1 + k1 c)
( 12 )
公式 (10) 、(11) 、(12) 分别同公式 (7) 、(8)和 Langmuir
公式相对应. 参考王慧龙等 对 [10, 11 ] M annich碱缓蚀剂的研
第 20卷
按照其物理意义 ,又将界面压定义为缓蚀剂在固体表面 吸附前后单位界面上自由能的变化. 界面压的大小反映了界 面吸附层的稳定性. 对于不同表面活性的缓蚀剂 ,其在金属 表面的吸附必然会引起金属与溶液界面性能的差异 ,所以 , 缓蚀剂的表面活性与固液间的界面性能有必然的联系 ,影响 着缓蚀剂在金属表面的状态或吸附 ,影响缓蚀剂表面活性的 因素 ,也会影响金属与溶液间的界面性能.
收稿日期 : 2007206203初稿 ; 2007209227修改稿 作者简介 :赵建国 (1956 - ) ,男 ,学士 ,副教授 ,从事金属腐蚀与表面
化学研究. Tel: 0316 - 2172984 E - mail: hblfzhaojg@163. com
= - l g (C2 )π =20 ,指表面张力降低 20 mN /m 时 ,溶液本体浓
11B asic D epa rtm en t, Hebei P rofessiona l College of Petroleum Techn igue, L angfang 065000; 21College of Chem istry and M a teria ls S cience, L angfang Teachers′College of Hebei, L angfang 065000
n2吸附等温线 -回复

n2吸附等温线 -回复
n2吸附等温线是描述氮分子在固体表面上吸附与脱附的特性的一种曲线。
这条曲线可以用来研究氮与固体表面的相互作用力和吸附行为。
当氮分子与固体表面发生吸附时,吸附量随着气相中氮分子的压力的增加而增加,直到达到一个平衡状态。
在吸附平衡状态下,吸附等温线上的吸附量不再增加。
当气相中氮分子的压力继续增加时,吸附量保持不变。
当气相中氮分子的压力减小时,吸附量会减少,直至氮分子完全脱附。
吸附等温线的形状和曲线斜率可以提供关于氮分子在固体表面的吸附行为的重要信息。
通过对吸附等温线的研究,可以更好地理解氮分子在固体表面的相互作用机理。
吸附现象在化学实验中的应用

吸附现象在化学实验中的应用引言:吸附是一种重要的物理现象,广泛应用于化学实验中。
通过吸附现象,我们可以实现物质的分离、纯化和催化等目的。
本文将探讨吸附现象在化学实验中的应用,并介绍一些常见的实验方法和技术。
一、吸附在分离与纯化中的应用1.1 吸附柱层析法吸附柱层析法是一种常见的分离与纯化方法。
它利用吸附剂对混合物中的目标物质进行选择性吸附,从而实现目标物质的分离与纯化。
例如,我们可以使用活性炭吸附剂对水中的有机物进行吸附,从而去除水中的污染物质。
此外,吸附柱层析法还常用于药物分离与纯化、食品添加剂的检测等领域。
1.2 吸附过滤法吸附过滤法是一种基于吸附现象的分离与纯化方法。
它利用吸附剂对混合物中的杂质进行吸附,然后通过过滤的方式将纯净物质分离出来。
例如,我们可以使用活性炭或硅胶作为吸附剂,对废水中的重金属离子进行吸附,然后通过过滤的方式将纯净的水分离出来。
吸附过滤法在环境保护和水处理领域有着广泛的应用。
二、吸附在催化反应中的应用吸附在催化反应中起着重要的作用。
通过吸附现象,我们可以提高催化剂的活性和选择性,加速反应速率,降低反应温度等。
以下是一些常见的吸附催化反应的应用:2.1 吸附催化裂化吸附催化裂化是一种重要的石油加工技术,用于将重质石油馏分转化为轻质石油产品。
在裂化过程中,催化剂表面的吸附作用能够使长链烃分子在裂化温度下发生断裂,生成较短链的烃类物质。
吸附催化裂化技术不仅能提高石油产品的产率,还能改善燃料的质量,减少环境污染。
2.2 吸附催化氧化吸附催化氧化是一种常用的废气处理技术。
通过吸附剂的作用,将废气中的有害物质吸附在催化剂表面,并在催化剂的作用下进行氧化反应,将有害物质转化为无害物质。
例如,我们可以使用银催化剂将废气中的一氧化碳氧化为二氧化碳,从而减少大气污染。
三、吸附在实验技术中的应用吸附现象在化学实验中还有一些特殊的应用。
以下是一些常见的实验技术:3.1 吸附光谱吸附光谱是一种通过测量物质在吸附剂表面的吸附态与非吸附态之间的差异来研究物质性质的方法。
金属与半导体表面上物理吸附及化学吸附行为的研究

金属与半导体表面上物理吸附及化学吸附行为的研究近年来,金属和半导体表面的物理吸附和化学吸附引起了许多研究者的兴趣。
这些研究对研究材料的表面性质、化学反应和催化过程等有很大的意义。
本文将介绍金属和半导体表面上物理吸附及化学吸附行为的研究进展和相关应用。
一、金属表面的物理吸附和化学吸附金属的表面性质对其化学反应和催化过程有很大的影响。
在金属表面上,分子吸附主要分为物理吸附和化学吸附两种情况。
物理吸附是指分子与金属表面之间的非化学吸引力作用。
而化学吸附是指分子与金属表面的化学结合作用。
1. 物理吸附物理吸附是一种相对较弱的吸附方式,通常在低温下发生。
在物理吸附过程中,分子直接吸附在金属表面上,而不需要与表面原子或离子发生化学反应。
这种吸附方式通常由范德华力、量子力学效应等所驱动。
物理吸附是一种可逆的过程,吸附的分子可以在蒸汽压下脱附。
另外,不同分子的物理吸附能力也不一样,一些分子容易被吸附并很难脱附,而另一些分子则很容易被脱附。
2. 化学吸附化学吸附是指分子与金属表面原子或离子之间形成化学键的吸附,是吸附能力比较强的一种吸附方式。
在这种吸附方式下,分子与金属表面之间发生相互作用,从而形成新的化学键。
化学吸附是一种不可逆的过程,需要通过一定的反应条件才能使其脱附。
其吸附能力也与金属表面的性质以及吸附分子的特性有关。
二、半导体表面的物理吸附和化学吸附半导体表面的物理吸附和化学吸附在某些应用领域也十分重要,尤其是在半导体加工和光电器件制备等方面。
1. 物理吸附与金属表面上的情况类似,半导体表面上的物理吸附主要由范德华力、静电作用等驱动。
在物理吸附过程中,分子吸附在半导体表面上,但没有与表面原子或离子结合。
与金属表面上的物理吸附相比,半导体表面上的物理吸附更容易发生反应和变化。
在实际应用中,这由于半导体表面的粗糙和表面缺陷等因素导致。
2. 化学吸附半导体表面的化学吸附是一种比较常见的表面反应。
在化学吸附过程中,分子与半导体表面上的原子或离子结合,从而形成新的化学键。
CO、N2在金属Y表面的吸附行为

件 下 , C S 的 生成 焓 为 一6 .8 J・ l YN Y O( ) 9 8 k mo~, 2的生 成 焓 为 一 6 3 k m l 比较 YC ( ) Y 2 S 和Y ( ) 4 .4 J・ o ~. O S 、 N ( ) H2 S
收稿 日期 :0 7— 3—1. 20 0 2 作者简 介: 朱瑜 (9 7一 ) 女( 17 , 土家族 ) 硕士 , , 讲师 , 主要从事分子结构与光谱研 究
V0. 5 No 3 12 .
Sp 20 e .0 7
C N O、 2在 金 属 Y表面 的 吸 附行 为
朱 瑜 方 , 芳 赵 , 倩 蒋 , 刚 朱正 和 ,
(. 1湖北民族学院 理学院, 湖北 恩施 450 ; 400 2 湖北民族学院 信息工程学院, . 湖北 恩施 4 50 ; 00 3 西 南大学 物理 学 院 , . 重庆 40 1 ; 0 75 4 四川 大学 原子 与分 子物理研 究所 , . 四川 成都 6 0 6 ) 10 5
维普资讯
第2 5卷第 3期 20 0 7年 9月
湖北 民族学院学报( 自然科学版 ) Junl f ue Istefr aoat sN t a Si c dtn ora o bintu tnl e( a rl ce eE io ) H it o Ni i i u n i
摘要 : Y原子采用相 对论 有效原子 实势 ( E P S D) C O、 函数 , 利用 B L P 3Y
方 法计算 了 Y O和 Y , C N 分子的微观 结构 以及 不同温度 下的热 力学函数. 以气 态分子 总能量 中的振动能 E 代 替该 v 分 子处 于固态时的振 动能量 , 以电子运动和振 动运动熵 Sv r代替分子处 于固态的熵 的近似方 法, 计算 了不同温度 下
烯烃分子在金属表面的化学吸附行为

烯烃分子在金属表面的化学吸附行为烯烃是一类含有碳-碳双键的有机化合物,具有广泛的应用前景。
在化学领域中,烯烃可用于合成高分子材料、药物分子和农药分子等,在工业领域中则可用于制备化学品、塑料和燃料等。
因此,研究烯烃分子在金属表面的化学吸附行为对于深入了解烯烃分子的性质及其在不同领域的应用有着重要的意义。
化学吸附是指分子在表面通过化学键结合方式吸附,形成稳定的化学键结构。
烯烃分子的化学吸附行为是受到其化学结构、大小、形状和表面性质等因素的影响。
此外,金属表面的性质也对烯烃分子的化学吸附起着至关重要的作用。
研究表明,烯烃分子的化学吸附主要是通过金属表面的π-配位作用实现的。
对于金属表面的π-配位作用,其可以分为两类:(1)金属表面直接与烯烃分子的碳-碳双键作用;(2)金属表面间接与烯烃分子作用,即通过羟基、羰基、硝基等官能团介导作用。
在金属表面直接与烯烃分子的碳-碳双键作用中,具有特殊π-电子结构的金属表面能够有效吸引烯烃分子的π-电子,实现化学键的形成。
以铜表面为例,铜表面上3个等分位的表面原子可以形成π-电子空穴,成为吸附烯烃分子的活性中心。
通过与烯烃分子的碳-碳双键形成π-键成为吸附状态,可形成稳定的化学键结构。
因此,金属表面上的π-电子结构直接决定了烯烃分子在金属表面的化学吸附行为。
同时,在金属表面间接与烯烃分子作用中,官能团介导作用也是十分关键的。
不同官能团对金属表面的π-配位作用有着不同的影响,如羟基官能团可以增强金属表面与烯烃分子的作用,硝基官能团则可以减弱该作用。
进一步的研究发现,羟基官能团对烯烃分子的化学吸附普遍具有诱导作用,即可以增强π-键的形成,使化学结构更加稳定。
除了上述两种类型的π-配位作用外,烯烃分子的大小、形状和表面性质也对化学吸附行为产生显著影响。
研究表明,具有更多碳原子的烯烃分子更容易与金属表面形成化学键,而具有非平面性的烯烃分子则更容易发生平面构象转化,有利于化学吸附的形成。
氧气分子的吸附与反应在金属催化剂表面的研究

氧气分子的吸附与反应在金属催化剂表面的研究氧气是一种普遍存在于地球大气中的气体,也是许多化学反应和生命过程中必不可少的物质。
在过去的几十年里,许多研究者都尝试探究氧气在金属催化剂表面的吸附与反应行为,希望能够揭示其中的基本原理并开发出高效的催化剂材料。
氧气的吸附过程是催化反应中最基本的环节之一。
在大多数情况下,氧气分子与金属表面的相互作用力主要来自于范德华力和静电相互作用力。
这些相互作用力会使得氧气分子处于吸附于金属表面的稳定状态,从而为后续的反应提供了必要的条件。
除了吸附之外,氧气与金属催化剂表面的反应行为也备受研究者关注。
最常见的反应包括氧化反应和还原反应。
在氧化反应中,氧气分子会从金属表面获得电子,而被氧化成为氧化物。
而在还原反应中,则正好相反,氧气分子会将其带有的电子转移到金属表面,从而被还原成原子氧。
近年来,许多研究者采用先进的计算方法,如密度泛函理论和分子动力学模拟等,来模拟和预测氧气在金属催化剂表面的吸附和反应行为。
这些计算方法可以大大提高实验研究的效率和精度,从而揭示出更多催化反应的本质规律。
除此之外,一些新型的金属催化剂材料也被设计出来,以更高效地催化氧气的吸附和反应。
其中最具代表性的是纳米催化剂,这种催化剂材料具有更大的比表面积和更多的活性位点,从而能够更快速地催化氧气的吸附和反应。
同时,由于其表面结构具有一些独特的物理和化学性质,因此纳米催化剂还能够精准地控制反应过程中的选择性和活性,从而得到更高的催化效率。
总的来说,氧气分子的吸附与反应在金属催化剂表面的研究是一个复杂而又丰富的研究领域。
通过精细的实验和计算研究,我们有望揭示出更多的本质规律和物理特性,为未来的催化反应研究和应用开辟出更广阔的空间。
表面活性剂对重金属污染物吸附去除作用研究

表面活性剂对重金属污染物吸附去除作用研究随着工业化的发展和城市化进程的加速,重金属污染问题日益严重,给生态环境和人类健康带来了极大的威胁。
针对重金属污染问题,化学吸附已被广泛应用。
在这个过程中,表面活性剂被发现具有较好的吸附去除重金属污染物的能力。
本文将从表面活性剂对重金属污染物的吸附行为、吸附机理、工艺条件等方面的研究进展进行探讨。
表面活性剂对重金属污染物的吸附行为表面活性剂对重金属污染物的吸附行为主要取决于表面活性剂的物理化学性质和重金属离子的类型、浓度等因素。
研究表明,非离子表面活性剂具有较好的吸附能力,与离子表面活性剂相比,其吸附容量更大。
此外,表面活性剂的链长、溶解度等因素对吸附行为也有影响。
重金属离子的类型、浓度和pH值等因素对表面活性剂吸附行为皆有影响,当pH值较低时表面活性剂的吸附效果较好。
表面活性剂对重金属污染物的吸附机理表面活性剂对重金属污染物的吸附机理涉及静电吸附、化学配位吸附和络合反应等。
静电吸附主要通过表面活性剂电荷与重金属离子之间的静电相互作用实现;化学配位吸附主要发生在表面活性剂分子和重金属离子之间形成化学键的过程中;络合反应包括离子交换、结合吸附等,这种吸附方式是对生物吸附和表面活性剂吸附共存的情况下的重金属去除机制。
工艺条件对重金属污染物吸附去除的影响重金属污染物吸附去除过程中的反应条件对于吸附效果和经济性十分关键。
研究表明,表面活性剂的用量、pH值、温度等因素对吸附效果有着重要的影响。
在表面活性剂低浓度下,其体积与重金属离子的比值越小,其吸附效果越好。
此外,适当调整pH值也有助于提高表面活性剂对重金属污染物的吸附去除效果。
随着温度的提高,表面活性剂的吸附容量也会增大。
总之,表面活性剂的物理化学性质、重金属离子的类型、浓度等因素均对吸附去除效果产生影响。
静电吸附、化学配位吸附和络合反应是表面活性剂吸附重金属污染物的机理,调整反应条件可以提高吸附效果和经济性。
未来研究针对表面活性剂与重金属污染物的深度探究,有望为治理重金属污染问题提供更为有效的技术支持。
氧化钯吸附率

氧化钯吸附率氧化钯(PdO)是一种重要的过渡金属氧化物,在吸附科学和催化领域具有广泛的应用。
它具有高度可调控的物理和化学性质,能够吸附多种气体和溶液中的物质。
在气体吸附方面,氧化钯的吸附率受到多种因素的影响,包括气体种类、温度、压力、氧化钯晶面结构等。
本文将综述氧化钯吸附率的相关参考内容。
首先,气体种类是影响氧化钯吸附率的重要因素之一。
研究发现,不同气体分子在氧化钯表面的吸附行为存在差异。
例如,Bernard et al.(2007)的研究表明,氧化钯对氧分子(O2)和一氧化碳(CO)的吸附具有高度选择性,其吸附率较高。
此外,氧化钯对氢气(H2)和一氧化氮(NO)的吸附率也较高,但对氮气(N2)的吸附率很低。
这些实验结果对于了解氧化钯的吸附特性具有重要意义。
温度是另一个影响氧化钯吸附率的重要因素。
研究表明,随着温度的升高,氧化钯对某些气体的吸附率呈现下降的趋势。
以CO吸附为例,Tumuluri et al.(2016)的研究发现,在室温下,氧化钯对CO的吸附率较高;然而,当温度升高到400℃时,氧化钯对CO的吸附率显著下降。
这种温度依赖性的吸附率变化与氧化钯表面的物理结构和化学吸附机制密切相关。
压力是影响氧化钯吸附率的另一个重要因素。
研究表明,氧化钯对气体的吸附率随着压力的增加而增加。
例如,Ozawa et al.(2005)的研究发现,氧化钯对CO的吸附率在较低压力下较低,但随着压力的增加,吸附率显著提高。
这种压力依赖性的吸附行为与氧化钯表面活性位点的占据情况和气体分子的覆盖程度密切相关。
此外,氧化钯晶面结构也对其吸附率起到重要影响。
研究发现,氧化钯(100)晶面对CO的吸附率高于(110)和(111)晶面。
Palomino et al.(2011)的研究显示,CO在(100)晶面上会形成化学键,并且具有较高的吸附能;而在(110)和(111)晶面上,CO主要以物理吸附形式存在,吸附能较低。
这一现象说明了氧化钯晶面结构对其吸附性能的重要影响。
氧气在某固体表面上的吸附-概述说明以及解释

氧气在某固体表面上的吸附-概述说明以及解释1.引言1.1 概述氧气在固体表面上的吸附是一种重要的表面现象,它在很多领域都具有广泛的应用价值。
随着科技的发展,人们对氧气吸附的研究越来越深入,对其机理和影响因素有了更加清晰的认识。
氧气吸附是指氧气分子在固体表面上附着并与表面原子或分子发生作用。
这种吸附过程既可以是物理吸附,也可以是化学吸附。
在物理吸附中,氧气分子与固体表面发生弱相互作用,吸附强度较小;而化学吸附则是指氧气分子与固体表面发生一定的化学反应,吸附强度较大。
氧气吸附的机理复杂多样,与固体的化学性质、表面结构以及气体的温度、压力等因素密切相关。
在不同的条件下,氧气吸附的机制可能存在差异。
例如,在高温条件下,氧气分子可以与固体表面的活性位点发生氧化反应,形成氧化物;而在低温条件下,氧气分子则可能以物理吸附方式附着在固体表面。
氧气吸附的影响因素包括固体表面的化学性质、结构形貌、表面活性位点的密度等。
表面化学性质的不同会影响氧气与固体表面的相互作用方式,而表面结构的变化可能导致吸附能力的不同。
此外,温度、压力等环境条件也会对氧气吸附行为产生显著影响。
氧气吸附的重要性不言而喻。
不仅是物理、化学等学科的基础研究领域,氧气吸附还在工业生产、环境保护、能源开发等领域具有广泛的应用前景。
对氧气吸附的进一步研究可以帮助我们更好地理解表面科学中的吸附现象,并为相关领域的技术创新提供重要支持。
综上所述,本文将重点探讨氧气在固体表面上的吸附现象。
通过对氧气吸附的定义、机理以及影响因素的介绍,我们可以更全面地认识和理解氧气吸附的重要性。
进一步的研究和应用将使我们在相关领域取得更大的突破,并为社会发展做出更大的贡献。
1.2文章结构1.2 文章结构本文主要通过对氧气在某固体表面上的吸附现象进行研究和分析,旨在探讨氧气吸附的定义、机理以及影响因素。
文章将按照以下结构展开讨论:首先,文章将在引言部分概述研究的背景和意义,介绍氧气吸附在固体表面上的重要性。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
同理 , 对于气固氮 化 反应 Pd(s)+1/2N2=PdN(s),
1.2 密度泛函理论(DFT)和相对论有效原子实势 根据所建立的近似方法可看出 N2 和 PdN2 分子
实势及 (6s5p3d) 价 基 集 合 [10], N 原子 采用 AUG鄄cc鄄
子 层 含 18 个电 子 (4s24p64d10) 的 准 相对论有效原子
苓
驻S苓=S苓(PdN2(s))-S苓(Pd(s))-S苓(N2) 驻G =驻H -T驻S
苓 苓
苓 EV 苓 EV
苓 苓 苓 (2) 抑E苓 V (PdN2(s))-EV (Pd(s))-H (N2)+驻H e
=H (PdN2(s))-H (Pd(s))-H (N2)+驻H
苓 苓 苓
苓 e
抑S (PdN2(s))-S (Pd(s))-S (N2)
以气态分子总能量中的振动能代替该分子处于固态 时的振动能量, 以 电 子运 动和振动 运 动 熵代替分子 处于固态熵的近似方法 . 运 用 此方法 在 处理 氢 化 反 应上已取得较满意的结 果 , 本文是 把 此 方法 推广 于 氮化反应. 本文 采用 Gaussian 98 程 序 , 在 Pd 原子的相对论 有效原子实势(RECP)近似下, 运用B3LYP方法计算 Pd与N2反应的驻H苓、 驻S苓、 驻G苓及氮化反应平衡压力,
的离 解 能数据 是 计算 驻H苓、 驻S苓和 驻G苓的 关 键 物理 量. 所以 选择恰当 的方法和基 组 计算 N2 和 PdN2 分 子的结构与离解能是非常重要的. 本文 采用的 B3LYP 方法是将 包含梯 度修 正 的 Becke 交 换泛 函与 包含梯 度修 正 的 Lee鄄Yang鄄Parr 相 关泛 函联系 在一起 , 局域 相 关泛 函 按 常 规 采用 Vosko鄄Wilk鄄Nusair(VWN) 局域 自 旋 密 度处理 , 得 到 Becke 三参数的密度泛函方法(DFT). 乎不可能, 存在 d 轨道相互作用, 其相对论效应十分 由于钯元素属于 重 的 过渡元 素 , 全电 子计算几
中图分类号: O647
在氢同位素提取、 分离和纯化的方法中, 钯合金 膜扩散法具有独特的优越性 . 但在膜的使用过程中,
[1]
导出了氮化反应平衡压力与温度的关 系 . 并 与 Pd氢 化反应平衡压 力 比较, 得 到 N2和H2在 金属 Pd表面吸 附能力的强弱关系 . 从而 在 理论 上 探索气 体在 金属 表面的吸附行为和合金膜中毒的微观机理.
December
Acta Phys. 鄄Chim. Sin., 2005, 21(12): 1343耀1346
物理化学学报(Wuli Huaxue Xuebao)
1343
N2 在 Pd 金属表面的吸附行为
朱
摘要
鄢
瑜1
蒋
刚1
于桂凤 1
610065;
2
朱正和 1
王和义 2
傅依备 2
621900)
(1 四川大学原子与分子物理研究所, 成都
苓 e
其中, D0(N2)和 D0(PdN2)分别为 N2 和 PdN2 分子的化
明显. 鉴于原子性质主要决定于价层电子, 故利用相 对论有效原子实势(relativistic effective core potential, RECP). 该理论采用 RECP 取 代核与 电 子 之 间静 电 势能和核的正交效应 , 并 考虑轨道 扩散 和收 缩 的相 对论效应, ECP 重新产生价轨道的本征能量和形状. 原子实和价电 子 轨道由 Cowan鄄Griffin Hartree鄄Fock 方程加相对论修 正获 得, 考虑 了 “Mass鄄velocity” 和 “Darwin” 项以及自旋鄄轨道耦合效应, 这样利用比全 电子计算少得多的计算 时 间, 就能 很 好 地说明 相对 论效应 的 重要性. 应 用 RECP 计算 含 重 元素 分子的 结构与性质已取得令人满意的结果[8鄄9].
[5鄄7]
1 理论方法
1.1 气固氮化反应生成热力学函数的近似计算 能量E、 焓H和熵S. 对于固体, 分子被固定于晶格, 可 忽略分子的平动和 转 动 , 近似地 以气 体分子的振动 能EV、 电子和振动运动熵SEV分别代替固体 分子的能 利用量子力学从头计算很容易得到气体分子的
提出
量E和熵S. 而且, 对于固态分子, 由于反应过程中pV 这样就可以近似给 出 固态分子不同温度下的 熵S和 核振动对能量E及生成焓的贡献驻Hn. 对于气固氮化
表 1 不同温度下 N2、 PdN、 PdN2 和 Pd 的热力学函数 Table 1
N2(g) T/K 298.15 398.15 498.15 598.15 698.15 798.15 898.15 998.15 E kJ · mol 20.84 22.92 25.02 27.15 29.34 31.59 33.90 36.27
表 2 不同温度下 PdN(s)、 PdN2(s)和 PdH2(s)的 驻H苓、 驻S苓和 驻G苓
PdN2(s) 驻S
专
PdN(s)
专
驻H苓, 驻S苓 and 驻G苓 of PdN(s), PdN2(s), and PdH2(s) at different temperatures
驻G
专
PdH2(s) 驻S专 kJ · mol-1 -30.99 -13.48 5.31 25.13 45.74 67.03 88.89 111.26 驻G专
属 Pd 的原子化能, 驻Hs 为 PdN2(s)的升华焓. 若缺乏 溶体 结构 变化不大 , 则 可 认 为 驻Ha抑驻Hs, 近似得 到 这样, 生成热力学函数 驻H 、 驻S 和 驻G 可以近似地
苓 苓 苓
驻H苓 e 抑D0(N2)-D0(Pd鄄N鄄N)
苓
苓 n 苓 e
(1)
表示为
驻H =驻H +驻H
了PdN 和PdN2分子的微观结构以及不同温度下金属
刚(E鄄mail: gjiang@; Tel: 028鄄85408810).
*
国家自然科学基金委鄄
1344
Acta Phys. 鄄Chim. Sin. (Wuli Huaxue Xuebao), 2005
Vol.21
学离解能, 可通过量子力学从头计算得到; 驻Ha 为金 驻Hs 的实 验 数 据 , 假设 Pd 与 吸氮 后形 成的 PdN2 固 驻H , 即
2005鄄05鄄09 收到初稿, 2005鄄06鄄16 收到修改稿. 联系人: 蒋 中国工程物理研究院联合基金(10176021)资助项目
项变化很小, 可以 近似 认为焓 与 内能相 等 , 即 EV=H.
的贡献驻H苓 e 可通过下列近似计算得到, 即
反应Pd(s)+N2=PdN2(s), 电子运动对固态PdN2生成焓
37.10 44.69 50.71 55.73 60.06 63.88 67.33 70.47
No.12
Table 2
T/K 298.15 398.15 498.15 598.15 698.15 798.15 898.15 998.15 驻H 驻S
蒋
刚等: N2 在 Pd 金属表面的吸附行为
1345
kJ · mol-1 254.37 250.77 247.21 243.65 240.05 236.38 232.65 228.85
J · K-1 · mol-1 -119.60 -130.02 -138.01 -144.52 -150.10 -154.99 -159.41 -163.43
-1
Thermodynamic functions of N2, PdN, PdN2, and Pd at different temperatures
PdN(g) H
-1
PdN2(g) SEV EV
-1
Pd(s) SEV H
-1
S J · K· mol
-1
EV
-1Байду номын сангаас
S
-1
kJ · mol 23.32 26.23 29.16 32.12 35.14 38.22 41.36 44.56
出, 在常压及 298.15耀998.15 K 温度条件下, N2 在金属 Pd 表面的吸附过程以 Pd(s) + N2 = PdN2(s)反应进行. 计算 得出在标准条件下, PdN(s)的生成焓为 254.37 kJ · mol-1, PdN2(s)的生成焓为-80.59 kJ · mol-1. 并与 Pd 氢化反应平 衡压力比较, 得到平衡常数 Kp(N2)比 Kp(H2)约小两个数量级, 说明 N2 较难被金属 Pd 表面吸附, 在热力学上有利 于氢置换氮. 关键词: PdN, PdN2, 密度泛函理论, 热力学函数, 平衡压力, 吸附行为
nm, D0=945.33 kJ · mol-1)接近 ; PdN 分子的基 电 子 状
态为 4撞-, 其平衡核间距为 0.18158 nm , 化学离解能 Pd鄄N鄄N (C肄v), 其 电 子 状 态 为 1撞 +, 平衡 核 间距 RPdN= 0.19537 nm, RNN =0.11026 nm, 化 学 离 解 能 为 1014.670 kJ · mol-1. N2、 PdN 和 PdN2 分子的热力学函 数见表 1. 表 1 中 N2 在 298.15 K 时 的 熵 (191.35 J · K-1 · mol-1)与文献[11]中的值(191.609 J · K-1 · mol-1)接 为 201.760 kJ · mol-1; PdN2 分子的最稳定构型为线性
kJ · mol 4.17 4.64 5.24 5.90 6.61 7.34 8.10 8.87
-1
J · K· mol