主动脉覆膜支架系统临床试验审评要点
ecmo评价技术审评要点

ecmo评价技术审评要点以ECMO评价技术审评要点为标题,本文将对ECMO评价技术的审评要点进行详细阐述。
ECMO,即体外膜氧合技术(Extracorporeal Membrane Oxygenation),是一种通过将血液引出体外进行氧合和二氧化碳排出的治疗方法,广泛应用于心肺功能衰竭等严重疾病的救治中。
ECMO治疗的有效性和安全性对于患者的生存和康复至关重要,因此对ECMO评价技术进行审评是非常必要的。
ECMO评价技术的审评要点主要包括以下几个方面:1. 技术可行性评价:评估ECMO技术在特定疾病或患者群体中的可行性。
这需要考虑ECMO设备的有效性、安全性和患者的适应性等因素。
同时,还需评估ECMO技术在不同年龄、病情严重程度和基础疾病等情况下的适用性。
2. 治疗效果评价:评估ECMO治疗对患者病情的影响和效果。
主要包括生存率、康复情况、症状缓解程度、并发症发生率等指标。
这需要进行临床观察和数据统计分析,对比ECMO治疗前后的差异,评估治疗效果的可靠性和可重复性。
3. 并发症评价:评估ECMO治疗过程中可能发生的并发症。
ECMO 治疗涉及到体外循环和抗凝等操作,患者可能出现出血、感染、血栓等并发症。
因此,需要对并发症的发生率、严重程度和影响因素进行评估,以便提前预防和处理。
4. 安全性评价:评估ECMO治疗过程中的安全性。
主要包括ECMO设备的安全性、抗凝剂使用的安全性、体外循环过程中的安全性等方面。
需要对可能出现的安全隐患进行评估,采取相应的措施保障患者的安全。
5. 费用效益评价:评估ECMO治疗的费用效益。
ECMO治疗是一种高技术、高成本的治疗方法,对医疗资源的利用情况和对患者生命质量的改善效果需要进行综合评估,以便更好地指导临床实践和决策。
在进行ECMO评价技术的审评时,需要注意以下几个方面:1. 综合评估:综合考虑不同评价指标的结果,进行综合评估。
不同的指标可能存在一定的冲突,需要权衡各个方面的因素,综合评估ECMO技术的优劣势。
主动脉半周覆膜支架生物相容性及封堵效果的实验研究

【 键 词 】 主 动 脉夹 层 ;半周 覆 膜 支 架 ; 入 治 疗 ; 物 相 容 性 关 介 生
中 图分 类 号 : 5 31 文 献标 志 码 : 文 章 编号 :o 87 4 2 1 )1 -8 30 R4 . B 1 o — 9 X( 0 0 一 10 8 —5
B o o p t i t n c l sv f c e c f s l- a e a r i afc ce se tg a : a x e i n a i c m a i l y a d o cu i e e i n y o e f b i i m d o t h l-y l t n - r f n e p rme t l c t
介入放射学杂志 2 1 0 0年 1 1月 第 1 第 1 期 9卷 1
JItre t ail 00, o.9 o1 nevn do 1 V 1 ,N .1 R 2 1
.— 8 -— - . 8 3- — - —
・
实 验研 究
E pr e t sac ・ xei na r e Nhomakorabeah m le
选用 1 5条 健 康 杂种 犬 , 自制 主 动脉 半 周 覆 膜 支 架规 格 为 1 m ×6 6m 0mm、 人工 血 管 长 度
为 4e 经 股 动脉 置 入 半 周 覆 膜支 架 , 机 封堵 左 或 右 侧 肾 动 脉 , 后 行 主 动脉 造 影 及 增 强 C m。 随 尔 T检 查 明 确 主动 脉 血 流情 况 及 肾脏 供 血 情 况 。术 后 1 4 8 1 , , ,2和 1 6周 , 机选 取 3条 犬 复 查 主 动脉 造 影 及 增 强 C 随 T, 随后 处 死 、 获取 主动 脉 标 本 , 别 对 正 常腹 主 动 脉 、 支 架 处 腹 主 动 脉 、 膜 支 架 处 腹 主动 脉 进 行 光 镜 及 分 裸 覆 电镜 的 观 察 。结 果 1 5条 犬 均成 功 地 置 入 主 动 脉半 周 覆 膜 支 架 ,分 别 封 堵 7条 右 肾动 脉 及 8条 左 肾 动
SFDA对血管内支架申报前临床试验指导原则

冠脉药物洗脱支架临床试验基本要求一、前言随着科学技术的不断发展,各类新型冠脉药物洗脱支架产品日益增多。
为了进一步规范该类产品上市前的临床试验,并指导该类产品生产企业在申请产品注册时临床试验资料的准备,特制定本临床试验基本要求。
需要强调的是,本临床试验基本要求虽然为该类产品的临床试验及生产企业在申请产品注册时临床试验资料的准备提供了初步指导和建议,但是不会限制医疗器械相关管理部门对该类产品的技术审评、行政审批以及企业对该类产品临床试验资料的准备工作。
它是一个动态的文件。
随着冠脉药物洗脱支架技术的发展、提高和相关法规政策、标准制定等情况的变化,本临床试验基本要求还会不断地完善和修订。
二、临床试验方案(一)、临床试验基本原则1、冠脉药物洗脱支架的临床试验应符合国家食品药品监督管理局颁布的《医疗器械临床试验规定》及其他相关法律、法规的规定。
2、进行上市前临床试验的冠脉药物洗脱支架应已经过相对科学的实验室研究和动物试验验证,研究结果可基本证明产品安全、有效。
(二)、临床试验目的临床试验目的应该清楚地表明产品名称(应注意与注册产品标准一致)、临床治疗的意义以及本次试验产品的评价目标(安全性、有效性)。
如果是新产品,生产者在进行临床试验前应考虑以下问题:1、所评价的治疗问题是否是当前临床上急需解决的治疗问题?2、问题的提出有无充分的实践基础和科学依据?3、其临床意义如何?4、拟进行的试验研究在原有的基础上有何改进与创新?5、试验题目是否明确、具体,能否反映试验的主要内容或方法?题目与内容是否相符合?如果已经有功能和原理相同或类似的产品上市,应遵照科学的安全有效的原则,试验产品与已上市产品在对照评价的基础上,明确相同临床治疗意义下的安全性和有效性。
就冠脉药物洗脱支架而言,其临床治疗意义是恢复冠状动脉狭窄患者的动脉血流;其安全性评价指标包括死亡、心肌梗死、支架内血栓形成和主要不良心脏事件(MACE);有效性评价指标包括靶病变血运重建(TLR)和靶血管血运重建(TVR)。
覆膜支架在主动脉缩窄归并动脉导管未闭时的应用指征

覆膜支架在主动脉缩窄归并动脉导管未闭时的应用指征【关键词】主动脉缩窄动脉导管未闭覆膜支架应用指征先本性主动脉缩窄指自无名动脉至第一对肋间动脉之间的主动脉管腔狭小,多为局限性,也可为长管状[1]。
主动脉缩窄占先本性心血管疾病发病率的1%~3%,本病可单独存在,但常归并其他心血管畸形,如室距离缺损、动脉导管未闭等[2]。
刘芳等报告的96例主动脉缩窄患者中归并室距离缺损与动脉导管未闭患者各占49%,而室距离缺损与动脉导管未闭同时并存者占23%[3]。
1主动脉缩窄外科手术医治存在不足长期以来,主动脉缩窄医治之外科手术为主,但外科手术医治存在以下不足:①患者术中及术后3个月内死亡率高(14%);②术后再缩窄发生率高(9.9%~33%);③动脉瘤形成发生率高(13%);④持续性高血压发生率高,文献报告达8.3%~43%不等[46]。
因此,探讨先本性主动脉缩窄新的有效医治方式始终是先心病介入医治领域研究的热点之一。
20世纪80年代,Mullins等[7]提出血管内支架可用于医治先本性主动脉缩窄等疾病。
1991年,O’Laughlin等[8]利用球囊扩张式血管内支架医治1例主动脉缩窄取得成功,标志着介入医治主动脉缩窄又一新方式的问世。
近十年来,先本性主动脉缩窄介入医治器材取得了不断改良与完善,BIB(balloon in balloon)球囊与CP(cheatham platinum)覆膜支架(covered stent)的问世及推送器材的不断改良,使支架置入医治主动脉缩窄的适应证不断拓宽。
覆膜支架是指支架内面或外脸部份或完全覆盖不透血液的膜性材料的人工血管内移植物[9],其在外周血管动脉瘤及动静脉瘘等的介入医治方面已取得了较好的疗效[10]。
覆膜支架置入医治青青年和成人主动脉缩窄,较球囊扩张术和裸支架置入术等介入医治手腕具有必然的优势。
第一,覆膜支架能够提供足够的支撑力,有效地抗击缩窄主动脉的弹性回缩;第二,覆膜支架置入术能够用来医治主动脉缩窄球囊扩张术或裸支架置入术后动脉瘤和动脉夹层等并发症。
胸主动脉覆膜支架产品手册Zenith胸主动脉瘤

Zenith ?胸主动脉瘤覆膜支架操作手册目录产品描述适应症警告防范措施 (3)潜在副作用 (4)治疗个性化 (4)支架的使用建议................................ . (4)胸主动脉瘤覆膜支架操作过程中的注意事项 ............ .7产品的提供............................................................. (7)患者随访………………………………………………… ... …… ..8 MR的安全性及兼容性................................. ..8产品描述Zenith?TX2? 胸主动脉瘤覆膜支架设计为两段型支架或单体支架。
近端支架可分为锥形支架和直筒支架。
不锈钢、自膨胀的 COOK-Z ?支架手工缝合与机织聚酯覆膜材料上。
Zenith?TX2?胸主动脉瘤覆膜支架, 提供最佳的支撑力及膨胀力, 确保了支架在释放过程中支架内腔完全的展开。
并且 COOK-Z ?支架提供与血管间最佳的贴壁。
为增强支架的固定性能,胸主动脉瘤覆膜支架标准型近端每间隔 2mm 有交错排列的倒钩。
远端支架远端固定依靠裸支架及倒钩。
利于在荧光屏下清晰的显示支架的位置, 这些标记距离覆膜边缘 1mm 。
适应症Zenith?TX2? 胸主动脉瘤覆膜支架及 H&L-B One- Shot? 输送系统,适用于动脉瘤位于降 主动脉的腔内介入治疗。
对于患者的解剖要求:髂动脉 /股动脉直径能够通过输送系统 主动脉弯曲半径〉35 mm 主动脉瘤颈:长度〉35 mm动脉壁外径在 24-38 mm 之间主动脉弓角度V 45? 警告根据通过的血管内径 (测量内径至内径) 和形态选择合适的血管入路, 以及输送系统的 规格 20Fr 或 22Fr 。
如果血管严重钙化,狭窄,曲折或血栓。
必须排除从股动脉进入输 送系统,否则将会增加栓塞可能。
心血管创新医疗器械技术审评要求解读

设计可行性试验方案时建议注意以下几点:
可行性试验中出现的任何不良事件应如实记录,对于严重不 良事件应按照法规要求及时上报;同时临床试验人员应当及 时做出临床判断,采取措施,保护受试者利益;必要时中止 临床试验
29
可行性试验结束后,申请人决定开展确证性试验或重新开 展可行性试验
30
支架平台由高分子材料/金属制成 生物可吸收支架作为一种“临时性支架”,早期维持有效的
血管支撑
4
预定的时间内降解
▪ 降低持续性机械牵拉,恢复血管对生理刺激的自然反应, 可能有助于血管的晚期扩张性,促进晚期血管良性重构
▪ 降低异物炎性反应等风险,有望减免长期服用双联抗血小 板药
▪ 不妨碍再次进行血运重建(PCI 和CABG) ▪ 与非侵入性影像诊断技术(MR/CT) 兼容,可应用这些技术
试验总样本量应在具有统计学意义基础上不少于1200例 两个临床试验的研究假设均需成立
33
除此之外,在随机对照试验中需预设一亚组
行光学相干断层扫描(OCT)观察产品的降解情况 该亚组样本量应不少于40对 产品注册时需提交亚组至少24个月的OCT分析数据
34
已在原产国/生产国获准上市情况下申请进入中国市场 若在境外已完成了设计良好的、前瞻性的临床试验,且在
动物实验研究中,建议对任何临床相关的安全性事件进行 观察,如血栓形成、心肌梗死、动脉瘤、穿孔等
动物实验中如发生动物死亡,应详细分析死亡的原因,分 析同器械相关性
动物实验结果能够充分地说明产品的安全性和初步可行性 时,方能开展首次人体试验
20
生物可吸收支架临床试验技术审评要求
21
用于《医疗器械注册管理办法》规定需在中国境内进行上市前 临床试验的生物可吸收冠状动脉药物洗脱支架
主动脉夹层覆膜支架植入术及其存在的问题(下)

主动脉夹层覆膜支架植入术及其存在的问题(下)2. 3 覆膜支架植入操作的方法和步骤手术最好由介入放射科、心血管外科和麻醉科医师共同完成。
具体如下:①患者取仰卧位于全麻或腰部硬膜外麻醉下行覆膜支架植入术。
穿刺左侧桡动脉并置入5F或6F桡动脉鞘。
以超滑导丝引导5F头端带有刻度标记的猪尾导管(金标猪尾导管)自桡动脉鞘经左锁骨下动脉送至升主动脉;②综合盆腔及双下肢血管螺旋CT或MRA 检查及体表血管检查结果,选择未受夹层累及一侧的髂股动脉进行皮肤切开和血管游离,直视下穿刺游离的股动脉并置入6F动脉鞘。
以1mg /kg 静脉推注肝素达到全身肝素化后,自鞘管送入0. 038inchⅹ260 cm超硬导丝并将其头端置于升主动脉内;③以CTA或MRA 上展示夹层破口及真假腔最好的角度行胸主动脉造影,一般为左前斜位,投照角度450~600之间。
造影视野中应包括升主动脉﹑主动脉弓、降主动脉、右无名动脉、左颈总动脉及左锁骨下动脉近端;④行胸主动脉造影,造影剂总量一般为35~ 45m ,l流率为20~ 25m l / s,采用DSA 或电影采集;⑤ 分析胸主动脉造影,首先应证实超硬导丝位于主动脉真腔内,如不明确需换其他投影角度造影。
如超硬导丝位于假腔,应在透视导引下改变路径将导丝送入真腔,并重新进行胸主动脉造影。
明确超硬导丝位于主动脉真腔内后,以金标猪尾导管不透X 线的刻度为标准,测量破口与左锁骨下动脉开口的距离以确定锚定区,测量锚定区主动脉弓部直径和长度,并结合CTA 或MRA 测量结果选定支架型号。
选择的支架应大于锚定区主动脉弓部直径10% ~ 15%内径的支架;⑥送入覆膜支架传输系统:再次证实加强导丝位于真腔后,即撤出股动脉鞘管并压迫止血和固定导丝,穿刺点股动脉切开(切口约5mm),将已选定好的覆膜支架传输系统沿超硬导丝送入真腔,并在透视下将其送到降主动脉近段;⑦麻醉师用硝普钠或其他降压药控制患者血压,一般收缩压控制在70~ 90mmHg 之间。
血管支架系统临床前研究申报资料准备技巧

– 材料为混合物的应明确比例 – 组成部分为分层分段结构的应分别描述制造材料 – 应与产品结构图示有明确的对应关系 – 明确材料符合的标准(如适用) – 可吸收材料需明确平均分子量/特性粘数、分子量分布、定性鉴别
常见问题
– 申报内容与其他资料不 一致
– 不应体现商品名称
证明性文件
• 境内申请人按照《创新医疗器械特别审批程序审批》申请 注册时,样品委托其他企业生产的,应当提供受托企业生 产许可证和委托协议。生产许可证生产范围应涵盖申报产 品类别
• 境外申请人注册地或生产地址所在国家(地区)医疗器械 主管部门出具的允许产品上市销售的证明文件、企业资格 证明文件
旋光度、降解等 – 药物:定性鉴别、定量分析、药物释放 – 输送系统的化学性能要求
– 其他:无菌、热原/细菌内毒素
注册检验报告和预评价意见
注册检验报告和预评价意见
• 检测样品的典型性
– 如疲劳测试样品
说明书和标签
• 不应有商品名称的表述 • 载明内容符合6号令的规定 • 标签载明内容不应指向原标签 • 说明书中相关信息应与其他资
械的相关性
• DES产品动物实验常见问题
– 载药密度/载药量的确定依据 – 药物释放、药代动力学的相关研究
• BVS产品动物实验相关要求
生产制造信息
生产制造信息
• 明确加工工艺
• 注明关键工艺和特殊工艺,并 说明其过程控制点
• 明确生产过程中各种加工助剂 的使用情况及对杂质(如残留 单体、小分子残留物等)的控 制情况
式(如植入、介入),以及适用范围等 • 相关信息不应指向其他资料
分支型主动脉覆膜支架系统临床试验指导原则

分支型主动脉覆膜支架系统临床试验指导原则The Clinical Trial Guidelines for Branch Aortic Arch Stent Graft SystemIntroduction:引言:In recent years, branch aortic arch stent graft systemshave emerged as a promising treatment option for patients with aortic arch aneurysms. These innovative devicesprovide an alternative to the traditional surgical approach of open repair, offering reduced invasiveness andpotentially shorter hospital stays. As these stent graft systems continue to evolve, it is essential to establish clinical trial guidelines that ensure safety, effectiveness, and reliable outcomes in their implementation. This article aims to outline the key principles that should guideclinical trials for branch aortic arch stent graft systems.Background:背景:Branch aortic arch stent graft systems consist of a main body and multiple branches or fenestrations that allow for the preservation of vital branch vessels in the thoracic aorta. Currently, there are several commercially available branch stent graft systems with different designs and techniques. However, due to their complexity and unique variations among patients, clinical trials are necessary to evaluate their efficacy and safety.Key principles:关键原则:1. Patient selection criteria:患者选择标准:Careful patient selection is crucial for the success of any clinical trial involving branch aortic arch stent graft systems. Patients with suitable anatomy, favorable operative risk profiles, and appropriate indications should be considered. Additionally, preoperative imagingassessments should be standardized to ensure accurate assessment of anatomical suitability and identification of potential contraindications.2. Multidisciplinary collaboration:多学科合作:Given the complexity of branch aortic arch pathology and the necessity for precise device placement,multidisciplinary collaboration is imperative during both the planning stage and actual implementation of clinical trials. Close collaboration between cardiovascular surgeons, interventional radiologists, anesthesiologists, and other relevant specialists will help optimize patient selection criteria, intraoperative decision-making, and postoperative care protocols.3. Standardized procedural techniques:规范化的操作技术:Establishing standardized procedural techniques is essential to ensure consistent outcomes and minimizevariability between different centers participating in clinical trials. This involves defining the optimal approach for device deployment, cannulation of branch vessels, and management of complications or adverse events. The development of a detailed procedural manual that takes into account various anatomical scenarios will provide guidance to operators and assist in reproducibility across centers.4. Long-term follow-up:长期随访:Adequate long-term follow-up is crucial to evaluate the durability and effectiveness of branch aortic arch stent graft systems. Regular monitoring should include imaging assessments, clinical evaluations, and quality-of-life assessments to assess the device's performance over time. The duration and frequency of follow-up visits should be determined based on the anticipated lifespan of the stent graft system.Conclusion:In conclusion, establishing clear clinical trial guidelines for branch aortic arch stent graft systems is paramount to ensure patient safety, optimize outcomes, and facilitate further advancements in this field. By carefully selecting patients, fostering multidisciplinary collaboration, standardizing procedural techniques, and ensuring long-term follow-up, clinical trials can provide valuable evidence on the efficacy and safety of these innovative devices.。
经导管主动脉瓣膜系统注册审查指导原则

经导管主动脉瓣膜系统注册审查指导原则本指导原则旨在指导注册申请人对经导管主动脉瓣膜系统注册申报资料的准备及撰写,同时也为技术审评部门对经导管主动脉瓣膜系统注册申报资料的技术审评提供参考。
本指导原则是对经导管主动脉瓣膜系统的一般要求,申请人应依据产品的具体特性确定其中内容是否适用。
若不适用,需具体阐述理由及相应的科学依据,并依据产品的具体特性对注册申报资料的内容进行充实和细化。
本指导原则是供注册申请人和技术审评人员使用的指导性文件,但不包括审评审批所涉及的行政事项,亦不作为法规强制执行,应在遵循相关法规的前提下使用本指导原则。
如果有能够满足相关法规要求的其他方法,也可以采用,但是需要提供详细的研究资料和验证资料。
本指导原则是在现行法规和标准体系以及当前认知水平下制定,随着法规和标准的不断完善,以及科学技术的不断发展,相关内容也将适时进行调整。
一、适用范围本指导原则适用于植入路径为经动脉途径的经导管植入主动脉生物瓣膜及其输送系统。
其他介入方式以及其他植入位置的人工心脏瓣膜系统可参考本指导原则中适用的内容。
按现行《医疗器械分类目录》,该类产品分类编码为13-07-06,管理类别为Ⅲ类。
二、注册审查要点注册申报资料宜符合国家药品监督管理局《关于公布医疗器械注册申报资料要求和批准证明文件格式的公告》中的要求,同时宜符合以下要求:(一)监管信息1.产品名称产品建议命名为经导管主动脉瓣膜系统,如产品植入路径为经静脉途径、经心尖途径等,需在产品名称中进行明确,并提供申报产品名称的确定依据。
2.注册单元划分经导管植入主动脉心脏瓣膜注册单元划分建议依据《医疗器械注册单元划分指导原则》进行,并着重考虑产品的技术原理、结构组成、性能指标及适用范围等因素,如:瓣叶材料、瓣架材料不同时,以及结构设计不同会导致产品安全有效性不同时,应划分为不同注册单元。
(二)综述资料1.器械及操作原理描述1.1提供产品(包括附件)的结构图示(建议提供工程结构图)和照片,图示中注意标注各部分名称、关键尺寸信息及测量位置,结合图示详细描述申报产品的结构组成、产品所有部件组成的工作原理/功能/作用机理,明确瓣膜各部分的连接情况。
经导管植入主动脉瓣膜系统注册审查指导原则

经导管植入主动脉瓣膜系统注册审查指导原则1. 引言:主动脉瓣膜(aortic valve,AV)疾病是一种常见的心脏瓣膜疾病,影响数以百万计的患者。
随着介入治疗技术的不断进步,经导管植入主动脉瓣膜系统(transcatheter aortic valve implantation,TAVI)已成为影响AV疾病治疗的重要方法之一。
为保障患者的安全和治疗效果,本指导原则主要针对TAVI系统的注册审查进行详细阐述,以明确TAVI系统符合注册审查的各项指导原则,规范TAVI系统的研究开发过程。
2. 适应症:目前,TAVI系统适用于高危和极高危AV疾病人群,包括以下情况:(1)症状性AV狭窄或关闭不全,且严重程度达到或超过美国心脏协会/美国心脏病学会(American College of Cardiology/American Heart Association,ACC/AHA)指南中相应的指示。
(2)存在严重共病或高危手术风险,例如肺部疾病、肾功能不全、冠状动脉疾病、阻塞性肺部疾病或近期中风等。
3. TAVI系统研究设计的考虑因素:在设计TAVI系统的研究过程中,需要考虑以下因素:(1)研究人群的选择标准,包括对患者疾病状态和手术风险评估的明确和一致的定义。
(2)研究终点和效果的定义,包括术后死亡率、主要不良事件(例如房室传导阻滞、卒中、器械相关性血栓形成等)的发生率、功能改善和生命质量的改善等。
(3)随访期的定义和安排,包括术后的不同随访时间点和检查、术后长期的随访安排和要求等。
(4)TAVI系统的技术规范,包括操作技术的标准化、器械的选择和优化以及围手术期管理等。
(5)研究的伦理考虑,包括对于患者知情同意的要求、数据安全和保护、研究中加入的监测和审计机制等。
4. TAVI系统的技术要求:为确保TAVI系统达到注册和临床应用的要求,需要满足以下技术要求:(1)TAVI系统能够稳定植入到正确的位置,达到良好的孔内位置和合适的心室流入道位置。
覆膜支架介入治疗b型主动脉夹层疗效观察

66World Latest Medicne Information(Electronic Version)2019Vol.19No.2•临床研究-覆膜支架介入治疗B型主动脉夹层疗效观察袁幸(贵州省人民医院心内科,贵州贵阳550000)摘要:目的对比分析采用常规治疗方法和覆膜支架介入治疗方法对B型主动脉夹层进行干预的效果。
方法于我院2016年4月至2018年4月接收的B型主动脉夹层患者中,将随机双盲法作为参考依据,选取42例患者入组研究,分为常规治疗组和覆膜支架介入组,分别实施常规治疗方法和覆膜支架介入治疗方法。
结果覆膜支架介入组患者的治疗有效率为85.71%,并发症发生率为4.76%,均优于常规治疗组。
结论应将覆膜支架介入治疗方法作为B型主动脉夹层的首选治疗方法,促进患者疾病的迅速康复。
关键词:覆膜支架;介入治疗;主动脉夹层中图分类号:R322.1+21文献标识码:A DOI:10.19613/ki.1671-3141.2019.02.039本文引用格式:袁幸.覆膜支架介入治疗B型主动脉夹层疗效观察[J].世界最新医学信息文摘,2019,19(02):66.0引言B型主动脉夹层具有发病急和病情凶险的特点,极容易导致患者死亡。
临床医学界通常将主动脉发育不良、主动脉硬化以及血压过高作为B型主动脉夹层的主要发病机制。
以往治疗该疾病通常以控制血圧、补充血容量和镇痛为主,不能够彻底治愈该疾病。
基于此,本研究分析了覆膜支架介入治疗B型主动脉夹层的效果,现阐述如下。
1资料与方法1.1 一般资料。
于我院2016年4月至2018年4月接收的B型主动脉夹层患者中,将随机双盲法作为参考依据,选取42例患者入组研究,分为常规治疗组和覆膜支架介入组,常规治疗组21例(女12例,男9例),年龄65-74岁,平均(70.18±0.92)岁。
覆膜支架介入组21例(女13例,男8例),年龄63-76岁,平均(70.97±0.23)岁。
国家药品监督管理局通告2019年第8号——关于发布主动脉覆膜支架系统等3项临床试验指导原则的通告

国家药品监督管理局通告2019年第8号——关于发布主动脉覆膜支架系统等3项临床试验指导原则的通告
文章属性
•【制定机关】国家药品监督管理局
•【公布日期】2019.02.25
•【文号】国家药品监督管理局通告2019年第8号
•【施行日期】2019.02.25
•【效力等级】部门规范性文件
•【时效性】现行有效
•【主题分类】药政管理
正文
国家药品监督管理局通告
2019年第8号
关于发布主动脉覆膜支架系统等3项临床试验指导原则的通
告
为加强医疗器械产品注册工作的监督和指导,进一步提高注册审查质量,国家药品监督管理局组织制定了《主动脉覆膜支架系统临床试验指导原则》《生物可吸收冠状动脉药物洗脱支架临床试验指导原则》《经导管植入式人工主动脉瓣膜临床试验指导原则》,现予发布。
特此通告。
附件:1.主动脉覆膜支架系统临床试验指导原则
2.生物可吸收冠状动脉药物洗脱支架临床试验指导原则
3.经导管植入式人工主动脉瓣膜临床试验指导原则
国家药监局
2019年2月25日。
经导管植入主动脉瓣膜系统注册审查指导原则

经导管植入主动脉瓣膜系统注册审查指导原则导管植入主动脉瓣膜系统(Transcatheter Aortic Valve Implantation, TAVI)是一种介入性手术技术,用于治疗严重的主动脉瓣狭窄。
该技术通过导管将人工瓣膜植入主动脉瓣位置,而无需开胸手术。
为了确保患者的安全和手术的有效性,各国都设立了注册和审查机制。
本文将介绍导管植入主动脉瓣膜系统注册审查的指导原则。
导管植入主动脉瓣膜系统注册审查应遵循国际标准和指南。
各国卫生部门或医疗机构应参考国际专业组织(如美国心脏协会、欧洲心脏病学会)发布的相关指南,制定本国的注册审查指导原则。
这些指南包括手术适应症和禁忌症、手术前评估和准备、手术技术和操作要求、术后管理和随访等内容。
导管植入主动脉瓣膜系统注册审查应考虑患者的安全和手术的有效性。
手术适应症和禁忌症是注册审查的重要内容之一。
患者的年龄、病情严重程度、合并症等因素应被评估,并根据专业指南制定适当的手术适应症和禁忌症。
手术前评估和准备包括患者的心脏功能评估、影像学检查、血液检查等,旨在确保手术的安全性和有效性。
第三,导管植入主动脉瓣膜系统注册审查应关注手术技术和操作要求。
手术技术和操作要求包括手术器械和设备的选择、手术操作步骤、手术中的监测和处理等。
手术器械和设备的选择应基于患者的解剖结构和病变情况,确保手术的顺利进行。
手术操作步骤应规范和标准化,以减少手术风险和并发症发生的可能性。
手术中的监测和处理是确保手术安全和有效性的重要环节,包括对患者的心电图、血压和氧饱和度等生命体征的监测,以及对可能发生的并发症进行及时处理。
导管植入主动脉瓣膜系统注册审查还应包括术后管理和随访。
术后管理包括对患者的护理和康复指导,以及对可能发生的并发症进行监测和处理。
随访是评估手术效果和患者生活质量的重要手段,应定期进行,并记录相关数据。
这些数据可以用于评估手术的长期效果和安全性。
导管植入主动脉瓣膜系统注册审查指导原则应遵循国际标准和指南,并关注患者的安全和手术的有效性。
《经导管植入式主动脉瓣膜临床试验审查原则》

《经导管植入式主动脉瓣膜临床试验审查原则》------------------------------------------作者xxxx------------------------------------------日期xxxx经导管植入式主动脉瓣膜临床试验审查原则(征求意见稿)经导管植入式人工心脏瓣膜(Transcatheter Aortic Valve Implantation,TAVI)产品在开展临床试验研究前,应完成必要的、科学的临床前研究(如性能验证、体外试验、生物学评价、动物实验研究等)。
由于该类产品的安全性和有效性尚需进一步观察,对于首次临床应用的新产品,在开展确证性临床试验前应首先开展可行性试验研究,该研究建议是前瞻性的小样本研究,对产品设计的安全性和性能进行初步评估,然后根据逐渐积累的结果对后期的确证性试验设计提供相应的信息。
可行性试验应有清晰和明确的研究目标。
可行性试验可为系列试验,可在单个中心完成。
单个可行性试验研究时病例数应不少于10例,观察时间应不短于30天。
观察指标为即刻器械成功率及手术成功率,以及30天的全因死亡。
在可行性研究过程中,出现失败病例时应即时进行分析,以确认是否可继续研究;可行性试验结束后,应对数据进行分析评估,为进一步设计临床试验方案(确证性试验或重新开展可行性试验)提供依据。
可行性试验中如出现不良事件时需按相关规定处理。
企业在开展确证性试验研究时,在临床试验方案制定中建议考虑以下因素,包括但不限于:一、患者人群的选择由于TAVI技术作为一种治疗主动脉瓣膜狭窄的全新技术,其安全性和有效性还需进一步进行评估,因此在开展临床试验时,对入组患者的选择应特别注重风险评估,建议建立由心血管内科和心血管外科专家、影像学专家、麻醉专家等组成的多学科心脏团队(心血管外科专家至少两名),对入组患者的风险进行充分评估。
在临床试验中,研究者应确保有足够数量且符合试验方案中入选标准的受试者进入临床试验。
临床试验主协议审核要点
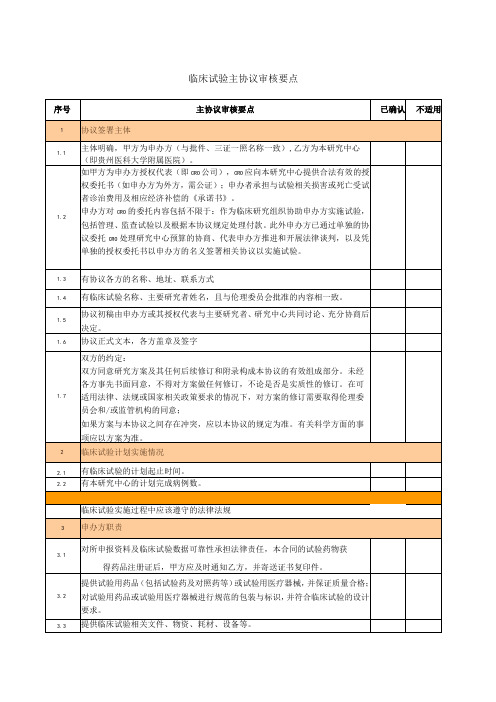
乙方研究者就研究结果在发表论文时,应事先征得甲方书面同意。乙方为多家研究机构时,通讯作者和/或第一作者所属单位为本试验的牵头单位,其他合作医院按贡献大小依次署名。
办方与研究中心共同委托第三方保存,研究中心对临床试验资料的真实性、完整性负责,申办方支付资料保管费用。
5.3
协助研究者接受申办者组织的监查和稽查,及药品监督管理部门的检查
5.4
对临床试验现场使用的试验用药品有管理责任
6
主要研究者职责
6.1
对临床试验数据真实性、完整性、规范性承担直接法律责任。
6.2
详细阅读、了解试验方案的内容,并严格按照方案执行。
注册类临床研究:
因本临床研究产生的任何与研究药物有关的发明、相关文件、原始数据、资料、信息、知识产权、技术秘密或商业利益归甲方所有。甲乙双方参与临床研究的人员依据各自的贡献,经甲方同意,可以作为专利申请的共同发明人。
乙方(包括乙方的合作者)同意为甲方得到专利提供必要的帮助,包括签署法律文件;乙方为学术研究发表本临床研究相关内容,包括但不限于对外发布报告、公开披露数据或者发表文章的,应事先征得甲方书面同意。在研究结果没有正式发表之前,乙方在学术会议上交流该临床研究结果时应事先征得甲方书面同意。
临床试验主协议审核要点
序号
主协议审核要点
已确认
不适用
1
协议签署主体
1.1
主体明确,甲方为申办方(与批件、三证一照名称一致),乙方为本研究中心(即贵州医科大学附属医院)。
1.2
医疗器械临床试验中的样本量如何确定?

医疗器械临床试验中的样本量如何确定?
答:
试验样本量以试验的主要评价指标来确定。
需在临床试验方案中说明确定样本量的相关要素和样本量的具体计算方法。
确定样本量的相关要素包括临床试验的设计类型和比较类型、主要评价指标的类型和定义、主要评价指标有临床实际意义的界值δ(如适用)、主要评价指标的相关参数(如预期有效率、均值、标准差等)、Ⅰ类错误率α和Ⅱ类错误率β以及预期的受试者脱落比例等。
主要评价指标的相关参数依据已公开发表的资料或探索性试验的结果来估算,需要在临床试验方案中明确这些估计值的来源依据。
如主动脉覆膜支架的非劣效试验设计,一般建议α取双侧0.05,β不大于0.20。
具体可参考《医疗器械临床试验设计指导原则》。
对于相关指导原则中对于样本量有明确规定的医疗器械,还需考虑按照指导原则中的相应要求。
审评三部供稿。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
主动脉覆膜支架系统临床试验审评要点
发布时间:2015-04-27 09:20 发布方:弗锐达医疗器械咨询机构浏览次数: 216
随着血管外科技术的不断发展,血管腔内修复主动脉疾病的各类主动脉覆膜支架日益增多。
为了进一步规范该类产品上市前的临床试验,并指导该类产品申请人在申请产品注册时临床试验资料的准备,特制订本审评要点。
本审评要点适用于直管型和腹主动脉分叉型主动脉覆膜支架,带主动脉弓分支、肾动脉分支的分支型主动脉覆膜支架除外。
主动脉覆膜支架产品在开展临床试验前,应当完成必要的临床前研究,如性能验证、体外试验、生物学评价、动物实验等。
对于在市场上尚未出现的全新设计的主动脉覆膜支架,在开展确证性临床试验前应先进行可行性临床试验研究。
可行性试验应有清晰和明确的研究目标,建议为前瞻性的小样本研究(样本量不少于10例,重点观察围手术期(30天)的主要不良事件,以对产品设计的安全性、有效性进行初步评估),并根据其逐渐积累的结果对后期的确证性试验设计提供相应的信息。
企业在开展确证性试验研究时,在临床试验方案制定中建议考虑以下因素,包括但不限于:
一、临床适用范围
产品的临床适用范围应从以下几个方面进行考虑。
(1)适用疾病(必要时,还应明确病程分期或分型):应明确该产品适用的具体临床适用疾病(如主动脉瘤、主动脉夹层等)。
对于主动脉夹层疾病,还应进一步明确其分型(如DeBakey分型的I/II/IIIa/IIIb型或Stanford分型的A型/B型)。
在考虑纳入多个临床适用疾病时,应充分考虑主动脉覆膜支架植入对自然病程的影响及疗效评价是否一致,能否纳入一并分析。
(2)适用部位:用于血管腔内修复的主动脉覆膜支架在进行临床试验时应考虑适用的主动脉部位,建议根据主动脉受累部位,对腹主动脉疾病和胸主动脉疾病分别开展临床试验验证。
(3)适用人群:由于主动脉覆膜支架植入作为一种血管内动脉疾病修复的技术,对其安全性和有效性还需进一步评估,且患者是否适合应用主动脉覆膜支架植入决定于多个临床和解剖因素,其中解剖因素可直接影响主动脉覆膜支架的技术结局和长期耐久性;临床因素如合并症会影响早期和晚期的并发症和死亡
率,因此在开展临床试验时,对入组患者的选择应基于风险受益评估,建议对以下因素进行充分评估:
是否有合适的血管入路;
主动脉疾病的大小和形态学(例如评估动脉瘤颈的长度、角度、形状以及近端、远端锚定区等);
患者年龄、预期寿命、是否适合外科手术;
该技术的短期和长期受益和风险(包括动脉瘤相关死亡率及手术死亡率的风险)。
二、临床试验中的研究终点
主动脉覆膜支架长期的安全性和有效性尚未完全明确,因此在进行临床试验设计的时候,研究假设要同时考虑主要安全性终点和主要有效性终点。
(一)安全性终点
1. 主要安全性终点
对于主动脉瘤或主动脉夹层疾病,主要安全性终点建议至少要考虑术后30天内的主要不良事件(Major Adverse Event, MAE)的发生情况。
2. 次要安全性终点
对于主动脉瘤或主动脉夹层疾病,其他安全性终点还可以考虑全因死亡、主动脉瘤(或主动脉夹层)相关死亡、输送安全性(并发症)等。
(二)有效性终点
1. 主要有效性终点
(1)主动脉瘤疾病:
对于针对主动脉瘤疾病的临床试验,建议主要有效性终点选择复合终点,建议至少应考虑12个月的动脉瘤治疗成功率。
动脉瘤的治疗成功率为至少应当包括主动脉覆膜支架的输送和展开成功、动脉瘤的生长速度、内漏、支架移植物移位发生情况等方面的复合终点。
(2)主动脉夹层疾病
对于针对主动脉夹层疾病的临床试验,主要有效性终点建议为12个月的复合终点,结合考虑临床结果(如即刻技术成功率、内漏、支架移植物移位、主动脉破裂等
发生情况)和主动脉重塑结果(如完全/不完全/无假腔血栓形成,真腔大小、假腔大小以及环主动脉直径的变化等)。
2. 次要有效性终点
对于主动脉瘤或主动脉夹层疾病,其他有效性终点还可以考虑术后即刻技术成功率以及内漏发生率、动脉瘤无继续生长(或主动脉夹层真腔、假腔大小变化)、动脉瘤破裂(或主动脉破裂)、转到手术组等情况。
如果主动脉夹层和主动脉瘤纳入同一研究中,应考虑两者有效性终点和安全性终点的差异,建议两组人群分别分析,均需符合统计学要求。
三、随访
随访时间和随访方式:必须进行长期的影像学随访来评估主动脉覆膜支架的有效性。
建议在术后30天、6个月、12月进行随访,在6个月、12个月时进行一次影像学评估;一年后应进行每年一次的随访,至少连续5年。
完成主要终点评估后可申请上市前注册。
四、试验设计
如条件许可,建议进行有良好设计的前瞻性、对照的多中心临床试验。
对照组建议采用已在中国上市,且国际公认疗效较好的同类器械。
企业应科学地确定试验设计类型,合理地计算样本量并提供样本量计算过程中相关参数的确定依据。
对于非劣效试验设计,一般建议α取双侧0.05,β不大于0.20,按1:1的比例入组,在符合统计学的基础上可评价病例不少于75对(总样本量150例),企业可根据实际情况合理考虑脱落率,计算出最终样本量。
如有合适的理由,也可采用单臂试验,但应进行合理设计,目标值的设定应依据当前充分、科学、合理的同类产品循证医学证据,同时需考虑该类产品技术的发展,目标值的设定应有前瞻性。
单臂试验的样本量在符合统计学原则的基础上应不少于120例可评价病例,企业可根据实际情况合理考虑脱落率,计算出最终样本量。
对于平行研究或单臂研究,建议连续入选所有符合入选/排除标准的病人,并采用基于互联网(IWRS)/电话(IVRS)/传真等计算机系统分配病例登记号,所有病例登记号不得二次使用。