AFLP双酶切和连接方法探讨
双酶切及连接

限制性内切酶的类型
根据限制酶的识别切割特性, 催化条件及是 否具有修饰酶活性可分为Ⅰ、Ⅱ、Ⅲ型三大 类。
限制性内切酶的类型
第一类(I型)限制性内切酶:能识别专一 的核苷酸顺序,它们在识别位点很远的地 方任意切割DNA链,但是切割的核苷酸顺 序没有专一性,是随机的。这类限制性内 切酶在DNA重组技术或基因工程中用处不 大,无法用于分析DNA结构或克隆基因。 这类酶如EcoB、EcoK等。
dna完全没有被内切酶切割原因对策内切酶活性下降内切酶稀释不正确dna不纯反应条件不佳内切酶识别的dna位点上的碱基被甲基化或存在其它修饰部分dna溶液粘在管壁上内切酶溶液粘度大取样不准酶切后dna粘末端退火由于反应溶液温度强烈振荡使内切酶变性过度稀释使酶活性降低反应条件不适识别位点两侧插入了可影响酶切效率的核酸顺序用510倍量过量消化用酶贮藏液或反应缓冲液稀释酶同上同上反应前离心数秒将内切酶稀释增大取样体积电泳前将样品置65保温510分钟取出后置冰浴骤冷使用标准反应缓冲液及温度避免强烈振荡适当稀释酶液反应液稀释的酶不能贮藏使用最佳反应体系加大酶量510倍问题二
限制性内切酶的类型
第二类(III型)限制性内切酶:也有专一的
识别顺序,在识别顺序旁边几个核苷酸对的固 定位置上切割双链。但这几个核苷酸对也不是 特异性的。因此,这种限制性内切酶切割后产 生的一定长度DNA片段,具有各种单链末端。 因此也不能应用于基因克隆。
限制性内切酶的类型
第三类(Ⅱ型)限制性内切酶:就是通常指的DNA限制性 内切酶. 它们能识别双链DNA的特异顺序,并在这个顺序内进行 切割,产生特异的DNA片段; Ⅱ型酶分子量较小,仅需Mg2+作为催化反应的辅助因子,识 别顺序一般为4~6个碱基对的反转重复顺序; Ⅱ型内切酶切割双链DNA产生3种不同的切口--5’端 突出;3’端突出和平末端。 正是得益于限制性的内切酶的发现和应用, 才使得人们能 在体外有目的地对遗传物质DNA进行改造,从而极大地推 动了分子生物学的兴旺和发展。
遗传标记技术中aflp

遗传标记技术中aflpAFLP(Amplified Fragment Length Polymorphism)是一种遗传标记技术,广泛应用于遗传学研究和种质资源保护。
本文将从AFLP的原理、操作步骤、优势和应用等方面进行介绍。
AFLP是一种基于多态性片段长度的遗传标记技术,它结合了PCR 扩增和限制性内切酶切割的特点。
首先,通过PCR扩增目标DNA 片段,然后利用限制性内切酶对扩增片段进行切割,生成多个DNA 片段。
这些片段在聚丙烯酰胺凝胶电泳中分离并检测,形成特定的DNA指纹。
通过比较不同个体或群体之间的AFLP指纹图谱,可以揭示出遗传变异的差异。
AFLP的操作步骤相对简单,主要包括DNA提取、PCR扩增、酶切反应、连接DNA适配体、选择性扩增和聚丙烯酰胺凝胶电泳等。
首先,需要从样品中提取高质量的DNA。
随后,利用特定引物对目标DNA进行PCR扩增,产生大量的扩增片段。
接下来,使用限制性内切酶对PCR产物进行酶切反应,得到多个DNA片段。
这些片段会与连接了适配体的引物结合形成适配体-片段复合物,随后进行选择性扩增,选择性引物将引导特定片段的扩增。
最后,将扩增产物进行聚丙烯酰胺凝胶电泳分离,并使用荧光染料或放射性同位素进行检测和分析。
AFLP技术具有以下几个优势。
首先,AFLP可以同时检测多个位点的多态性,相比传统的基因标记技术,其检测效率更高。
其次,AFLP技术不需要事先了解目标基因序列,适用于未知基因组的研究。
此外,AFLP可以检测到较低频率的遗传变异,对于研究物种间或个体间的遗传关系非常有用。
最后,AFLP技术具有较高的重复性和稳定性,结果可靠性较高。
AFLP技术在遗传学研究和种质资源保护中得到了广泛应用。
在遗传学研究中,AFLP可以用于揭示物种间或个体间的遗传关系、群体遗传结构的分析以及基因组演化的研究。
在种质资源保护中,AFLP可以用于遗传多样性的评估和遗传资源的鉴定,为保护和合理利用遗传资源提供科学依据。
双酶切连接反应之全攻略(

双酶切连接反应之全攻略(双酶切连接(Double Digestion)是一种常用的分子生物学技术,用于在DNA分子上选择性地切割两个特定的限制性内切酶位点。
它可以用于构建重组DNA,进行基因克隆,等等。
下面是一个全面的双酶切连接反应的攻略,包括实验前的准备工作,实验步骤和注意事项。
实验前的准备工作:1.获得限制性内切酶:选择两个互不相容的限制性内切酶。
确保这两个酶能够在相同的反应缓冲液中活性工作。
2.准备DNA底物:获得需要连接的DNA片段。
可以通过PCR扩增,限制性消化或DNA合成等方法获得。
3.选择连接载体:选择合适的连接载体,如质粒。
确保载体具有想要插入的目标基因的适当特性,如选择性标记物(如抗生素抗性基因)和启动子等。
4.验证限制酶位点:使用限制性内切酶图谱检测DNA片段和连接载体中的限制酶位点。
这有助于确定两个限制酶是否能够溶解目标DNA片段。
实验步骤:1.提取DNA:从细菌培养基中提取所需的DNA片段。
可以使用商用试剂盒或自制提取方法。
2.酶切反应:在适当的反应条件下,将需要连接的DNA片段和连接载体分别与两个限制性内切酶一起孵育。
反应条件包括酶的浓度,缓冲液的类型和pH值,反应温度和孵育时间。
3.酶停止反应:通过加入酶停止缓冲液或加热短暂孵育,停止酶切反应。
这样可以避免过度消化和限制酶反应继续进行。
4.凝胶电泳:将切割后的DNA片段经过琼脂糖凝胶电泳分析。
这一步骤可以检测酶切效率和特异性。
将反应样品和相应的对照样品(未经酶切)加载到琼脂糖凝胶上,然后运行电泳以分离DNA片段。
5.库仑凝胶纯化:根据所选的DNA片段大小,可以选择不同浓度的琼脂糖凝胶切片进行纯化。
将所需大小的DNA片段切割下来并进行库仑凝胶分离。
6.连接反应:将纯化的DNA片段与连接载体进行连接反应。
可以使用商业化的连接试剂盒,其中包含待连接DNA和连接载体之间的连接酶,以及其他必要的试剂。
7.转化:将连接后的DNA样品转化到合适的宿主细胞中。
双酶切连接反应的注意要点

双酶切连接反应的注意要点1.选择适当的酶切位点:在进行双酶切连接反应之前,需要选择适当的酶切位点。
这些酶切位点应该满足以下几个要求:-位点不应该在目标DNA序列中出现,以避免酶切产生剪切产物;-两种酶切位点应该在目标DNA序列中相对靠近,以确保连接的有效性;-酶切位点的序列应该被两种酶同时识别和切割。
2.协议的优化:双酶切连接反应的协议需要进行优化,以确定最适合的条件。
一些重要的实验条件包括反应缓冲液的成分和浓度、酶的浓度和反应温度。
对于每个反应参数,应该进行范围的优化实验,以确定最佳的条件。
3.应用正确的酶切酶:双酶切连接反应需要同时使用两种酶来进行切割。
这些酶应该是互相兼容的,并且能够在相同的反应缓冲液中活性。
此外,酶的纯度和活性也应该得到保证,以确保酶切的效果。
4.反应的时间和温度:双酶切连接反应的时间和温度都需要进行优化。
反应时间应该足够长,以确保两种酶都能充分切割目标DNA序列,并且不会出现过度切割的情况。
反应温度也应该适中,通常在酶的推荐温度范围内选择。
5.质量控制:在完成双酶切连接反应之后,应该进行质量控制以确保反应的成功。
常用的方法包括琼脂糖凝胶电泳和DNA测序。
通过这些方法,可以检测连接产物的大小和纯度,并确认连接的正确性。
6.反应产物的处理:根据实验需要,对双酶切连接反应的产物进行处理。
这可能包括:-凝胶电泳分离:使用琼脂糖凝胶电泳分离不同大小的连接产物;-提取纯化:通过凝胶电泳或商业化学试剂盒,从琼脂糖凝胶中提取并纯化连接产物;-DNA测序:对连接产物进行测序,以确认连接的正确性。
总之,双酶切连接反应是一种常用的分子生物学技术,但在实验中需要注意一系列要点。
选择适当的酶切位点、优化实验条件、正确选择酶切酶、时间和温度的控制,以及进行质量控制和反应产物的处理,对于确保双酶切连接反应的成功至关重要。
这些注意要点的遵守可以确保实验结果的准确性和可靠性。
双酶切连接反应常见问题分析

前一阵子一直在做双酶切质粒重组,失败了很多次,不过很快改善了实验方法,用2周重组了14个质粒。
现就自己的体会,谈一下质粒重组的一些个人经验。
1. 回收PCR产物:在进行PCR扩增时候,给引物两端设计好酶切位点,一般说来,限制酶的选择非常重要,尽量选择粘端酶切和那些酶切效率高的限制酶。
选好酶切位点后,在各个酶的两边加上保护碱基。
双酶切时间及其体系:需要强调的是很多人建议酶切过夜,其实完全没有必要,我一般酶切3个小时,其实1个小时已经足够。
应用大体系,如100微升。
2. 纯化问题:纯化PCR产物割胶还是柱式,我推荐柱式,因为割胶手法不准,很容易割下大块的胶,影响纯化效率。
现在的柱式纯化号称可以祛除引物,既然如此,酶切掉的几个碱基肯定也会被纯化掉了。
所以,PCR产物和双酶切产物的纯化均可应用柱式纯化。
3. 酶量的问题:对1单位酶的定义如下:在50μl 反应液中,30℃温度下反应1小时,将1μg 的λDNA完全分解的酶量定义为1个活性单位(U)。
而该酶浓度约为15单位/微升,在除外酶降解的因素外,该酶可分解15μg的DNA,而一般从1-4m l菌液提出的DNA约为3μg,而PCR纯化后的产物(50体系)约为3μg,所以即便全部加进去,只要纯化的质量好,酶切完全切得动。
4. 酶切、回收后的PCR产物与载体的连接:摩尔比的计算,很多人凭经验也可以。
但对于初学者从头认真计算则非常有必要。
回收的载体片段:回收的PCR 产物片段=1:10,一般取前者,后者取。
pmol为单位的DNA转换为为μg单位的DNA:(X pmoles×长度bp×650)/ 1,000,000(注:长度bp×650是该双链DNA的分子量)所得数值即为μg,也可以直接用这个公式套。
1pmol 1000bp DNA=μg,如载体是5380bp,则为××=μg。
5. 测DNA浓度:测DNA浓度可以在专用机子上测,注意OD值,一般约另外,如果嫌麻烦,也可用MARKER进行估测,如MARKER 2000,5微升的 MARKER每个条带约50ng。
AFLP实验方案

AFLP实验方案1.实验试剂、接头和引物限制性内切酶MseI和EcoRI,T4连接酶,Taq DNA 聚合酶,dNTPs,Mg2+,引物,接头。
2.主要试剂的配置1M Tris-HCL(pH=8.0):800 mL ddH2O +Tris碱121.1g 溶解,用浓HCL调pH 到8.0定容至1L,常温保存。
0.5M EDTA (乙二胺四乙酸二钠)(pH=8.0):800 mL ddH2O + EDTA 186.1g,用NaOH调pH至8.0,定容至1L,室温保存。
TE缓冲液:1mL的1M Tris-HCL(pH8.0) + 800 mL ddH2O + 200 μL的0.5M EDTA,4℃保存备用CTAB溶液:NaCl 82g + CTAB 20g + 100 mL Tris-HCL + 40 mLEDTA(0.5mM),用ddH2O定容至1L,65℃,水浴溶解。
5×TBE溶液:Tris-碱54g + 硼酸27.5g + 0.5 M的EDTA 20mL(pH=8.0),加水定容至1L。
10%过硫酸铵(APS)溶液:1g过硫酸铵,加水定容至10 mL,4℃保存(可用范围4天内)40%丙烯酰胺:丙烯酰胺38g,甲叉双丙烯酰胺2g,然后加水定容至100 mL,0.45μM滤膜过滤,4℃保存。
Urea Buffer:尿素529.4g + 235 mL 5×TBE + 定容至1L(加水)(37℃助溶)3M NaAc:8mL H2O,4.801g 乙酸钠50×TAE:Tris-碱242g,EDTA-Na2 37.2g + 800 mL 水,搅匀,然后加57.1g冰醋酸充分溶解,定容至1L(加水)亲水剂:亲水硅烷300 μL + 冰醋酸160 μL + 95%乙醇30mL疏水剂:剥离硅烷1mL + 95%乙醇2mL固定液:无水乙醇200 mL,冰醋酸10 mL,1790 mL的水(可用4次)染色液:同固定液,另加4g AgNO3 避光保存(可用4次)显影液:水2000 mL,NaOH 60g,使用前8 min加4 mL的甲醛变性剂:去离子甲酰胺100 mL,1g 过硫酸钠,4℃保存(可用范围4天内)AFLP实验步骤:基因组DNA的酶切与连接AFLP使用两种限制性内切酶:MseI 和EcoRI(1)酶切MseI /EcoRI双酶切反应体系:MseI酶(10U/μL)0.5 μLEcoRI酶(10U/μL)0.5 μL20×NEB buffer4 2 μL模板DNA 2 μLddH2O 15 μL总体积20 μL37℃酶切3h(1h,2h,3h,4h,5h,6h,12h和24h,检测最佳酶切时间),65℃灭活20 min(2)连接MseI-接头:(+)5’-TACTCAGGACTCAT-3’(-)3’-GAGTCCTGAGTAGCAG-5’EcoRI-接头:(+)5’-CTCGTAGACTGCGTACC-3’(-)3’-CATCTGACGCATGGTTAA-5'AFLP连接体系:MseI酶(10U/μL)0.5 μLEcoRI酶(10U/μL)0.5 μL10×NEB Buffer4 2 μLMseI-adaptor(10 μM)0.5 μLEcoRI-adaptor(10 μM)0.5 μLT4连接酶(400 U/μL)0.1 μL酶切产物10 μLddH2O 5.9 μL总体积20 μL16℃过夜(12-16h)连接,65℃灭活15 min(3)预扩增预扩增引物:MseI-C:5’-CATGAGTCCTGAGTAAC-3’EcoRI-A:5’-GACTGCGTACCAATTCA-3’AFLP预扩增反应体系:10×PCR Buffer(不含Mg2+) 2 μLMgCl2 (10 mM) 1.2 μLdNTP(2.5 mM each) 1.5 μLMseI-C(10 μM)0.5 μLEcoRI-A(10 μM)0.5 μLTaq酶(5 U/μL)0.2 μL酶切连接产物 4 μLddH2O 10.1 μL总体积20 μLPCR扩增反应:94℃ 3 min94℃40 s56℃45 s 24个循环(18,20,22,24,26,28,30寻找最佳)72℃ 1 min72℃10 min反应完后,PCR产物用1.5%的琼脂糖凝胶电泳检测(4)选择性扩增1:20稀释取10 μL预扩增产物,加入190 μL的dd H2O,将预扩增产物稀释20倍,作为选择性扩增的模板选择性扩增的引物序列:MseI-CNN:5’-GATGAGTCCTGAGTAAC-3’EcoRI-ANN:5’-GACTGCGTACCAATTCA-3’扩增产物置于-4℃保存AFLP选择性扩增的体系:10×PCR Buffer(不含Mg2+) 2 μLMgCl2 (10 mM) 1.2 μLdNTP(2.5 mM each) 1.5 μLMseI-CNN(10 μM)0.5 μLEcoRI-ANN(10 μM)0.5 μLTaq酶(5 U/μL)0.25 μL酶切产物 2.5 μLddH2O 11.55 μL总体积20 μL选择性扩增反应:94℃ 5 min;94℃30 s65℃30 s(-0.7℃/cycle)每个循环降低0.7℃13个循环72℃ 1 min94℃30 s56℃30 s 23个循环72℃ 1 min72℃ 5 min选择性扩增的引物序列(5)变性选择性扩增反应结束后,加入变性剂,在PCR仪上95℃变性处理5 min,后迅速取出放置在冰上,于-20℃保存备用。
双酶切步骤

双酶切步骤
嘿,朋友们!今天咱就来讲讲双酶切步骤。
你可别小瞧这双酶切,
它就像是一场精细的手术,每一步都得小心翼翼,不然可就出岔子啦!
首先呢,你得准备好你的“手术工具”,也就是各种试剂和酶啦。
这
就好比厨师要准备好食材和调料才能做出美味佳肴一样。
然后,就是要把你的 DNA 片段给找出来。
这 DNA 片段就像是一个宝贝,得好好对待它,不能让它有一点点损伤哦。
接下来,把酶和 DNA 放在一起,让它们开始“亲密接触”。
这时候
你就得像个守护天使一样,在旁边好好看着,确保一切都按计划进行。
在这个过程中,温度可很重要哦!就像人洗澡水不能太烫也不能太
冷一样,得恰到好处。
不然酶宝宝可不高兴,就不好好工作啦。
时间也是关键啊!不能太短,太短了酶切不完全;也不能太长,太
长了可能会有其他问题出现。
这就好像烤蛋糕,时间掌握不好,蛋糕
不是没熟就是烤焦了。
等酶切结束后,可不能就不管啦。
得检查检查,看看切得怎么样。
这就像是考试结束后要检查一遍试卷一样,可不能马虎。
要是切得好,那当然开心啦,就像考了个好成绩一样。
要是切得不好,也别灰心,找找原因,下次再来。
总之,双酶切步骤虽然听起来有点复杂,但只要你认真对待,就一
定能做好。
就像学骑自行车一样,一开始可能会摔倒,但多练习几次
就会啦。
所以啊,大家别怕困难,大胆去尝试吧!相信自己,一定能掌握好
双酶切步骤,在生物学的世界里畅游无阻!这双酶切啊,真的是很神
奇的一个过程,能让我们看到生命的奥秘一点点被揭开。
大家加油哦!。
AFLP的原理及其应用

基础理论 Basic researchA FLP的原理及其应用王 斌 翁曼丽(中国科学院遗传研究所 100101)提 要:AFLP是检测DNA多态性的一种新的分子标记技术。
对其起源、基本原理、技术程序和应用范围及前景进行了介绍和描述。
关键词:分子标记技术 AFLP 原理 应用1 分子标记技术的快速发展在过去10a中,分子标记技术得到了突飞猛进的发展,至今已有10余种分子标记技术相继出现,并在各个研究领域得到了应用。
其中在植物分子生物学领域中应用最广泛的是RF LP和RA PD。
R FL P 的结果稳定可靠,重复性好,特别适应于建立连锁图,例如水稻RF LP连锁图的建立〔1,2〕。
RF L P比较作图进一步揭示了在主要粮食作物中,R FL P标记在染色体上的排列具有类似的顺序〔3,4〕。
因此R FL P 自出现至今虽然已有10多a了,但它仍是当今应用最广泛的一种分子标记。
然而,R FL P必须经过滤膜转移和So uthern杂交,费时、费力、周期长。
另外, RF L P对DN A多态性检出的灵敏度不高,RF L P连锁图上还有很多大的空间区,这限制了它的进一步发展。
PCR对分子标记技术的发展产生了巨大的推动作用,迄今所用的分子标记技术尽管可以分为多种类型,其实除了以传统的Souther n杂交为基础的RF L P外,其它各类分子标记都涉及P CR。
R AP D就是以随机引物为模板通过P CR扩增进行DN A多态性研究的,与RF LP相比,它较便宜,方便易行,非常灵敏,DN A用量少,而且不需要同位素,安全性好。
继RF L P之后,RA PD是应用最广泛的,特别是在寻找与目的基因连锁的分子标记方面,近年来报道了大量的与各种目的基因连锁的R AP D标记。
近来西红柿P to基因和水稻Xa21基因的成功分离就是首先找到了与目的基因紧密连锁的RA PD标记,然后通过M BC(M ap based clo ning)方法克隆了目的基因〔5,6〕。
AFLP 技术的基本原理
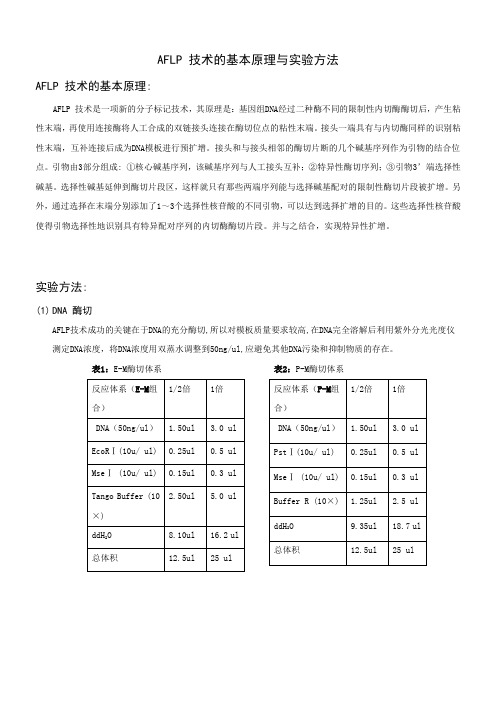
AFLP 技术的基本原理与实验方法AFLP 技术的基本原理:AFLP 技术是一项新的分子标记技术,其原理是:基因组DNA经过二种酶不同的限制性内切酶酶切后,产生粘性末端,再使用连接酶将人工合成的双链接头连接在酶切位点的粘性末端。
接头一端具有与内切酶同样的识别粘性末端,互补连接后成为DNA模板进行预扩增。
接头和与接头相邻的酶切片断的几个碱基序列作为引物的结合位点。
引物由3部分组成: ①核心碱基序列,该碱基序列与人工接头互补;②特异性酶切序列;③引物3’端选择性碱基。
选择性碱基延伸到酶切片段区,这样就只有那些两端序列能与选择碱基配对的限制性酶切片段被扩增。
另外,通过选择在末端分别添加了1~3个选择性核苷酸的不同引物,可以达到选择扩增的目的。
这些选择性核苷酸使得引物选择性地识别具有特异配对序列的内切酶酶切片段。
并与之结合,实现特异性扩增。
实验方法:(1)DNA 酶切AFLP技术成功的关键在于DNA的充分酶切,所以对模板质量要求较高,在DNA完全溶解后利用紫外分光光度仪测定DNA浓度,将DNA浓度用双蒸水调整到50ng/ul,应避免其他DNA污染和抑制物质的存在。
表1:E-M酶切体系表2:P-M酶切体系表3:P-T酶切体系表4:E-T酶切体系表5:M-S酶切体系表6:T-S酶切体系※先将模板吸入PCR板中,再将内切酶、Buffer、双蒸水配制为Mix(因内切酶用量很少或者因少数枪头质量问题,每次吸取内切酶时一定要注意观察吸取是否足量),将配制好的Mix 加入到模板中,最后在配好的反应体系中加入矿物油覆盖离心,放入PCR仪或水浴锅中37℃条件下5-6小时,然后立即转入65℃条件下1小时完全酶切。
一般做1/2倍体系即可。
目前本实验室拥有的内切酶有:EcoRⅠ;MseⅠ;PstⅠ;TaqⅠ;SacⅠ,以上内切酶均为Fermentas 公司生产。
(2)连接表8:连接体系※在连接的过程中不同的内切酶都有其对应的接头(表7),不同的酶切组合就用相应的接头组合。
《双酶切及连接》课件

• 双酶切技术简介 • 双酶切的实验步骤 • 双酶切的应用 • 双酶切的注意事项 • 双酶切技术的发展趋势
01
双酶切技术简介
酶切技术的定义
01
02
03
酶切技术定义
酶切技术是一种利用酶的 专一性对特定底物进行切 割的生物技术。
酶的专一性
酶只对特定的底物起作用 ,切割位点具有高度专一 性。
双酶切技术的改进与创新
新型限制性核酸内切酶的 开发
随着生物技术的不断发展,新型限制性核酸 内切酶不断涌现,为双酶切技术提供更多选 择和灵活性。
自动化双酶切系统的研发
通过自动化技术实现双酶切的快速、高效和 标准化操作,提高实验效率并减少人为误差
。
双酶切技术的发展前景
01
双酶切技术在基因克隆和基因治 疗等领域的应用前景广阔,未来 将继续发挥重要作用。
05
双酶切技术的发展趋势
双酶切与其他技术的结合
双酶切与PCR技术的结合
通过双酶切技术将目的基因和载体进行酶切,再利用PCR技术进行扩增和鉴定,提高基 因克隆的效率和准确性。
双酶切与基因编辑技术的结合
将双酶切技术与CRISPR-Cas9等基因编辑技术结合,实现对特定基因的敲除、敲入和 定点突变等操作,为基因功能研究和基因治疗提供有力工具。
02
随着生物技术的不断进步,双酶 切技术将与其他技术不断融合创 新,为生命科学研究提供更多有 力工具。
THANKS
感谢观看
酶切技术的分类
单酶切技术
使用一种限制性内切核酸酶对 DNA进行切割。
双酶切技术
使用两种不同的限制性内切核酸酶 对DNA进行切割,通常用于产生 具有不同黏性末端的DNA片段, 便于后续的连接反应。
分子标记――AFLP原理和操作步骤

分子标记――AFLP原理和操作步骤一、原理AFLP也是通过限制性内切酶片段的不同长度检测DNA多态性的一种DNA 分子标记技术。
但AFLP是通过PCR反应先把酶切片段扩增,然后把扩增的酶切片段在高分辨率的顺序分析胶上进行电泳,多态性即以扩增片段的长度不同被检测出来。
实验中酶切片段首先与含有与其共同粘末端的人工接头连接,连接后的粘末端顺序和接头顺序就作为以后PCR反应的引物结合位点。
实验中,根据需要通过选择在末端上分别填加了1~3个选择性核苷的不同引物,可以达到选择性扩增的目的。
这些选择性核苷酸使得引物能选择性地识别具有特异配对顺序的内切酶片段,进行结合,导致特异性扩增。
二、实验试剂Taq酶 EcoRI/ MseIEcoRI/ MseI接头 E+A引物M+C引物T4DNALigaseE和M引物琼脂过硫酸胺丙烯酰胺尿素硝酸银甲酰胺 dNTPs 二甲苯青冰醋酸玻璃硅烷50bpMark三、操作步骤(一)基因组DNA提取和纯化A 、参考实验一的大量提取DNA实验方法,B、DNA的纯化:用0.8%琼脂糖凝胶(含EB0.5µg/ml)电泳检测片段大小,取出其中的1/3已提取的基因组DNA进行纯化,首先用TE缓冲液补满至总体积50ul,再等体积苯酚/氯仿/异戊醇(25:24:1)、氯仿/异戊醇(24∶1)各抽提一次, 离心吸上清液于Eppendorf管中,加入1/1 0体积的NaAC和二倍体积预冷的无水乙醇,-20℃放置2h以上,10000g 离心10min,用70%的乙醇漂洗DNA沉淀2次 ,风干后溶于30μlTE缓冲液中,UV-2401PC(岛津)紫外分光光度计检测A260、A280值并定量,再用0.8%琼脂糖凝胶(含EB0.5µg/ml)电泳检测片段大小。
注:0.1-0.2g组织可用100ul溶液E溶,0.5g组织,溶液E可增加至300u l,此时DNA浓度大约为100ng/ul。
(二)限制性酶切及连接在0.2ml离心管中加入:模板量约为250ng,2.5μl 10×酶切缓冲液, 2. 5μl 10×T4DNA连接酶切缓冲液,5U EcoRⅠ, 5U MseⅠ,2U T4连接酶,50pmol MseⅠ接头(序列见表2),双蒸水补至25μl。
AFLP实验操作指南(修改后)

AFLP实验操作指南(修改后)AFLP实验操作指南一、实验操作流程1.提取DNA样本2.DNA酶切、连接同时进行(1)反应体系(50ul)成分体积(ul)双蒸水35.810*Tango buffer 5EcoRI(10u/ul) 0.5MseⅠ(10u/ul) 0.5Ea (5pmol/ul) 1Ma (50pmol/ul) 110mM ATP 1T4DNA连接酶(5U/ul)0.2模板DNA(100ng/ul) 5总体积50(2)反映程序37℃反应3~5个小时,最后保存于4℃。
(3)检测取4-6ul酶切液进行酶切效果检测,跑出的带以弥散状、无明显主带为好。
(4)稀释按1:10的比例稀释,已稀释的和未稀释的均可长时间储存于-20o C备用。
3.预扩增(1)反应体系成分体积(ul)双蒸水 4.4Premix taq 10EA00(100ng/ul) 0.3MC00(100ng/ul) 0.3稀释后已连接DNA模板 5总体积20(2)反映程序a、94℃2minb、94℃30s、56℃30s、72℃1min共24个循环c、72℃5mind、保存于4℃(3)检测取未稀释的预扩增液4-6ul进行琼脂糖电泳检测。
(4)稀释取部分预扩增后溶液按1:25的比例稀释。
将已稀释的和未稀释的保存于-20℃储存。
4.选择扩增(4)反应体系成分体积(ul)双蒸水 4Premix taq 10EA--(100ng/ul) * 0.5MC--(100ng/ul) * 0.5稀释后已连接DNA模板 5总体积20*:EA--,MC—后面两个碱基因不同引物对而不同。
迅速漩涡混匀10s,或者离心4000r/min,10s。
(2)反应程序a、94℃4minb、第1~13个循环:94℃30s, 65℃30s, 72℃1min(退火温度每隔一个循环降低0.7℃);c、第14~36个循环:94℃30s, 56℃30s, 72℃1min;d、72℃5min, 4℃保存。
酶切连接经验之谈

酶切本实验室条件下酶切连接经验之谈1、PCR产物可以切胶回收后酶切,用Elution buffer溶解即可,不影响后续实验。
2、50ulPCR产物切胶回收后用35ul Elution buffer溶解后只需取一半体积用于后续实验即可满足要求。
3、PCR产物可以用乙醇沉淀法获得DNA用于后续反应,但需取少于一半的量用于后续实验,否则会不能完全切开而导致实验失败。
4、双酶切时,若2种酶不是同一厂家时,可以根据Thermo厂家该2种酶的共同buffer,选用Tango缓冲液,一般50ul体系各加1ul酶,而快切酶只需0.5ul,切3小时即可。
5、添加酶切试剂时,应先将buffer和样品振荡均匀后再加入相应的酶,轻弹混匀即可。
6、观察酶切后的载体片段会比质粒大很多,PCR产物双酶切后的片段也比未酶切时要大,并且酶切后产物有时会呈现稍微弥散的宽带,由此可以判断是否切开。
7、部分限制性内切酶对甲基化的DNA不能切割,如FbaI和MboI等,一般生物公司提供的内切酶说明中均有说明。
大多数酶切位点的甲基化不影响切割,而有些会影响,如XbaI, BclI等。
而且甲基化只发生在特定序列,以XbaI为例,只有在位点序列旁出现GA或TC,该XbaI位才会被甲基化。
而要解除这种限制修饰作用通常有两种方法:(1)选用上述酶的同功酶,如Sau3AI,DNA识别切割位点与MboI相同;但不受甲基化影响;(2)利用甲基化酶缺失的受体细胞进行DNA的制备,如E.coli JM110和链霉菌等,前者Dam和Dcm甲基化酶已敲出,而后者细胞内本就没有甲基化酶,从这些细胞中抽提的DNA就能被上述酶切割。
8:E.coli JM110要排除dam,dcm甲基化的影响,需要用特定的dam-,dcm-的菌株,如JM110如果由JM110或SCS110等甲基化缺失的菌株产生的质粒,则不会被甲基化.若酶切不成功可以考虑以下因素的影响a)有些内切酶对PCR产物酶切效率较低b)双酶切无共同buffer时,可以采用分步酶切c)PCR产物直接双酶切不成功,可以选择先做TA克隆后再双酶切d)当载体的2个酶切位点很接近,或者其中一个酶切效率很差时,可以对载体进行去磷酸化,该酶为牛小肠碱性磷酸酶,在大多数限制酶缓冲液中均有活性e)导致“星星活性”可能是体系中甘油浓度过高、高PH、较低的离子强度所致。
菠菜AFLP反应体系初探

菠菜AFLP反应体系初探梅燚;侯雷平;崔艳玲;陈海丽;孟淑春【摘要】提取菠菜嫩叶DNA,运用改良CTAB法提取6份菠菜品种的DNA,对AFLP反应体系的DNA用量、酶切连接、预扩、选扩等试验条件进行了优化分析,初步建立适合于菠菜作物的AFLP反应体系.结果表明:①在PCR仪中酶切的效果比水浴的要好,酶切反应对酶切时间和DNA的浓度要求不敏感.②菠菜AFLP预扩选扩体系的反应体积为20 μL,Mg2+ (25 mmol/L) 1.2 μL,dNTPs (2.5 mmol/L) 1.6 μL,Taq-polymerase (5 U/μL) 0.2 μL最佳.为菠菜AFLP分子标记的品种亲缘关系鉴定和遗传育种等提供一定的理论基础.【期刊名称】《华北农学报》【年(卷),期】2010(025)004【总页数】5页(P111-115)【关键词】菠菜;AFLP;影响因子【作者】梅燚;侯雷平;崔艳玲;陈海丽;孟淑春【作者单位】山西农业大学,园艺学院,山西,太谷,030801;北京市农林科学院,蔬菜研究中心,北京,100097;山西农业大学,园艺学院,山西,太谷,030801;北京市农林科学院,蔬菜研究中心,北京,100097;北京市农林科学院,蔬菜研究中心,北京,100097;北京市农林科学院,蔬菜研究中心,北京,100097【正文语种】中文【中图分类】S636.1AFLP分子标记技术结合了RFLP技术和RAPD技术的特点,既克服了RAPD可靠性低、重复性差的缺点,又避免了RFLP操作复杂和安全等的问题[1,2]。
它具有很好的重复可靠性和PCR技术的高效性[2,3],目前已被广泛应用在遗传图谱的构建[4,5],基因定位及其克隆[6,7],物种的遗传多样性分析[8],种质和种子纯度的鉴定[9]等方面,是一种高效、安全、可靠、投入少的分子标记。
菠菜,别名波斯草,是藜科菠菜属一年生或二年生草本,原产波斯,2000年前已有栽培,中国最晚在唐代已有菠菜的栽培。
实验推荐分子标记技术AFLP

实验推荐分子标记技术AFLP 引言:分子标记技术是现代遗传学和分子生物学研究中不可或缺的工具之一。
由于其高度多态性和高分辨率的特点,AFLP(Amplified Fragment Length Polymorphism,扩增片段长度多态性)技术在种质资源评价、基因组构建和进化生态学研究等领域得到广泛应用。
本文将介绍AFLP 技术的基本原理、实验步骤及数据分析方法,旨在为研究人员提供一种高效、准确的分子标记技术。
一、AFLP技术的原理AFLP技术利用限制性内切酶对基因组DNA进行特异性切割,然后通过PCR扩增和电泳分离等步骤,获得多态性的DNA序列。
其原理流程如下:1. DNA提取:从不同样本(如植物、微生物等)中提取总DNA,保证所需的DNA质量和浓度。
2. 酶切反应:选择适当的限制性内切酶对DNA进行切割,生成特定的DNA片段。
3. 适配体连接:将适配体序列连接到DNA片段的末端,使其成为PCR扩增的起始模板。
4. 扩增反应:利用引物对连接的DNA片段进行PCR扩增,生成大量的AFLP产物。
5. 电泳分离:将扩增的AFLP产物进行电泳分离,根据片段的大小而呈现不同的迁移距离。
6. 成像和数据分析:利用凝胶图像获取AFLP分析结果,并进行数据分析和解读。
二、AFLP实验步骤AFLP实验的具体步骤如下:1. 样本准备:选择适当数量的样本,根据研究目的确定样品的种类和数量,并进行相应的预处理。
2. DNA提取:采用合适的DNA提取方法提取样本中的总DNA,并进行质量和浓度检测。
3. 酶切反应:选择合适的限制性内切酶对DNA进行酶切反应,生成DNA片段。
4. 适配体连接:将适配体序列连接到DNA片段末端,以便进行扩增反应。
5. 扩增反应:利用PCR技术对连接的DNA片段进行扩增,生成大量的AFLP产物。
6. 电泳分离:将扩增的AFLP产物进行凝胶电泳分离,根据片段大小进行排序,形成AFLP图谱。
7. 图谱分析:根据AFLP图谱进行数据分析,解读样品之间的遗传关系。
AFLP分子标记技术的改进——内切酶EcoRⅠ/TruⅠ组合与EcoRⅠ/MseⅠ组合的比较

中 图分 类 号 : 73 1 Q 8 . 文 献 标 识 号 : A 文 章 编 号 :0 1 44 ( 08 0 0 o o 10 — 9 2 2 0 )9— 04一 4
I p o e e t0 m r v m n f AFLP o e u a a k r Te h i u M lc l r M r e c n q e
要: 通过对 E o /Tu1 E o /Ms 两个双酶切组合的 比较 , cRI r 与 cR I eI 证明在 A L F P标记体系中 , 完全
可 以用内切酶 TuI 代昂贵 的内切酶 M eI。E o T I可同时加入 , r 替 s cR I与 m 在不 同温度下分段 进行酶切 : 3 ̄ 7C酶切 4h 然后 6 c , 5c酶切 2h 可达到分步酶切的效果。 ,
Ab ta t I hss d h f c ftersr t n e zme c mbn t n E o I T u IfrAF P wa sr c n ti t y.teef to et ci n y o iai c R / r o L s u e h i o o
山东农 业科 学
20 ,: 69 0894~ ,
AL F P分子 标 记 技术 的改进
内切 酶 E o /TuI 合 与 E o /M eI cRI r 组 cR I s 组合 的 比较
徐 文玲 , 王淑芬 , 牟晋华 , 王翠花 , 贤娴 刘
( 山东省农业科学院蔬菜研究所 , 山东 济南 摘 20 0 ) 5 10
c mp rd w t h o iainEc R I/Ms o ae i tec mbn t o h o eI.I hsmeh d,Ec R Ia dT u 1w r d e g te t n ti to o n r eea d d t eh rwi o h
AFLP技术的基本原理

AFLP技术的基本原理
AFLP 技术的基本原理与实验方法
AFLP 技术的基本原理:
AFLP 技术是一项新的分子标记技术,其原理是:基因组DNA经过二种酶不同的限制性内切酶酶切后,产生粘性末端,再使用连接酶将人工合成的双链接头连接在酶切位点的粘性末端。
接头一端具有与内切酶同样的识别粘性末端,互补连接后成为DNA模板进行预扩增。
接头和与接头相邻的酶切片断的几个碱基序列作为引物的结合位点。
引物由3部分组成: ①核心碱基序列,该碱基序列与人工接头互补;②特异性酶切序列;③引物3’端选择性碱基。
选择性碱基延伸到酶切片段区,这样就只有那些两端序列能与选择碱基配对的限制性酶切片段被扩增。
另外,通过选择在末端分别添加了1~3个选择性核苷酸的不同引物,可以达到选择扩增的目的。
这些选择性核苷酸使得引物选择性地识别具有特异配对序列的内切酶酶切片段。
并与之结合,实现特异性扩增。
实验方法:
(1)DNA 酶切
AFLP技术成功的关键在于DNA的充分酶切,所以对模板质量要求较高,在DNA完全溶解后利用紫外分光光度仪测定DNA浓度,将DNA 浓度用双蒸水调整到50ng/ul,应避免其他DNA污染和抑制物质的存在。
表1:E-M酶切体系
表2:P-M酶切体系
上一页下一页。
aflp技术的原理与应用

AFLP技术的原理与应用1. 简介AFLP(Amplified Fragment Length Polymorphism,扩增片段长度多态性)技术是一种基于PCR扩增的分子标记技术,用于研究基因组的遗传多样性和基因型差异。
它结合了限制性内切酶的切割特点和PCR扩增的高效性,可以快速而高效地分析大量DNA样本上的多态性。
2. 原理AFLP技术将DNA分子进行限制性内切酶切割,得到的片段通过两次PCR扩增反应使其增加特异性引物序列,然后通过聚丙烯酰胺凝胶电泳分析DNA片段的长度差异,从而确定遗传多样性。
AFLP技术的主要步骤包括:2.1 DNA样本的制备首先,需要从研究对象中提取DNA样本,并通过质量检测确保样本的质量和纯度。
通常使用酚/氯仿法、磁珠法等方法提取高质量的DNA。
2.2 限制性内切酶切割将提取的DNA样本与适当的限制性内切酶一起孵育,选择性切割DNA片段。
不同的内切酶会产生不同的切割模式和DNA片段长度。
2.3 适配体连接适配体是特定序列的引物,通过连接适配体,使得限制性内切酶切割的DNA 片段获得特异性序列。
适配体连接反应包括两步PCR反应,第一步是单核苷酸加酶反应(T4 DNA核苷酸转移酶),第二步是锚定PCR。
2.4 扩增和荧光标记经过适配体连接后的DNA片段即可进行扩增反应。
采用带有荧光染料的引物进行PCR扩增,荧光标记的引物可用于不同长度的DNA分子之间的区分。
2.5 凝胶电泳分析将扩增产物进行凝胶电泳分析,通常使用聚丙烯酰胺凝胶,根据DNA片段的长度差异,通过荧光检测仪或射线照射,获得DNA的长度信息。
3. 应用AFLP技术具有高通量、高灵敏度和高分辨率的特点,广泛应用于生物学和农学等领域。
以下是AFLP技术的一些应用:3.1 遗传多样性研究AFLP技术可以用于测定不同个体、种群或物种之间的遗传多样性,从而了解基因组的遗传变异情况。
3.2 基因型鉴定AFLP技术可以用于鉴定不同个体或物种之间的基因型差异,以区分各个个体或物种。
连接双酶切

连接双酶切连接双酶切PCR鉴定目的片段是否连接上表达载体可能的原因,1.表达载体双切不充分,载体量太大,而酶又加的太少,酶切时间短2.连接最好是12~16小时,因为你的目的片段和载体的浓度比,不是很合适3.回收胶的问题今天酶切时没有和空载体对比,TE,含有EDTA,会抑制连接酶的活性洗下来后要先确定一下浓度,浓度太低可定连不上1:在使用 Wash Buffer 洗涤前,在柱子中加入少量 (200ul) 溶胶液。
室温3-5 分钟后,离心。
再接标准洗涤及以后操作。
该操作针对胶块大的情况。
2:加入 Wash Buffer 后,静置 1-3 分钟后,再离心。
3:增加 Wash Buffer 的洗涤次数 (少量多次)。
胶的残留导致的问题不是单纯的胶残留问题,同时也会导致溶胶液中盐的残留 - 这似乎对连接影响更大。
最好的解决方法是 1;当然,可能会导致得率的小量下降,但质量绝对好。
为了充分去除残留,总结,1.多加一步200ul溶胶液,2.加 Wash Buffer 前空柱离心 30 sec,3.Wash Buffer洗涤时静置及多次柱子允许胶通过的量是有限的,胶越多或是浓度太大,残留就会更明显,所以切胶时应该尽量要小,假如过大的话分到两个柱子里应该比较好,另外就是溶胶液宁可多加也不能少加首先要保证待测质粒的纯度,除PCR外,可以对重组质粒做两个双酶切反应:1.ECoR I与Xh0 I双酶切; 2.选一个插入片段中特异存在的切点酶和质粒ECoRI下游到Xba1间的合适位点酶(插入片段中没有的),进行双切,结果可以预测到.每个双切反应包括酶1单切,酶2单切,双切三个反应,别忘了以空载体做对照.试试看.如果没有得到重组质粒只好再连接反应了.酶是TAKARA的,方法就是按说明书上做的,酶各1微升,buffer 2微升,质粒5微升,20微升体系.遇到双酶切可是两个酶没有共用buffer时两种办法:一是低盐buffer的酶先切,然后乙醇沉淀回收,然后高盐buffer的酶再切,乙醇沉淀或切胶回收。
双酶切连接全攻略

双酶切连接反应之全攻略前一阵子一直在做双酶切质粒重组,失败了很多次,不过很快改善了实验方法,用2周重组了14个质粒。
现就自己的体会,结合丁香园战友的宝贵经验,谈一下质粒重组的一些个人经验。
1、回收PCR产物:在进行PCR扩增时候,给引物两端设计好酶切位点,一般说来,限制酶的选择非常重要,尽量选择粘端酶切和那些酶切效率高的限制酶,如BamHI,HindIII,提前看好各公司的双切酶所用公用的BUFFER,以及各酶在公用BUFFER里的效率。
选好酶切位点后,在各个酶的两边加上保护碱基,其原则可参照:/upload/2006/08/13/31219184.pdf。
双酶切时间及其体系:需要强调的是很多人建议酶切过夜,其实完全没有必要,我一般酶切3个小时,其实1个小时已经足够。
应用大体系,如100微升。
纯化问题:纯化PCR产物割胶还是柱式,我推荐柱式,因为割胶手法不准,很容易割下大块的胶,影响纯化效率。
现在的柱式纯化号称可以祛除引物,既然如此,酶切掉的几个碱基肯定也会被纯化掉了。
所以,PCR产物和双酶切产物的纯化均可应用柱式纯化。
我用的是TAKARA的纯化柱试剂盒酶量的问题:以TAKARA的为例,其对1单位酶的定义如下:在50 μl 反应液中,30℃温度下反应1小时,将1 μg 的λDNA完全分解的酶量定义为1个活性单位(U)。
而该酶浓度约为15单位/微升,在除外酶降解的因素外,该酶可分解15μg的DNA,而一般从1-4ml 菌液提出的DNA约为3μg,而PCR纯化后的产物(50体系)约为3μg,所以即便全部加进去,只要纯化的质量好,酶切完全切得动。
2、酶切、回收后的PCR产物与载体的连接摩尔比的计算,很多人凭经验也可以。
但对于初学者从头认真计算则非常有必要。
回收的载体片段:回收的PCR产物片段=1:10,一般取前者0.03pmol,后者取0.3pmol。
pmol为单位的DNA转换为为µg单位的DNA:(X pmoles×长度bp×650)/ 1,000,000 (注:长度bp×650是该双链DNA的分子量)所得数值即为µg,也可以直接用这个公式套.1pmol 1000bp DNA=0.66μg,如载体是5380bp,则0.03pmol为0.03×5.38×0.66=0.106524µg。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
植物保护2003年12月塑垫鲞蔓!塑!丛蔓!堕堕旦坚!型堕竺.200t!堕.!!。
奠!:!
M:Marker(北京鼎国生物技术公司)1:EcoRI(02pL)/M删I(02pL)2:EcoRI(O.3PL)/MseI(03止)3:67一『YJRI.(04£t1.)/MmI(0.4vL)4:F“,RI(0.5vL)/M∞I(05/LL)5:EcoRI(06pl。
)/M.∞I(0.6pL)6:EcoRI(f)7fm)/MseL(07“L)7:EcoRI(O8¨L)/M,el(08gL)8:EcoRI(O9vL)/MseI(09tzL)
圄1小麦基因组DNA不同酶切用量的酶切效果
从图1可看出,5泳道的EcoRI(0.6"L)和Msel(0、6“L)的酶切效果比较理想,基因组100~l800bp问的DNA片段都有出现。
EvoRI和MseI两种酶用量各为0.2uL时酶切DNA片段为700~I800bp之间,各Hj0.3“L的酶切片段在500~一I800bp之间,各用0.4“L的酶切片段在400~1800bp之间,各用0.5"L的酶切片段在250~1800bp之间。
由此可见,由于限制性内切酶』甘量太少,酶切不完全,基因组多态性不丰富。
在6、7、8泳道则酶用量太多,既造成浪费,也容易出现酶切过头现象,产生许多低于100bp的DNA片段,导致接头连接不上,PCR反应失败的结果。
6、7、8泳道的两种酶总用量分别是1.4、1.6、1.8HL,都接近或超过总反应体系15止的l/10(1,5止)。
显而易见,一般两种酶的总量不能超过反应体积的l/10。
2限制性内切酶的缓冲液选择
在双酶切过程中,如果两种酶反应温度一致而缓冲液不同时,可查阅内切酶供应商提供的各种酶在不同缓冲液中的话力表。
如果一种缓冲液能同时使两种酶的活力都超过70%,便可用这种缓冲液作为反应缓冲液。
如果两种酶厂家不同,可比较其缓冲液成份,倘若相似,可各取一半中和,也可使用通用缓冲液。
例如,作者在进行小麦抗锈基因的AFLP标记研究中,选用E∞Rl(上海生工)和MseI(纽英伦生物有限公司)两种限制性内切酶,分别查阅并比较了卜述公司的酶在不同缓冲液中的活力表,确定用BufferY+/TANGO…(表1)。
从表1可见,EcoRI和M”I两种酶在2倍Buffery+/TANCr)TM中括力均为100%,而且没有过度消化现象。
因而选用2倍液Buffery+门"ANGO"‘”
寰1限制性内切酶EcoR[和MseI在苇同缓冲液中韵酶活力情况
1)HufferB+(蓝)Ⅲ10IIml/LTrisHCI(pH75)。
10mmol/LMgCl2,0.1mg/mLBSA;2)BufferG+(绿):10mmo]/I,TrisHCI(pH75).10mmol/LMgCl2,50mmo]/I。
NaCI,0.1mg/mLBSA;3)BufferO+(橘):10mmo[/LTris—HCI(pH75)・10nmaol/LMgCIz,100mmo[/l—N《1.01mg/ml。
BSA;4)BufferR’(红):iommol/LTrisHCI(pH85),10mmol/LMgClz,100mmolA—NaC[。
01mg/mLBSA;5)BuHe“+,l'ANGOⅢ:33mmol/LTris8cetate(pH79),10mmol/I,Mgaeetste,66mmol/LKReState,01rag/mLFKqA。
作为2种内切酶的通用缓冲液。
3酶切时间
酶切时间是双酶切时较易忽略的问题,但却是酶切成败的关键。
酶切时问过长,许多酶可能出现新的活性,就不能在原定的酶切位点切割,使接头连接不上,PCR反应无结暴。
在作琼脂糖检测时,Marker100bp条带下方出现一片弥散,很可能就是酶切时间太长所致。
因为AFLP经过PCR反应扩增出的产物主要在1000bp左右,范围可在100~1500bp之间,小于100bp的小分了量片段,接头可能连接不r。
这是引起PCR扩增不出结果的原因之一。
酶切时间太短,酶坷J不完全,酶切片段就不能覆盖整个基因组,影响PCR扩增的多态性。
一般来讲,酶切时间长短与酶的性质有关,具体
植物保护2003年12月
釜垫鲞箜i塑
旦!:垒盟!P—ROTEC—TION
些!.!!!!上!型.!!!坠生.!
要参看其说明。
在进行第一次酶切时,可进行梯度实验。
每隔半小时或一小时点样观察一次,增加Marker,注意酶切分子量的大小。
作者在使用
EcoRI(上海生工)和MseI(纽英伦生物有限公司)进行双酶切时,设计以下时间段,获得了不同的酶切效果(图2)。
M:Marker(北京鼎国生物技术公司);1酶切1h;2酶切4h;3酶切7
h
4酶切10h;5酶切13h;6.酶切14h;7酶切17h
圈2小麦基因组DNA不同酶切时间的酶切效果
从图2可看出,5、6泳道酶切13h和14h的效果比较理想。
1、2、3、4泳道的酶切时间太短,酶切不完全,多态性不丰富;7泳道酶切时间太长(17h),出现酶切过头现象,使接头连接不上,造成PCR反应失败。
4反应温度及分步酶切
如果两种酶的反应温度不同或两种酶的缓冲液成份相差较大则必须分步酶切。
第一种酶切后用酚抽提、电泳或加热等方法使酶失活,再进行下一次酶切反应。
厂家目录一般都有相应的附录以供查『列各种酶的反应温度。
第一次酶切后,通过电泳确证酶切效果,从胶中吲收DNA片段进行下次酶切,也可在第一种酶切后直接用PCR产物回收试剂盒纯化酶切样品(约需
5
mln),保留少量样品进行电泳分析,其余进行第二
次酶切。
亦可用离心纯化管或离心浓缩管,将酶切样品进行离心,过滤盐等成分,而核酸则被截留在柱子中间的膜上,加入适量ddH,O,离心洗去膜r残留的杂质,再加几微升ddH:O在膜上,将柱子反转插入新的Eppendorf管上离心,即可分离获得DNA片段,做下一次酶切。
5其他注意事项
(1)双酶切时如果两种酶的酶切位点靠得很近,必须注意酶切顺序。
因为有的限制性内切酶要求其识别序列的两端至少保留有若干个碱基才能保证酶的有效纠割。
有的酶要求识别序列两端有多个碱基,则必须先切,否则就可能造成酶切失败。
例如,质粒pLITMUS29的多克隆位点中BarnHI旁边有
H/ndⅢ位点,如果先切BamHI再切H/ndⅢ时就会出现H/ndHI切不开的情况。
(2)DNA酶切完成后,经加热将限制性内切酶失活。
其失活温度和时间根据酶的种类而定。
例如,EcoRI的失活温度是65℃,MseI失活温度是80℃。
因此,酶切后,一般采用80℃水浴20rain的方式使之失活。
(3)酶失活后,将酶切片段连接到特定的接头上。
加热酶切有时并不能达到很好的效果,可能会对连接产生影响,在此情况下可采用酶切后沉淀的方法,具体操作如下。
(a)加1/10体积,pH5.2的NaAc和2倍体积无水乙醇沉淀,在20℃放至2h以上。
(b)4℃,15000r/min或以上,离心15rain。
(c)倒上清液,70%乙醇洗2~3遍。
(d)干燥机干燥5~7
rain。
(e)加TE复溶,即可用于连接反应。
(4)用T4连接酶进行连接反应时,注意T4连接酶的最适反应温度为22℃,连接8~12h,习惯于连接过夜。
参考文献:
[1]是晓梅.孙忐栋,王学德.AFLP标记在植物研究上应用[J]
浙江万里学院学报,2000,13(2):24~26
72]翁曼丽,谢纬武.新一代分子标记技术一AFLP[]]应用与
环境牛物学报,1996,2(4):424~429
[3]工斌,翁曼丽AFLP的原理及其应用[J]杂空水稻,1996,
(5):27~30.
[4]萨姆布鲁克J,弗里奇,EF,曼尼阿蒂斯T分子克隆宴验指南
(第2版)[M]金冬雁.黎孟枫译北京:科学出版社.1996。