上,下运动神经元疾病
运动神经元病的定义及表现症状和治疗措施

运动神经元病的定义运动神经元病(MND)是以损害脊髓前角,桥延脑颅神经运动核和锥体束为主的一组慢性进行性变性疾病。
临床以上或(和)下运动神经元损害引起的瘫痪为主要表现,其中以上、下运动神经元合并受损者为最常见。
运动神经元病与癌症、艾滋病齐名。
只要患了这种病,先是肌肉萎缩,最后在病人有意识的情况下因无力呼吸而死。
所以这种病人也叫“渐冻人”。
【运动神经元病的临床表现】起病缓慢,病程也可呈亚急性,症状依受损部位而定。
由于运动神经元疾病选择性侵犯脊髓前角细胞、脑于颅神经运动核以及大脑运动皮质锥体细胞、锥体束,因此若病变以下级运动神经元为主,称为进行性脊髓性肌萎缩症;若病变以上级运动神经元为主,称为原发性侧索硬化;若上、下级运动神经元损害同时存在,则称为肌萎缩侧索硬化;若病变以延髓运动神经核变性为主者,则称为进行性延髓麻痹。
临床以进行性脊肌萎缩症、肌萎缩侧索硬化最常见。
本病主要表现,最早症状多见于手部分,患者感手指运动无力、僵硬、笨拙,手部肌肉逐渐萎缩,可见肌束震颤。
四肢远端呈进行性肌萎缩,约半数以上病例早期呈一侧上肢手部大小鱼际肌萎缩,以后扩展到前臂肌,甚至胸大肌,背部肌肉亦可萎缩,小腿部肌肉也可萎缩,肌肉萎缩肢体无力,肌张力高(牵拉感觉),肌束颤动,行动困难、呼吸和吞咽障碍等症状。
如早期病变性双侧锥体束,则可先出现双下肢痉挛性截瘫。
运动神经元疾病有哪些表现及如何诊断?根据受损最严重的神经系统部位而定,临床症状也根据病变部位不同而各异,具体分型如下:1. 肌萎缩性侧索硬化症(ALS):最常见。
发病年龄在40~50岁,男性多于女性。
起病方式隐匿,缓慢进展。
临床症状常首发于上肢远端,表现为手部肌肉萎缩、无力,逐渐向前臂、上臂和肩胛带发展;萎缩肌肉有明显的肌束颤动;此时下肢则呈上运动神经元瘫痪,医学教`育网搜集整理表现为肌张力增高、腱反射亢进、病理征阳性。
症状通常自一侧发展到另一侧。
基本对称性损害。
随疾病发展,可逐渐出现延髓、桥脑路神经运动核损害症状,舌肌萎缩纤颤、吞咽困难和言语含糊;晚期影响抬头肌力和呼吸肌。
执业医师考试之精神神经系统,大苗老师讲义,非常精彩!!

精神神经系统,大苗老师讲义,非常精彩!!神经病学概述神经系统病变包括运动障碍和感觉障碍。
运动神经为传出通路,运动障碍时病变在前,感觉神经为传入通路,感觉障碍时病变在后。
运动系统由上运动神经元和下运动神经元组成。
上运动神经元:1.终止于脊髓前角细胞2.上运动神经元损伤导致中枢瘫又叫硬瘫,表现为肌张力增强,腱反射亢进,病理征阳性(巴宾斯基征-最有意义)下运动神经元1.起始于脊髓前角细胞2.下运动神经元损伤导致周围瘫又叫软瘫,表现为肌张力下降,腱反射减弱或消失,病理征阴性,最常见的疾病是小儿麻痹。
定位诊断:上运动神经元1.皮层:破坏导致对侧单瘫;刺激导致为杰克逊癫痫2.内囊:对侧三偏。
对侧偏身感觉障碍、对侧偏瘫、偏盲3.脑桥、脑干:同侧面部周围瘫,对侧躯体硬瘫。
(TMDQ)(瞳孔没问题,因为动眼神经在中脑)。
4.脊髓:1)颈膨大以上损伤导致上下肢均为硬瘫。
2)颈膨大损伤导致上肢的软瘫,下肢的硬瘫。
3)胸髓受损导致下肢的硬瘫,上肢正常。
4)腰膨大受损导致下肢软瘫,上肢正常。
(只要上面出问题,下面全是硬瘫,只要自己出问题就是软瘫)记忆:皮层受损是单瘫,内囊对侧三偏见,脑干受损交叉瘫,上软下硬颈膨大,颈大以上是硬瘫,对浅同深是半切。
5.脑神经所在部位:3.4中,5-8桥,9、10、12在延脑下运动神经元损伤导致节段性迟缓性瘫痪感觉障碍1.浅感觉:指痛、温觉和粗触觉。
传导束:躯体皮肤黏膜痛温觉周围感受器→脊神经节(第一神经元)→脊神经后根后角细胞(二级神经元)灰质前联合交叉→脊髓丘脑侧束→脑干丘脑核团(第三级神经元)→丘脑皮质束→经内囊后肢→大脑皮层中央后回中上部。
2.深感觉:震动觉、位置觉。
传导束:深部感受器和精细触觉感受器→脊神经节(I)→脊神经后根入脊髓后索组成薄束/楔束上行→延髓薄束核和楔束核(Ⅱ)→发出纤维交叉到对侧形成内侧丘系→丘脑核团(Ⅲ)→丘脑皮质束→内囊后肢→大脑皮层中央后回中上部。
3.脊髓半切:对侧浅感觉,同侧深感觉障碍(同学情深)椎体外系管理肢体的协调运动,使拮抗剂协调收缩。
下运动神经元综合症怎么诊断

如对您有帮助,可购买打赏,谢谢下运动神经元综合症怎么诊断
导语:下运动神经元综合症是威胁人们健康的一种恶性疾病,这种疾病的治愈率不高,一旦患上了下运动神经元综合症就会造成患者行动困难,让患者在生
下运动神经元综合症是威胁人们健康的一种恶性疾病,这种疾病的治愈率不高,一旦患上了下运动神经元综合症就会造成患者行动困难,让患者在生活中变得不方便,然而于这种疾病类似的疾病较多,诊断方面是比较难的,需要一些特别的方法来操作。
那么,下运动神经元综合症怎么诊断呢?
1、脑干:一侧脑干病变既损害同侧该平面的脑神经运动核,又可累及尚未交叉至对侧的皮质脊髓束及皮质延髓束,因此引起交叉性瘫痪,即一侧脑神经下运动神经元病瘫痪和对侧肢体上运动神经元病瘫痪。
如一侧中脑病变出现同侧动眼神经或滑车神经瘫痪,对侧面神经、舌下神经及上、下肢的上运动神经元病瘫痪。
2、皮质:这也是运动神经元病要做的检查的一种。
局限性病变仅损伤其一部分,故多表现为一个上肢、下肢或面部瘫痪,称单瘫。
当病变为刺激性时,对侧肢体相应部位出现局限性抽搐(常为阵挛性),皮质病变多见于肿瘤的压迫、皮层梗死、动静脉畸形等。
3、脊髓:这是一种运动神经元病要做的检查方法,横贯性损害可累及本平面脊髓前角细胞和双侧锥体束,故高颈髓(颈1~4)病损,产生四肢上运动神经元瘫,常伴呼吸肌障碍。
颈膨大(颈5~胸2)病损产生上肢下运动神经元瘫痪,下肢上运动神经元瘫痪。
胸髓病损产生双下肢上运动神经元病瘫痪。
腰膨大病损(腰1~骶2)产生双下肢下运动神经元病瘫痪。
生活中我们会遇到非常多的疾病,有些疾病是我们没有办法治疗的,预防疾病常识分享,对您有帮助可购买打赏。
运动神经元疾病诊疗规范2023版

运动神经元疾病诊疗规范2023版运动神经元疾病(motorneurondisease,MND)是一组选择性侵犯上、下运动神经元而引起脊髓前角细胞、下位脑干运动神经核、大脑运动皮质细胞及锥体束进行性变性的神经退行性疾病。
临床表现为不同组合的肌无力、肌萎缩、延髓麻痹及锥体束征,而感觉不受累。
【病因】病因不明,90%为散发性,5%~10%有家族遗传史(常染色体显性遗传为主)。
现已定位的致病基因有SO。
/、TDP43、FUS.C9q∕72基因等25个。
其中SO。
/基因最早发现,约占家族性患者的20%o2008年发现的C9α∕72基因突变在高加索人群中占家族性患者的40%,散发性患者的5%-20%,被认为是全球最常见的致病基因,但亚洲患者中仍以SOD1.突变最多。
免疫异常、环境因素、病毒感染、外伤等可能是散发性患者发病的危险因素。
欧洲及美国的发病率每年(1.5~2)/10万,患病率为(4~8)/10万。
【病理】肉眼可见脊髓萎缩变小。
显微镜下见脊髓前角细胞及延髓、脑桥的脑神经运动核变性破坏,肌肉呈失神经支配性萎缩;脊神经前根轴索断裂、髓鞘脱失;运动皮质的锥体BetZ细胞及其发出的皮质脑干束和皮质脊髓束亦变性破坏。
细胞内或轴索近端出现含泛素或TDP-43的胞质包涵体。
【临床表现】发病年龄大多50~60岁,男女比例约1.5:Io按病损部位不同通常分为四种亚型:肌萎缩侧索硬化(上下运动神经元受累)、进行性(脊)肌萎缩(下运动神经元受累)、进行性延髓麻痹(延髓受累为主)、原发性侧索硬化(上运动神经元受累)。
各型之间随疾病进程可能会转化,多数最终发展为典型的肌萎缩侧索硬化。
1.肌萎缩侧索硬化(amyotrophic1.atera1.sc1.erosis,A1.S)此型最常见。
脊髓前角、脑干运动神经核及锥体束均受累,出现上、下运动神经元损害并存的特征。
典型患者多从一侧上肢远端起病,出现手部肌力减退、肌肉萎缩及肉跳,逐步向近端发展。
运动神经元病临床诊断治疗指南

运动神经元病临床诊断治疗指南【概述】运动神经元病(motor netlron disease,MND)为一组病因不清的系统性疾病。
主要累及上、下运动神经元的慢性进行性变性疾病。
根据受损的病变部位不同而分为进行性脊肌萎缩症、进行性延髓麻痹、原发性侧索硬化和肌萎缩侧索硬化数种类型。
一、肌萎缩侧索硬化【临床表现】肌萎缩侧索硬化(amyotrophic lateral sclerosis,ALS)可分为三型,即散发型(或称经典型)、家族型(5%~10%)和关岛型。
散发型表现为中年或中年以后起病,40岁以下起病者亦不少见。
国内报告最早发病为18岁。
男性多于女性。
起病隐袭,呈慢性进展病程,部分患者为亚急性病程,少数起病后呈急剧进展,可于病后半年左右死亡。
早期出现肌肉无力、肌肉萎缩及肌纤颤。
常自上肢远端手部肌肉开始,可自一侧手肌开始,数月后可波及对侧;也可双侧手肌同时受累,随后波及前臂肌肉及上臂和肩胛部肌肉。
部分患者可以三角肌或冈上、下肌无力开始,造成肩胛下垂,抬肩或臂上举无力。
少数患者可以下肢起病,表现为下肢无力、沉重、走路无力,骨盆带肌肉受累后可有上台阶、楼梯、蹲下起立困难,下肢肌肉萎缩。
随着病情的发展,肌无力和萎缩可延至颈部、躯干、面肌及延髓支配的肌肉,表现为抬头困难、转颈障碍,呼吸肌受累出现呼吸困难,延髓支配肌肉受累则有吞咽困难、咀嚼费力、舌肌萎缩、发音障碍等。
延髓麻痹通常出现于疾病晚期,但也可于手肌萎缩不久后出现,少数情况下为首发症状。
肌纤颤为常见的症状,可在多个肢体中发生。
在舌体由于肌膜薄而可看到肌纤维颤动。
本病很少有感觉障碍,客观感觉异常少见,有少数患者可有痛性痉挛。
绝大多数患者无括约肌障碍。
本病的生存期随临床类型而有不同,短者数月,长者可十余年,最长可至35年,平均生存3年左右。
严重者多因延髓麻痹或呼吸麻痹而死亡。
本病患者在肌肉萎缩的同时出现上运动神经元损害体征,表现为腱反射亢进,手部可引出Hoffmann及Rossolimo等病理征。
运动神经元病PPT课件

• 神经病学 (第8版)
运动神经元病
临床表现
4. 原发性侧索硬化
➢ 首发症状为双下肢对称性僵硬、乏力,行走呈剪刀步态,缓慢进展,逐渐累及双上肢 ➢ 四肢肌张力呈痉挛性增高,腱反射亢进,病理反射阳性,一般无肌萎缩和肌束颤动,感觉
无障碍,括约肌功能不受累 ➢ 可出现假性球麻痹表现
• 神经病学 (第8版)
• 神经病学 (第8版)
运动神经元病
病理
➢ 肉眼:脊髓萎缩变细 ➢ 光镜:脊髓前角细胞、大脑皮质运动区锥体细胞变性、脱失;脑干运动神经核变性 ➢ 泛素化包涵体:存在于患者的神经元细胞胞质内,其主要成分为TDP-43,是ALS的特征
性病理改变
• 神经病学 (第8版)
运动神经元病
病理
➢ 脊神经前根变细,轴索断裂,髓鞘脱失,纤维减少 ➢ 锥体束自远端向近端发展,出现脱髓鞘和轴突变性 ➢ 肌肉呈现失神经支配性萎缩 ➢ 晚期,体内其它组织如心肌、胃肠道平滑肌亦可出现变性改变
• 神经病学 (第8版)
运动神经元病
病因与发病机制
1. 感染和免疫 ALS患者CSF免疫球蛋白升高 血中T细胞数目和功能异常,免疫复合物形成 抗神经节苷脂抗体阳性
2. 金属元素 MND患者有铝接触史,血浆和CSF中铝含量增高
• 神经病学 (第8版)
运动神经元病
病因与发病机制
3. 遗传因素 铜(锌)超氧化物歧化酶基因 TAR DNA结合蛋白基因突变
1. 颈椎病:肌萎缩常局限于上肢,多见手肌萎缩,客观检查常有感觉障碍,可有括约肌 障碍,无延髓麻痹表现 2. 延髓和脊髓空洞症:节段性感觉分离现象,MRI检查可鉴别脊髓或延髓空洞症、脑干 或颈髓肿瘤以及颈椎病
• 神经病学 (第8版)
上下运动神经元的特点

middle cerebral artry 大脑中动脉
parieto-occipital sulcus 顶枕沟
choroids plexus 脉络丛
internal capsule 内囊
caudate head 尾状核头部
.
12
foramen of Monro 室间孔
middle cerebral artry 大脑中动脉
额中回
inferior frontal
gyrus
额下回
central sulcus
中央沟
superior parietal
gyrus
顶上回
postcentral gyrus
中央后回
precentral gyrus
中央前回
centrum semiovale
半卵圆中心
.
4
superior frontal
3、4——脑中桥脑腹;外侧综合征
5-8——(脑M桥il;lard-Gubler综合征) 9-12——展延神经髓、。面神经
锥体束损害:对侧中枢性瘫痪
如累及到脊髓丘脑束:对侧偏身感觉障碍
.
16
上运动神经元瘫痪定位
Dejerine综合征
舌下神经损害:同侧舌肌瘫痪及萎缩 锥体束损害:对侧中枢性瘫痪 内侧丘系损害:对侧肢体感觉障碍
.
17
上运动神经元瘫痪定位
脊髓损害
Brown-Seguard综合征: 病变同侧锥体束征 和深感觉(内侧丘系)障碍, 对侧浅感觉(脊髓丘脑侧束)障碍。
急性全横贯损害: 双侧锥体束受损,出现截瘫或四肢瘫
.
18
上运动神经元瘫痪定位
癔病性瘫痪
1.不符合上述部位和瘫痪的特点。 2.鲜明的心理因素。 3.性别、年龄特征。 4.人格特征。 5.暗示有效。
渐冻症的病理特征与疾病分型

渐冻症的病理特征与疾病分型渐冻症(Amyotrophic Lateral Sclerosis,ALS),又称运动神经元病,是一种神经退行性疾病,主要损害运动神经元,导致肌肉无力和萎缩,严重影响患者的运动功能。
本文将从病理特征与疾病分型两个方面介绍渐冻症。
一、病理特征1. 神经元损害:渐冻症主要侵害中枢神经系统的上运动神经元和下运动神经元。
上运动神经元位于中枢神经系统皮质脊髓束及其下行途径,对横纹肌肉起控制作用;下运动神经元主要位于脊髓前角,通过周围神经与肌肉相连。
渐冻症患者会出现上运动神经元和下运动神经元同时受损的情况。
2. 胶质细胞活化:胶质细胞是中枢神经系统的支持细胞,其在渐冻症中发生了活化。
活化的胶质细胞会释放出炎症因子和氧自由基等有害物质,进一步损伤神经元,加剧疾病的进展。
3. 纤维化变化:渐冻症引起周围神经及脊髓灰质变性,使组织纤维蛋白沉积增加,出现纤维化改变。
这种纤维化改变对神经传导起到阻碍作用,导致肌肉无力和萎缩。
4. 蛋白质异常:有研究表明,渐冻症患者的脑脊液中蛋白质的含量会发生变化,其中一种重要的蛋白质是超氧化物歧化酶(Cu/Zn SOD)。
这种异常的蛋白质会导致氧自由基以及与神经元损害相关的炎症反应。
二、疾病分型根据临床病情和受累部位的不同,渐冻症可分为以下几类:1. 运动神经元病:这是最常见的渐冻症类型,主要受累的是运动神经元。
患者呈现进行性肌肉无力和萎缩,常伴有肌肉痉挛和病理反射。
病情逐渐加重,最终导致全身肌肉功能完全丧失。
2. 前角细胞变性:该型渐冻症主要影响脊髓前角的运动神经元,而上行运动神经元较少受累。
患者表现为肌肉无力和萎缩,但相对而言病情进展较为缓慢,且智力和感觉功能受累较少。
3. 脑神经病变:这种类型的渐冻症会累及脑神经核心区域,如舌下核、眼动神经核等。
患者会表现出吞咽困难、说话困难以及眼球运动异常等症状。
4. 呼吸肌麻痹型:该型渐冻症主要受累的是呼吸肌群,治疗上具有一定的特殊性。
上运动神经元瘫痪名词解释

上运动神经元瘫痪名词解释
上运动神经元瘫痪是一种神经系统疾病,通常由中枢神经系统的损伤或病变引起。
这种疾病会导致肌肉瘫痪、运动功能障碍等症状,严重的情况下甚至会影响呼吸和吞咽功能。
以下是一些有关上运动神经元瘫痪的常见术语和解释:
1. 上运动神经元:指位于大脑皮层或脊髓的神经元,负责控制身体的运动和姿势。
2. 瘫痪:指肌肉失去了运动能力,不能自主收缩和舒张,导致肢体无法移动或运动功能受限。
3. 张力障碍:指肌肉过度收缩,导致肢体僵硬、运动不协调,可能出现痉挛和震颤等症状。
4. 肌肉萎缩:指肌肉组织逐渐变得瘦弱、减少,失去了原本的力量和精细动作能力。
5. 运动功能障碍:指身体无法完成正常运动功能,例如走路、握物、抬手等,可能会导致日常生活和工作的困难。
6. 神经元退行性变:指神经元受到损伤或病变,逐渐退化和死亡,导致神经系统功能障碍。
7. 多发性硬化症:一种自身免疫性疾病,会影响中枢神经系统和周围神经系统,导致多种神经系统症状,包括上运动神经元瘫痪。
8. 脊髓性肌萎缩症:一种常见的神经系统疾病,主要影响脊髓的神经元,导致肌肉萎缩和运动功能障碍。
- 1 -。
运动神经元病的种类

运动神经元病的种类运动神经元病是一类罕见的神经系统疾病,它主要影响运动神经元,导致肌肉无法正常运动。
根据病因和症状的不同,运动神经元病可以分为多种不同的类型。
1. 肌萎缩侧索硬化症(ALS)肌萎缩侧索硬化症是最为常见的一种运动神经元病,它主要累及中枢神经系统的上运动神经元和周围神经系统的下运动神经元。
患者会出现进行性肌肉无力和萎缩,以及进行性运动功能障碍。
ALS病因复杂,可能与遗传、环境和代谢因素有关。
2. 亨廷顿舞蹈症亨廷顿舞蹈症是一种遗传性运动神经元病,它由基因突变引起。
患者会出现进行性的舞蹈样动作和不自主运动,伴随认知功能下降和行为异常。
亨廷顿舞蹈症的病程较长,通常在成年后才会出现症状。
3. 脊髓性肌萎缩症(SMA)脊髓性肌萎缩症是一种由基因突变引起的运动神经元病,它主要累及脊髓前角的下运动神经元。
患者会出现进行性的肌肉无力和萎缩,可能导致呼吸和吞咽困难。
SMA的严重程度和病程因基因突变的不同而有所差异。
4. 多发性硬化症(MS)多发性硬化症是一种自身免疫性疾病,它主要累及中枢神经系统的髓鞘,导致神经传导障碍。
患者会出现多样化的症状,包括肌肉无力、痉挛、平衡障碍和运动协调障碍。
MS的严重程度和病程因个体差异而有所不同。
5. 肌阵挛性侧索硬化症(PLS)肌阵挛性侧索硬化症是一种罕见的运动神经元病,它主要累及中枢神经系统的上运动神经元。
患者会出现进行性的肌肉僵硬和痉挛,但无肌肉萎缩。
PLS病程较长,但进展缓慢。
6. 神经肌肉病神经肌肉病是一类运动神经元病的总称,包括多种不同的亚型。
这些疾病主要累及周围神经系统的下运动神经元,导致肌肉无力和萎缩。
神经肌肉病的病因和症状多样,常见的亚型包括家族性周期性瘫痪、家族性高血钾性瘫痪和肌营养不良等。
以上是运动神经元病的一些常见种类,每种病都有其特定的病因和临床表现。
虽然这些疾病目前尚无特效治疗方法,但针对不同种类的运动神经元病,可以采取支持性治疗和康复治疗,以提高患者的生活质量和功能恢复。
运动神经元疾病分类

运动神经元疾病分类引言运动神经元疾病是一类神经系统疾病,主要影响运动神经元的功能,导致肌肉无力、肌张力异常等运动障碍。
本文将对运动神经元疾病进行详细分类和探讨,以增加对这些疾病的理解和认识。
运动神经元疾病分类上运动神经元疾病上运动神经元疾病是指主要损害大脑皮质皮质运动区的疾病,其特点是肌张力增高、肌力减弱和病理反射阳性。
常见的上运动神经元疾病有以下几种:1.肌萎缩性侧索硬化症(ALS)–病因:大多数ALS病例是散发性的,病因仍然不明确。
–症状:早期症状包括四肢乏力、肌肉萎缩和痉挛,逐渐发展为进行性肌无力。
–治疗:目前尚无治愈ALS的方法,但可以通过药物和治疗手段来减缓病情。
2.原发性侧索硬化症(PMA)–病因:PMA是一种进展缓慢的、只累及上运动神经元的疾病,病因尚不明确。
–症状:主要表现为进行性肌无力和肌肉萎缩,但与ALS不同的是没有痉挛。
–治疗:治疗方法主要包括物理疗法和药物治疗,帮助缓解症状并提高生活质量。
下运动神经元疾病下运动神经元疾病是指主要损害脊髓前角细胞或脑干运动神经核的疾病,其特点是肌张力减低、肌力减弱和病理反射减弱或消失。
常见的下运动神经元疾病有以下几种:3.脊髓性肌萎缩症(SMA)–病因:SMA是一种常染色体隐性遗传疾病,主要是由于脊髓前角细胞生存和功能减退所引起的。
–症状:主要表现为进行性运动功能障碍、肌力减退和肌肉萎缩。
–治疗:目前尚无治愈SMA的方法,但可以通过支持性护理和药物治疗来改善症状。
4.运动神经元病(MND)–病因:MND是一组罕见疾病的总称,包括多种不同类型的运动神经元损害。
–症状:症状取决于受累的神经元类型,可能导致肌无力、肌肉萎缩、运动功能障碍等。
–治疗:治疗方法主要包括药物治疗、康复治疗和支持性护理。
诊断和治疗诊断运动神经元疾病主要依靠病史、临床表现、神经系统检查和辅助检查。
治疗方法根据不同类型的疾病而有所不同,主要包括以下几种:药物治疗药物治疗主要针对疾病症状的缓解和控制,例如使用肌松药物来减轻肌肉痉挛和痛苦。
上下运动神经元鉴别要点

上下运动神经元鉴别要点1.引言1.1 概述概述神经元是神经系统中最基本的功能单元,负责传递和处理信息。
在运动控制中,上下运动神经元扮演着至关重要的角色。
上运动神经元负责控制身体的上半部分肌肉的运动,包括手臂、肩膀和脖子等部位的动作。
下运动神经元则负责控制身体的下半部分肌肉的运动,包括腿部和髋部的动作。
对于神经科学研究人员和临床医生而言,鉴别上下运动神经元的要点至关重要。
这些要点的准确理解和应用,可以帮助我们更好地理解神经元的功能和疾病的产生机制,从而为神经系统疾病的诊断和治疗提供指导。
本文将深入探讨上下运动神经元的鉴别要点,帮助读者更好地理解和应用相关知识。
在下一节中,我们将详细介绍上运动神经元的鉴别要点。
1.2文章结构文章结构的主要目的是为了让读者可以清晰地理解文章的整体框架和内容安排。
本文的结构主要包括引言、正文和结论三个部分。
引言部分主要是对本文主题进行概述,介绍上下运动神经元鉴别要点的重要性和研究背景。
通过引言可以引起读者的兴趣和吸引力,为后续的内容铺垫。
正文部分是全文的核心部分,涵盖了上运动神经元鉴别要点和下运动神经元鉴别要点两个方面的内容。
在正文中,我们将从解剖特征、生理功能、病理变化等多个角度进行阐述,详细介绍上下运动神经元鉴别要点的具体知识。
通过对每个要点的分析和说明,读者可以深入了解和掌握上下运动神经元鉴别的关键要点。
结论部分是对全文内容的总结和归纳。
在结论中,我们将总结上文所论述的上下运动神经元鉴别要点,强调其重要性和实际应用价值。
同时,结论部分还可以对未来可能的研究方向和发展前景进行展望,为读者提供思考和探索的空间。
通过以上的文章结构,读者可以清晰地了解全文的基本组织方式,有助于更好地理解和消化文章的内容。
文章1.3 目的部分的内容可以按照以下方式编写:目的:本文旨在介绍和探讨上下运动神经元的鉴别要点。
随着神经科学研究的不断深入,对于上下运动神经元的认识也在逐渐加深。
准确地鉴别上下运动神经元对于我们理解神经系统的运作机制,特别是运动控制的神经调节是至关重要的。
简述上下运动神经元瘫痪的鉴别

简述上下运动神经元瘫痪的鉴别你知道什么是上运动神经元瘫痪吗?那什么又是下运动神经元瘫痪呢?下面就跟着店铺一起来看看吧。
上运动神经元性瘫痪上运动神经元性瘫痪,亦称中枢性瘫痪,是由皮层运动投射区和上运动神经元径路(皮层脊骨髓束和皮层脑干束)损害而引起。
因瘫痪肌的肌张力增高,故又称痉挛性瘫痪或硬瘫。
因为纤维束的纤维和细胞排列得相当紧密,故上运动神经元瘫痪多为广泛性的,波及整个肢体或身体的一侧。
病因病理病机凡皮层运动投射区和上运动神经元径路受到病变的损害,均可引起上运动神经元性瘫痪,常见的病因有颅脑外伤、肿瘤、炎症、脑血管病、变性、中毒、以及内科某些疾病,如糖尿病、血卟啉病、大红细胞性贫血及维生素B12缺乏等。
临床表现由于病变损害的部位不同,在临床上可产生不同类型的瘫痪,如单瘫、偏瘫、截瘫、四肢瘫等,尽管瘫痪的表现不同,但它们都具有相同的特点,即瘫痪肌肉张力增高、腱反射亢进、浅反射消失、出现所谓连带(联合)运动和病理反射,瘫痪肌肉不萎缩,电测验无变性反应。
诊断及鉴别诊断(一)短暂脑缺血发作(transient ischemic attaks.TIA) 是指一时性脑缺血引起的一种短暂而局限的脑功能丧失。
其上运动神经元性瘫痪的特点是症状突起又迅速消失,一般持续数分钟至数十分钟,并在24小时内缓解,不留任何后遗症,可反复发作。
(二)脑出血(cerebral hemorrhage) 是指原发于脑实质内血管破裂引起的出血。
出现典型的上运动神经元性瘫痪,患者有高血压和动脉粥样硬化病史,以55岁以上中老年人居多,多在动态和用力状态下发病。
出现前数小时至数日常有头痛、眩晕及意识混浊的先兆症状。
起病急,进展快,常出现意识障碍、偏瘫、早期呕吐和其他神经系统局灶症状。
脑脊液压力增高,80%脑脊液中混有血液,50%患者呈血性外观,CT 检查可见颅内血肿高密度阴影。
(三)脑血栓形成(cerebral thromobosis) 是急性脑血管病中最常见的一种。
运动神经元疾病

成人慢性脊肌萎缩征: 18-60岁 男女比例明显高于ALS, 7.5:1 vs 1.3:1 病程长于ALS,
整理版ppt
37
(2)下运动神经元损害的 症状和体征: 肌无力,肌萎缩,肌束震颤, 肌张力及腱反射减低,多起于上肢
远端。 构音障碍,吞咽及呼吸困难
无感觉障碍,括约肌功能不受累。
整理版ppt
38
电生理检查 electrophysiological Examinations
神经传导速度测定 measurement of nerve conduction velocity: 通过刺激周围神经不同点, 在肌肉或神经上记录到电反应。
肌电图 electromyography: 记录肌肉在静止状态、主动收缩和周围 神经受刺激时的电活动。
运动神经元疾病
Motor neuron disease, MND
整理版ppt
1
定义
运动神经元疾病是指以
肌无力(muscular weakness)
肌萎缩( muscular atrophy)和
锥体束征(corticospinal tract signs)
不同组合为临床表现、选择性侵犯:
脊髓(spinal cord)
减低,呈弛缓性瘫痪 减弱或消失 阴性 显著,且早期出现
可有 周围神经病,脊髓前角 灰质炎
整理版ppt
6
整理版ppt
7
根据症状和体症的特殊组合 将运动神经元疾病分为几个亚型 Motor system disease is subdivided into several types on the basis of the particular grouping of symptoms and signs
运动神经元疾病分类

运动神经元疾病分类运动神经元疾病是一类累及运动神经元的神经系统疾病,其特点是运动障碍和肌无力。
这些疾病可以分为两大类:上行性运动神经元疾病和下行性运动神经元疾病。
一、上行性运动神经元疾病上行性运动神经元(UMN)位于中枢神经系统,包括大脑皮质的锥体束和脑干的皮质脊髓束。
当这些上行性运动神经元受损时,会导致肌张力增高、反射亢进和肌肉抽搐等临床表现。
1. 脊髓性肌萎缩(spinal muscular atrophy,SMA)脊髓性肌萎缩是一种常见的遗传性上行性运动神经元疾病,主要特征是进行性的肌无力和肌萎缩。
这种疾病通常在婴儿期或儿童早期发生,并且会导致呼吸困难和吞咽困难等严重并发症。
2. 运动神经元病(motor neuron disease,MND)运动神经元病是一组罕见的上行性运动神经元疾病,包括肌萎缩侧索硬化症(amyotrophic lateral sclerosis,ALS)和原发性进行性肌萎缩等。
ALS是最常见的运动神经元病,其特征是进行性的上行性和下行性运动障碍。
二、下行性运动神经元疾病下行性运动神经元(LMN)位于脊髓前角和脑干的脑神经核。
当这些下行性运动神经元受损时,会导致肌张力降低、反射减弱或消失以及肌无力等临床表现。
1. 肌萎缩侧索硬化症(amyotrophic lateral sclerosis,ALS)ALS在上述上行性运动神经元疾病中已有所提及,在此再次强调其同时累及了上行性和下行性运动神经元。
这种疾病通常在成年期发生,并且会导致进行性的肌无力、肌萎缩和呼吸困难等严重并发症。
2. 帕金森氏综合征(Parkinson's disease,PD)帕金森氏综合征是一种常见的下行性运动神经元疾病,其特征是进行性的肌张力降低、震颤和运动缓慢。
这种疾病通常在中老年人发生,并且会导致平衡障碍和语言障碍等临床表现。
3. 肌萎缩性侧索硬化(progressive muscular atrophy,PMA)肌萎缩性侧索硬化是一种罕见的下行性运动神经元疾病,其特征是进行性的肌无力和肌萎缩。
执业医师-神经病学
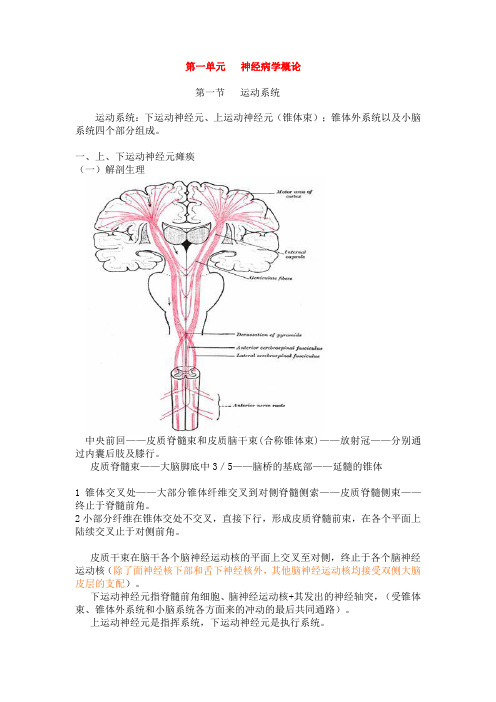
第一单元神经病学概论第一节运动系统运动系统:下运动神经元、上运动神经元(锥体束);锥体外系统以及小脑系统四个部分组成。
一、上、下运动神经元瘫痪(一)解剖生理中央前回——皮质脊髓束和皮质脑干束(合称锥体束)——放射冠——分别通过内囊后肢及膝行。
皮质脊髓束——大脑脚底中3/5——脑桥的基底部——延髓的锥体1锥体交叉处——大部分锥体纤维交叉到对侧脊髓侧索——皮质脊髓侧束——终止于脊髓前角。
2小部分纤维在锥体交处不交叉,直接下行,形成皮质脊髓前束,在各个平面上陆续交叉止于对侧前角。
皮质干束在脑干各个脑神经运动核的平面上交叉至对侧,终止于各个脑神经运动核(除了面神经核下部和舌下神经核外,其他脑神经运动核均接受双侧大脑皮层的支配)。
下运动神经元指脊髓前角细胞、脑神经运动核+其发出的神经轴突,(受锥体束、锥体外系统和小脑系统各方面来的冲动的最后共同通路)。
上运动神经元是指挥系统,下运动神经元是执行系统。
(二)临床表现值得注意的是,在急性严重的病变(如急性脑血管病或急性脊髓炎),脊髓休克期,可以表现为软瘫。
休克过后即逐渐转为硬瘫。
例题1:符合中枢性瘫痪的临床特征是(2000)ZL】A.肌群瘫痪为主B.有肌萎缩C.肌张力增高D.腱反射消失E.无病理反射答案:C例题2:周围性瘫痪也称为(2004)【ZL】A.周围神经损害性瘫痪B.脊髓前角细胞损害性瘫痪C.皮质运动中枢损害性瘫痪D.下运动神经元损害性瘫痪E.脊髓损害性瘫痪答案:D(三)定位诊断1.皮层:损伤——单瘫(对侧对应部位):病变刺激性,对侧相应部位阵发性抽搐(相应部分扩散——杰克逊)2.内囊锥体束——对侧偏瘫,丘脑皮质束,故该处损害——对侧偏身感觉减退,及视放射——对侧同向偏盲,称“三偏”征。
例题1:右侧内囊后肢(后脚)ABC.双眼左侧半视野偏盲DE答案:C解析:内囊后肢受损,可出现三偏症-----病变对侧偏瘫、偏身感觉障碍(深浅感觉障碍)。
偏盲。
例题2:内囊出血所致的对侧肢体运动障碍(偏瘫)AB.皮质红核束CDE答案:A解析:内囊后肢有皮质脊髓束、丘脑辐射和视放射及听放射,其损伤时表现为三偏征对侧肢体运动障碍、感觉障碍及同向性偏盲。
神经病学名词解释整理

1.上运动神经元锥体系:包括上、下两个运动神经元。
上运动神经元胞体主要位于大脑皮质躯体运动中枢的锥体细胞,这些细胞的轴突组成下行的锥体束,下行至脊髓的称皮质脊髓束,至脑干运动神经核的纤维为皮质核束。
上运动神经元损伤可引起痉挛性瘫痪,肌张力增高,深反射亢进,可出现病理反射,早期不出现肌萎缩。
2.下运动神经元:下运动神经元胞体位于脑干脑神经运动核和脊髓前角运动细胞,它们的轴突分别组成脑神经和脊神经,支配全身骨骼肌的随意运动。
下运动神经元受损时,出现弛缓性瘫痪,肌张力降低、肌萎缩,深反射消失。
3.视神经萎缩(optic atrophy ):是指任何疾病引起视网膜神经节细胞和其轴突发生病变,致使视神经全部变细的一种形成学改变,为病理学通用的名词,一般发生于视网膜至外侧膝状体之间的神经节细胞轴突变性。
4.一个半综合征(one-and-a-half syndrome ):又称脑桥麻痹性外斜视,若一侧桥脑侧视中枢(外展旁核)及双侧内侧纵束同时受到破坏,则出现同侧凝视麻痹(一个),对侧核间性眼肌麻痹(半个),即两眼向病灶侧注视时,同侧眼球不能外展,对侧眼球不能内收,向病灶对侧注视时,对侧眼球能外展,病灶侧眼球不能内收,两眼内聚运动仍正常。
5.帕里诺综合征(Parinaud 综合征):又称上丘脑综合征、中脑顶盖综合征、上仰视性麻痹综合征。
由中脑上丘的眼球垂直同向运动皮质下中枢病变而导致的眼球垂直同向运动障碍,累及上丘的破坏性病灶可导致两眼向上同向运动不能。
6.阿-罗瞳孔(Argyell-Robertson 瞳孔):发生在神经梅毒病人中,在半数左右的脊髓痨或者麻痹性痴呆病人中出现,主要是瞳孔缩小、光反射消失、而调节反射存在(调节反射是在看东西有远向近时,两眼向中间辐辏、瞳孔缩小)。
7.艾迪瞳孔:又称强直性瞳孔,其临床意义不明。
表现一侧瞳孔散大,只在暗处用强光持续照射时瞳孔缓慢收缩,停止光照后瞳孔缓慢散大。
调节反射也缓慢出现喝缓慢恢复。
运动神经元病针灸治疗进展

运动神经元病针灸治疗进展运动神经元病(Motor Neuron Disease,MND)是以脊髓前角细胞、脑干后组运动神经元和锥体束为主要病变部位的,一种上、下运动神经元受损且逐渐进展的神经系统变性疾病[1]。
其主要临床表现为肢体肌肉痿软无力、肌束震颤、言语不利、咀嚼无力及吞咽、呼吸困难等,根据其不同的临床表现,可分为原发性侧索硬化(primary lateral sclerosis,PLS)、进行性肌肉萎缩(Progressive muscular atrophy,PMA)、进行性延髓麻痹(Progressive Bulbar Palsy,PBP)、肌萎缩侧索硬化(amyotrophic lateral sclerosis,ALS)四种临床类型,其中最为常见的是ALS[2]。
MND病因不明,致死性高,为神经科疑难杂症之一,目前临床上尚无有效的治疗方法。
中医文献中并无MND相应病名的记载,但是可根据其临床表现不同,将其归为“痿证”、“瘖痱”、“痿痹”等疾病的范畴。
邓铁涛教授认为本病与脾肾密切相关,应用自拟汤剂强肌灵配合捏脊手法治疗该病可稳定病情并提高病人生存质量。
丁元庆教授认为人的呼吸、运动、代谢、言语、吞咽、感觉、二便等功能活动是营卫的外在表现形式,并提出痿证初期的病变脏腑在肺、脾,日久进而累及肝肾;营卫失常、宗气不利、使道不通是痿证发病之根本病机[3]。
冯瑞华通过对文献进行整理分析得出,MND的病机为肝脾肾亏虚,以气虚、精亏及血虚为主,标实为湿、热、痰、瘀等[4]。
大量临床研究发现针灸方法能够有效改善MND患者临床症状,提高患者生活质量,整理如下。
1.针灸疗法:1.1普通针刺:石学敏院士通过醒脑开窍针法及足阳明经筋排刺等手法,有效调整MND患者萎缩肌群经筋生理功能,改善肌肉运动功能。
石教授重视针刺手法(独创凤凰展翅法)及其量效关系,认为其是疗效的关键[5]。
1.2电针疗法:孙远征通过对肌萎缩侧索硬化(ALS)hSOD1G93A转基因小鼠进行研究,发现夹脊电针能够促进星形胶质细胞升高基因-1表达,调节PI3K/Akt信号通路相关蛋白的表达,发挥运动神经元保护作用[6]。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
上,下运动神经元疾病
一些病因不明的疾病,其特征是皮层脊髓束,脊髓前角细胞和/或延髓运动核的进行性变性.
这些疾病的命名都是根据受损最严重的神经系统部位而定,临床症状也根据病变部位不同而各异.发病年龄的中位数是55岁,男性发病率高于女性;5%的病例为家族性,属常染色体显性遗传.
肌萎缩性侧索硬化症肌肉无力,肌萎缩以及前角细胞功能障碍的体征最常先开始出现在手部,较少从足部先开始.发病的部位是随机的,进展呈不对称性.常见肌肉痛性痉挛,可能出现在肌肉无力之前.肉眼可见的肌肉束颤,强直,亢进的深腱反射,伸性划跖反射以及皮层脊髓束受损的体征很快相继出现.呐吃与吞咽困难是由于脑干运动核及通路被累及所致.感觉系统,随意的眼球运动以及膀胱括约肌功能一般不受波及.极少数病人能存活30年;50%病人在发病后3年内死亡;20%能存活5年;10%能存活10年.
进行性延髓瘫痪主要受到影响的是颅神经与皮层延髓束支配的肌肉,因此咀嚼,吞咽与讲话愈来愈困难.可能发生假性延髓型情绪反应,表现为情绪易变与无故哭笑.吞咽困难造成预后特别差;常见病人在1~3年内死亡(往往是死于呼吸道并发症).
进行性肌萎缩症约10%病例具有常染色体显性遗传因素.前角细胞病变进展的速度超过皮层脊髓束的病变,病程进展比较良性.肌肉萎缩与显著的无力开始于手部,然后扩展至臂部,肩部与腿部,最后成为全身性.肌肉束颤可能是疾病最早出现的临床表现.本病可在任何年龄发病,病人可能存活≥25年.
原发性侧索硬化和进行性假性延髓瘫痪临床表现为缓慢进展的强直性肌肉无力,在原发性侧索硬化中是肢体远端部位的肌肉无力,在进行性假性延髓瘫痪中则以后组颅神经支配的肌肉的无力症状为主.肌肉束颤与肌肉萎缩可能发生在许多年以后.这些疾病通常在进展若干年以后才造成病人活动能力的全部丧失.
脊髓灰质炎后综合征很久以前患过脊髓灰质炎的病人可以在灰髓炎恢复后隔了许多年(至少15年)新出现进行性疲乏,疼痛与无力.在某些病例中,还可发生脊髓灰质炎后肌萎缩,与进行性肌萎缩症相似.不过,在大多数病例中,新出现的症状并非是由于远隔的脊髓灰质炎的重新进展,而是由于附加发生的第二种情况,例如糖尿病,椎间盘脱出或关节变性疾病.
诊断和治疗
诊断要点是发病于中,晚年的进行性全身运动功能障碍,不伴感觉功能异常.神经传导速度在疾病进入晚期之前保持正常.肌电图是最有用的辅助检查,甚至在没有临床症状的肢体内也可以查到纤颤,正相波,束颤与巨大动作电位.必须排除肌病,颈椎关节强硬,椎间盘脱出,脊髓肿瘤,脊髓空洞症,颈椎先天性畸形以及糖尿病性肌萎缩症.在原发性侧索硬化与进行性假性延髓瘫痪的鉴别诊断中,必须除外多发性硬化,脊髓压迫症与脑肿瘤.
对这类疾病尚无特殊治疗.理疗有助于肌肉功能的维持.对咽部肌肉无力的病人,喂食必须极其小心,可能需要施行胃造口术.巴氯芬可有助于减轻强直症状,苯妥英钠可用于减轻痛性痉挛.具有强力抗胆碱能作用的阿米替林10mg口服,每日4次,可用于减少唾液分泌.在进行性延髓瘫痪的病例中,旨在改善吞咽功能的手术措施成功率有限.到一定时间,护理主要都是支持性.
脊肌萎缩症
一组可起病于婴儿期或儿童期的疾病,其特征是由脊髓前角细胞与脑干内运动核进行性变性引起的骨骼肌萎缩.
大多数病例都属常染色体隐性遗传,看来都是第5号染色体上一个单独的基因位点上的等位基因突变.有四种主要的变型.
Ⅰ型脊肌萎缩症(Werdnig-Hoffmann病) 在胎儿中已存在或在出生后2~4个月出现症状.大多数患病婴儿在出生时就有肌张力过低的表现;在6个月龄期前,所有患病婴儿都已表现出明显的运动功能发育的延缓.95%的病孩在1岁前后死亡,没有病例能存活超过4岁的,通常都是死于呼吸衰竭.
Ⅱ型(中间型)脊肌萎缩症患儿大多数是在6~12个月期间出现症状,在2岁以前所有病例都已有明显症状.不到25%的病例能学会坐,但没有能走或能爬的.所有患儿都显出肌张力过低,伴松弛性肌肉无力,腱反射消失与肌肉束颤,后者在幼儿中不容易察觉.可有吞咽困难.患儿往往因呼吸道并发症在早年夭折,但也有病情进展自发停顿的,使患儿处于永久性非进展性的无力状态中.
Ⅲ型脊肌萎缩症(Wohlfart-Kugelberg-Welander病) 在2~30岁期间发病.病理变化及遗传方式与前两种变型相似,但病情进展较为缓慢,预期寿命也较长.腿部的无力与肌萎缩最为显著,以股四头肌与髋关节屈肌最早出现症状.较后可累及臂部.无力现象往往从近端向远端扩展.某些家族性病例可能是继发于特殊的酶的缺陷(例如氨基己糖苷酯酶缺乏).
Ⅳ型脊肌萎缩症遗传方式不定(常染色体隐性,常染色体显性,性联),成年期发病(年龄30~60岁),病情进展缓慢.可能无法将其与肌萎缩性侧索硬化症的下运动神经元型病例作鉴别.
诊断和治疗
若肌电图检查发现有失神经支配现象,而神经传导速度检查正常说明失神经支配并非由周围神经病变所引起,则通常可以证实临床诊断.偶尔需作肌肉活检.血清酶(肌酸激酶,醛缩酶)可略见增高.羊膜穿刺不能作出产前诊断.
对这类疾病无特殊治疗.对病情静止或进展缓慢的病例,理疗,支架以及特殊的矫正器材在防止脊柱侧凸与关节挛缩方面可起相当作用.。