系统发育树的编辑
Word编辑法修改系统发育树序列标签教程

Word编辑法修改系统发育树序列标签教程
系统树在文章发表时一定要符合学术常规,如系统树上每个分枝的序列标签名。
这些序列标签名一般都包含菌的登录号和菌的拉丁名等。
当二者放在一起时,登录号需要用正体,而菌名的拉丁文需要用斜体来表示。
(注:或许你会发现在某些已发表的文章中有些作者却忽略了这样的处理)。
你是否也遇到了如何编辑系统树中序列标签名的问题呢?
今天的教程给大家带来另一种系统树编辑的简单方法(Word编辑法),这种方法对于一些同学可能是有些陌生的。
原因在于我们构建完系统树后,保存成的jpg、pdf、tif等格式图往往不知道如何对图片上的标签名进行更改。
Word编辑系统发育树标签名(登录号等正体、拉丁文斜体)操作:
例:MEGA建树
1.采用MEGA软件构建完成系统发育树如下图:
2.然后点击MEGA工具栏里的Image按钮,会看到如下图的保存类型的选项。
系统发育树构建教程(PHYLIP)

系统发育树构建教程(PHYLIP)PHYLIP网址:/phylip.html(一)序列的前期准备1.用ENTREZ或SRS搜索同源DNA/蛋白质序列(same sequence in different organisms) 2.用CLUSTALX进行多条序列比对,在output format option选定PHY格式,构建进化树需要这个phy文件。
Figure 4.1 用clustalx进行多条序列比对3.解压缩phylip-3.68.exe,得到三个文件夹,doc文件夹里是关于所有PHYLIP子程序的使用说明,exe文件夹里是直接可以使用的各个子程序,src文件夹里是所有程序的源文件。
4.打开exe文件夹,双击SEQBOOTt子程序(SEQBOOT是一个利用bootstrap方法产生伪样本的程序),输入刚刚生成的phy文件的路径,点击enter。
5.所有PHYLIP程序默认的输入文件名为infile, 输出文件名为outfile。
如果在exe文件夹里找不到默认的输入文件,会提示can’t find input file “infile”。
Figure 4.2 seqboot程序起始界面6.进入程序参数选择页面(Figure 4.3)。
第一列中的D、J、%、B、R、W、C、S等代表可选的参数。
想改变哪个参数,就键入此参数对应的字母,并点击回车键,对应参数将会发生改变。
当我们设置好所有参数后,(这里我们可以不做任何修改),键入Y,按回车。
此时程序询问“random numbe r seed? <must be odd>”,这是询问生成随机数的种子是多少,输入一个4N+1的数,点击回车程序开始运行,输出结果到文件outfile,保存在当前文件夹里。
.Figure 4.3 seqboot程序参数选择页面主要参数解释:D: 数据类型,有Molecular sequence、discrete morphology、restriction sites和gene frequencies4个选项。
系统发育树构建

1、找模式菌株
Blast后,在LPSN内先找到属,再找到种,点击序列号,fasta后,复制文档至记事本,备注名称为(储藏所编号,序列号)
2、比对
打开GENEDOC,file-import-下载的序列,project-edit sequences list 删除不需要的序列,edit-pairwise alignment-align,edit-clear gap columns,+—号,人工比对(左键添加gaps,右键删除gaps),掐头去尾(edit-select columns-选择需要删除的末尾列-edit-delete all data)-file-export
输出的序列----最上面一行输入序列长度,然后将文档中的>和点号替换掉,除blast登录号外,其他的删除。
3、建树
打开TREECONW,找到treeconw exe打开,distantce estimation –start distantce estimation-找到文档,选择所有文件-打开目标文件-select all-taken into account,yes-ok-boots samples-1000, 后面选择YES,OK。
最后draw phylogenetic tree,点file下的空白。
标尺0.1,统计学50%。
File-copy-任一word文档
4、修改名字
种名(斜体),保藏号,模式菌株加上标T,括号blast序列号。
构建系统发育树的流程

构建系统发育树的流程
1.选择系统学单元:确定研究对象,比如可以是种、属、科、门等分类单元。
2. 收集分类学特征:通过对系统学单元进行形态学、生理学、生态学等多方面的观察和分析,收集其分类学特征,并对这些特征进行编码。
3. 建立相似性矩阵:将不同系统学单元之间的特征相似性进行比较,并将比较结果编码成相似性矩阵。
4. 构建分子系统发育树:利用分子生物学技术,如DNA条形码、基因序列分析等,构建分子系统发育树。
5. 应用系统发育树:利用系统发育树进行分类、进化以及生物多样性研究。
树形图表达了不同物种之间的关系,可以更好地理解生物界的多样性和进化历程。
6. 更新和修订系统发育树:随着研究的不断深入,对系统发育树进行更新和修订,以更好地反映生物系统间的亲缘关系。
- 1 -。
系统发育树的编辑

系统发育树的编辑与图形输出* 可用于系统发育树编辑与图形输出的软件很多,可根据研究目的和使用习惯选择其中的一种或几种使用。
与系统发育分析软件一样,各种编辑系统发育树的软件也可在一下网址下载:/phylip/software.html#Plottinghttp://bioinfo.unice.fr/biodiv/Tree_editors.htmlDendroscope的使用* 该软件可免费下载,地址是:rmatik.uni-tuebingen.de/software/dendroscope* 该软件安装后必须注册,否则部分功能无法解锁,注册密码可在下载软件的网页内的“Dowload”条目下点击“Obtain a license key online”[Use of the program requires a license. Academic licenses are freely available to all academic users. Usage in non-academic settings requires a free commerical license. All uses of the program must be acknowledged by citing our paper on Dendroscope.Obtain a license key online.]Your personal licence-key is:fuiceGUANGXI NORMALfuicewww@11590779一、树的导入和选择运行“Dendroscope”,点击“Fil e→Open”,在弹出的“打开”对话框中指定要打开的树文件,点击“确定”按钮即可导入树文件。
如果一个文件中包含多棵树,可用“Tre e→Next Tree”和“Tre e→Previous Tree”菜单命令选择。
用“Fil e→Recent Files”可直接打开最近编辑过的树文件。
教程:如何给系统发育树的不同部位添加颜色渐变

教程:如何给系统发育树的不同部位添加颜色渐变
系统树的美化可以使用MEGA、Figtree、Treeview等软件,这些软件可以调整系统树中分支的上下位置等(但不影响其系统学关系)。
调整好树形后小编认为PS处理是相当方便的(包括拉丁文斜体,颜色添加等),这也是本人比较喜欢的系统树处理方法。
通过对比以下两张图大家就可以知道哪种背景更合适发文章啦!
大家也许在期刊文章中经常见到系统树中的重要部分添加了背景颜色,但这些背景颜色从左到右均是相同程度的(见下图)。
而在文章中使用渐变样式的背景较为好看(见下图),那么如何添加一种从左到右颜色渐变的背景呢?
今天的教程给大家分享一下如何将系统树中重要分支部分添加颜色渐变。
系统树的美化处理可以让系统树的外观看起来更顺眼,更美观。
首先我们来对比一下两张系统树的图:
下面这个系统树是没有经过添加背景颜色的系统树:
而下面这张图是经过背景颜色渐变之后的系统树:
接下来小编将教大家如何添加背景颜色渐变:
注:当我们把系统树的分支上的bootstrap值添加及标签中拉丁文斜体后我们就可以进行颜色渐变处理了。
步骤:
1.新建一空白图层(操作见下图)
2.点击左侧工具栏中的矩形选框工具,将需要添加渐变背景的区域用矩形选框
画出,见下图红色框标出的区域:。
贝叶斯法构建系统发育树

贝叶斯法构建系统发育树1.打开PAUP软件,打开目标文件和primates文件,将目标文件修改成primates文件格式。
2. 用modeltest3.7软件分析模型参数。
3. 打开mrbayes软件,文件输入。
命令:>execute 文件名.nex4. 设置参数,模型(上面modeltest3.7软件分析模型参数)。
命令:>lset nst=6/2 rates =gamma/invgamma/propinv,若要检查模型的参数,输入命令showmodel。
若设定lset nst=2,需输入命令report tratio=dirichlet。
3.1 >mcmc ngen=100000(1000000) (samplefreq=10(100)),注意:代数可以先设为10000,以便估计时间的长短。
>help mcmc来确认设置。
3.2 运行结束前,标准误差要小于0.01,否则增加代数,继续运行4.1 >sump burnin=250(2500);抽样的25%划为老化样本,舍去。
PSRF值需约等于1.0,否则要运行更长时间。
4.2 >sumt burnin=250(2500),输出所得的进化树,可用treeview打开.Modeltest 3.7基本操作步骤(中文)Moedltest是进行似然法计算必须的软件之一,它可以帮助大家为所获数据选择最佳的模型进行计算,得到最优的结果。
目前该软件的这里介绍一下Modeltest3.7的基本操作步骤:1. 下载Modeltest3.7软件和模型文件modelblockPAUPb10.txt;2. 将序列同源排序后保存为XXX.nex文件;全部拷贝到C盘。
3. 打开模型文件,将文件内容拷贝到XXX.nex文件的末尾,可以将该文件另存为XXX.test.model.nex,保留原来的*.nex文件;;4. 打开PAUP4.0应用程序,将XXX.test.model.nex文件拖入PAUP窗口,然后在命令行输入:execute XXX.test.model.nex,回车后PAUP就开始对数据进行模型估计,结果将保存为model.scores文件和modelfit两个文件,文件位于PAUP4.0软件的文件夹中;5. 将model.scores文件拷贝到Modeltest3.7.win.exe所在的文件夹中。
分子系统发育树构建的简易方法

分子系统发育树构建的简易方法
分子系统发育树的构建是根据分子序列的差异来推断不同物种之间的进化关系。
下面是一个简易的分子系统发育树构建方法:
1. 选择目标基因序列:选择与所研究物种相关的基因序列(如核糖体RNA或蛋白质编码基因)作为目标序列。
2. 数据收集:收集各个相关物种的目标基因序列数据。
可以通过公共数据库(如NCBI)或研究文献中的已有数据进行获取。
3. 序列比对:使用序列比对软件将收集到的序列进行比对,找出相同和不同的碱基或氨基酸位置。
常用的比对软件有CLUSTALW和MAFFT。
4. 构建进化树:根据序列比对结果,使用进化树构建软件(如MEGA)进行系统发育树的构建。
常用的进化树构建方法包括最大简约法(UPGMA)和最大似然法(ML)。
5. 进化树评估:对构建的系统发育树进行评估,可以使用Bootstrap方法进行支持值分析,提高树的可靠性。
6. 结果解读:根据构建的系统发育树,可以解读不同物种之间的进化关系和群体间的分化程度。
需要注意的是,分子系统发育树是基于目标基因序列的进化关系推断,仅仅代表目标基因的进化历史,并不一定能完全反映
整个物种的进化历史。
因此,在研究中还需要综合考虑其他重要因素,如形态特征和生态行为等。
MEGA-系统发育树-快速入门

系统发育树
1.软件准备
DNAman、MEGA
2.序列文件转换格式
2.1先准备一个txt记事本文件,在序列的上一行添加字符>和名称(如>R31-ITS1),然后用MEGA打
开seq格式的序列文件,复制序列到名称下一行
2.2将所需要的比对的所有序列以相同方式写入同一个txt文件中
2.3在MEGA中用ALIGN功能打开准备好的txt文件,选择create a new alignment,数据类型选DNA,
然后从编辑edit中导入新的序列文件(即txt文本)即可导入所需序列
2.4删除无关序列后,先对序列进行分析然后再把序列对齐,类型选DNA,参数默认
颜色一致即为对齐,不一致的就是突变的位点。
然后通常需要把首尾两端没有对齐的序列删掉(只处理首尾两端未对其的序列)
对齐部分
未对齐的删掉
2.5处理完后保存文件并关闭当前窗口,如果不是连续使用的话,切换不同功能时一般点close date
关闭之前的数据
3.构建系统发育树
3.1邻接法构建系统发育树。
构建系统发育树的方法

构建系统发育树的方法
构建系统发育树的方法
一、定义
系统发育树(Phylogenetic Tree)又称为系统种群学树,是一
种描述物种演化的树型结构,从根节点开始描述物种主要进化分支结构,树上的每条边则表示两个物种在进化的历史中距离彼此更近或来自同一进化祖先的关系。
二、建立系统发育树的方法
1.收集数据:系统发育树的建立首先要收集数据,作为建立树的基础,这些数据一般是利用各种实验技术来收集,比如遗传学实验和物种形态的实验。
2.选取特征:从收集的大量数据中,应选取尽可能多的可靠特征,作为建立树的材料,这些特征要有规律性,有可靠性,可以容易发现物种之间的内在关系,有利于在研究中可靠地比较各物种之间的相似程度。
3.分类比较:将所有待比较的物种或实体按照类似的特征进行分类,根据同一物种种的特征之间的差异,可以比较出物种之间的相似度,确定出有利于建立树的特征。
4.描绘树枝:根据比较的结果,可以依次将物种分类编码,从根节点开始,逐级分细枝条,最后得出系统发育树的图形结构。
5.校正树枝:检查系统发育树的构建结果,如果发现有一些物种不太符合物种演化过程的规律,可以根据其他数据和结果来校正树枝,
从而得出最终的发育树结构。
MEGA 系列软件系统发育树构建方法

MEGA软件——系统发育树构建方法1)序列文本构树之前先将每个样品的序列都保存在同一个txt文本文件中,序列只包含序列字母(ATCG或氨基酸简写字母)。
文件名名称可以已经您的想法随意编辑(不能有中文)。
保存为fasta格式2)右键点击fasta文件,打开方式,mega3、全选,点击alignment,algin by culstx(按钮W),OK4、关闭此窗口,点击Yes保存5、再次点击Yes保存,6、点击cancel取消7、选择是否为编码蛋白质的核酸序列8、选择是否用mega打开文件9、点击YES,激活mega,此时mega的菜单栏与刚开始打开的菜单栏有区别。
10、系统发育树构建原理不讲了,此处以构建NJ树为例。
点击工具栏上的phylogeny,construct phylogeny,neighbor joining (NJ).出现如下界面(注意几个绿颜色的小方块):点击第一个小绿方块,选择,小绿方块会变成四个点的省略号,再点击出现如下页面:选择Bootstrap,后面的replication改为1000,点击对勾。
然后点击第三个小绿方块,这个时候对于蛋白质序列以及DNA序列,两者模型的选择是不同的。
对于蛋白质的序列,多选择Poisson Correction (泊松修正)这一模型。
而对于核酸序列,多选择Kimura 2-parameter (Kimura-2参数) 模型。
所有设置完毕之后,点击compute,雏形的树就出来了:可以对此树做出一些修改,比如线条粗细,树的形状等等,此处自己多试试。
6)树的修饰建好树之后,往往需要对树做一些美化。
这个工作完全可以在word中完成,达到发表文章的要求。
点击image,copy to clipboard。
新建一个word文档,选择粘贴。
见下图:在图上点击右键,就可以对文字的字体大小,倾斜等做出修饰。
见下图:PDF,见下图:将打印出来的PDF保存在桌面上,打开,如下图:此时,点击工具,高级编辑工具,裁剪工具,如下图所示:选择需要的区域以删除周围的空白区,双击发育树,会出现下图:点击确定,出现下图(把空边切掉了):点击文件,另存为,在保存类型一栏中选择TIFF格式,点击确定后会生成下面这个图片,所生成图片绝对可以满足文章的发表:OK,结束了,自己玩一把吧。
系统发育树构建的三种方法

系统发育树构建的三种方法
系统发育树(Systems 发育 Tree,简称Stree)是一种用于描述生物系统进化的图形化工具,通常用于模拟生物系统行为的演化过程。
以下是三种构建系统发育树的方法:
1. 基于规则的方法:这种方法使用预定义的规则和偏好来构建
系统发育树。
例如,可以使用遗传算法或人工神经网络等机器学习方法,来预测一个物种的遗传特征或行为演化轨迹。
这种方法需要大量
的人工工作,但可以生成较为准确的演化树。
2. 基于统计方法的方法:这种方法使用统计学方法来推断物种
之间的演化关系。
例如,可以使用最大似然估计或贝叶斯推断等方法,来预测一个物种的遗传特征或行为演化轨迹。
这种方法不需要人工工作,但需要更多的计算资源和时间,才能得到比较准确的演化树。
3. 基于模型的方法:这种方法使用已经建立的模型和数据来构
建系统发育树。
例如,可以使用层次结构模型(如生物进化树、社会网络模型等)来预测一个物种的遗传特征或行为演化轨迹。
这种方法可
以快速构建系统发育树,但需要更多的人工工作来验证模型的准确性。
叶绿体基因组系统发育树的构建

叶绿体基因组系统发育树的构建
叶绿体基因组是一种常用的分子标记,可用于构建植物的系统发育树。
构建叶绿体基因组系统发育树的步骤如下:
1. 叶绿体基因组测序:首先进行叶绿体基因组的测序,获取叶绿体基因组的完整序列信息。
2. 叶绿体基因组序列比对:将测得的叶绿体基因组序列与已知的参考序列进行比对,找出相似的序列区域。
3. 序列选择:根据不同研究目的和物种特点,选择一些适当的叶绿体基因组序列作为研究对象。
一般选择高度保守的基因,如rbcl、matK等。
4. 序列比对:将选定的叶绿体基因组序列进行多序列比对,找出序列间的相似性和差异性。
5. 构建系统发育树:利用多序列比对结果,采用系统发育学分析的方法,如最大似然法、最小进化法等,构建叶绿体基因组的系统发育树。
6. 树的评估和解读:对构建的系统发育树进行评估,包括支持值(bootstrap值)和相似度等。
根据系统发育树的拓扑结构和分支长度,解读物种间的亲缘关系和进化历史。
需要注意的是,叶绿体基因组系统发育树的构建是一种较为复杂和精细的分析过程,需要较高的数据分析能力和专业知识。
同时,样本选择、序列比对和系统发育分析方法的选择也会对结果产生一定影响,因此需要进行合理的设计和操作。
如何制作系统发育树

主要的数据库资源
➢ 数据库是生物信息学的主要内容,各种数据库几乎覆盖了生 命科学的各个领域。
➢ 核酸序列数据库主要有GenBank, EMBL, DDBJ等. ➢ 蛋白质序列数据库有SWISS-PROT, PIR, OWL, NRL3D,
数据库检索:Blast、Entrez 序列分析:序列对位排列、同源比较、进化分析。 统计模型:如隐马尔可夫模型(hidden Markov model, HMM)--
基因识别、药物设计。最大似然模型(maximun likelihood model, ML)、 最大简约法(Maximun Parsimony, MP)--分子进化分析。 算法:如自动序列拼接、外显子预测和同源比较、遗传算法、 人工神经网络(artificial neural network)。
NCBI gi: 995614
FEATURES
Location/Qualifiers
source 1..539
/organism="Rattus norvegicus"
/strain="OLETF, LETO and Zucker"
/dev_stage="differentiated"
/sequenced_mol="cDNA to mRNA"
GenBank由位于马里兰州Bethesda的美国国立卫生研 究院下属国立生物技术信息中心建立,与日本DNA数 据库(DNA Data Bank of Japan,DDBJ)以及欧洲 生物信息研究所的欧洲分子生物学实验室核苷酸数据 库 ( European Molecular Biology Laboratory , EMBL)一起,都是国际核苷酸序列数据库合作的成 员。
系统发育树构建(“序列”文档)共10张

• PHYLIP
• MEGA
• PHYML
• PAUP • BEAST
系统发育树构建软件
• Figtree (树形显示软件)
• TreeView (树形显示软件)
6
系统发育树构建的基本方法
Distance-based methods 基于距离的方法
Unweightedpair group method using arithmetic average (UPGMA) 非 加权分组平均法
系统发育树构建
分子系统发育分析
• 系统发育分析是研究物种进化和系统分类的一种方法, 研究对象为携带遗传信息的生物大分子序列,采用特 定的数理统计算法来计算生物间的生物系统发生的关
系。并用系统进化树来概括生物间的这种亲缘关系。
2
分子系统发育分析
• 系统发育进化树( Phylogenetic tree)
用一种类似树状分支的图形来概括各种生物之间的亲缘关系。 • 系统进化树的主要构成: NCBI——BLAST——输入序列对比——记录好以下几方面: 结点(node):每个结点表示一个分类单元(属、种群)。 文 献: TITLE Circumpolar synchrony in big river
系统发育分析是研究物种进化和系统分类的一种方法,研究对象为携带遗传信息的生物大分子序列,采用特定的数理统计算法来计算生物间的生
• Clustalx比对结果是构建系ห้องสมุดไป่ตู้发育树的前提
具体步骤
• 根据需要,选定要比对的菌株及相应的序 列。将序列COPY至记事本
• 1)File Load sequences 找序列 • 2)Alignment Do complete alignment
MEGA软件——系统发育树构建方法
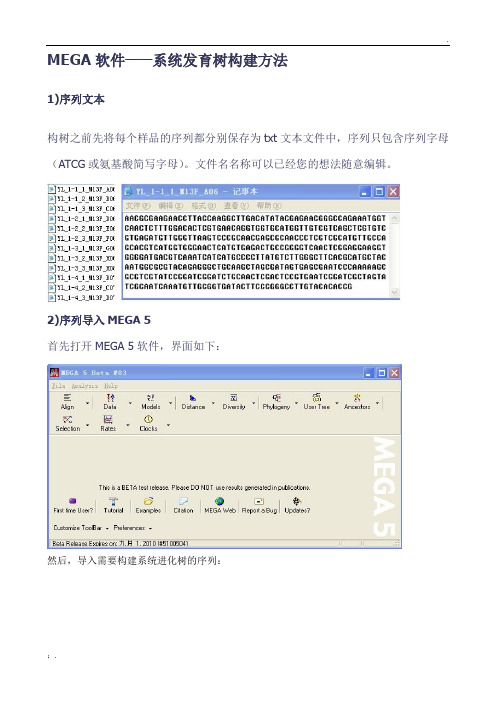
MEGA软件——系统发育树构建方法1)序列文本构树之前先将每个样品的序列都分别保存为txt文本文件中,序列只包含序列字母(ATCG或氨基酸简写字母)。
文件名名称可以已经您的想法随意编辑。
2)序列导入MEGA 5首先打开MEGA 5软件,界面如下:然后,导入需要构建系统进化树的序列:点击OK出现新的对话框,创建新的数据文件导入成功3)序列比对分析点击W,开始比对。
比对完成后删除序列两端不能完全对其的碱基。
系统分析然后,关闭该窗口,在弹出的对话框中选择保存文件,文件名随便去,比如保存为1。
4)系统发育树构建以NJ为例Bootstrap选择1000,点Computer,开始计算计算完毕后,生成系统发育树。
以下“系统发育树树的修饰”方法沿用斑竹brightfuture01的方法5)树的修饰建好树之后,往往需要对树做一些美化。
这个工作完全可以在word中完成,达到发表文章的要求。
点击image,copy to clipboard。
新建一个word文档,选择粘贴。
见下图:在图上点击右键-编辑图片,就可以对文字的字体大小,倾斜等做出修饰。
见下图:这个时候可以通过Adobe professional 对其进行图像导出:先将此word文档打印成PDF,见下图:将打印出来的PDF保存在桌面上,打开,如下图:此时,点击工具,高级编辑工具,裁剪工具,如下图所示:选择需要的区域以删除周围的空白区,双击发育树,会出现下图:点击确定,出现下图(把空边切掉了):点击文件,另存为,在保存类型一栏中选择TIFF格式,点击确定后会生成下面这个图片,所生成图片绝对可以满足文章的发表:OK,结束了,自己玩一把吧。
用PHILIP软件构建系统发育树操作(新)

序列文件准备第一行序列总数量最长序列的bp数第二行>序列名称第三行无空格的序列第一个序列往往是外群然后是自己的序列最后是外源序列Clustal 2.0应用程序File→load sequences 选多序列mode→multiple Alignment modeAlignment→iteraction→iterate each alignment stepAlignment→output formut options→phylip format(选中)→close→do complete Alignment→align找出相似bp的头部,把前边的从文本中切掉,后边的类似(n尽量少)File→load sequencesAlignment→output formut options→phylip format(选中)→close→do complete Alignment→align将生成的.phy文件复制到简单路径中1、phylip→exe→seqboot(应用程序),输入文件的路径名(.phy)改参数:Y 回车101 回车(随机数)生成“outfile文件”改名为:12、prodist(应用程序)输入1改参数:P回车M 回车选D100 回车Y 回车生成“outfile文件”改名为:23、neighbor(应用程序)输入2改参数:M 回车100 回车随机数回车(见文件)O 回车输入外群序列位置回车Y 回车生成“outfile文件”改名为:3“outtree文件”改名为:44、consense (应用程序)输入4改参数:O 回车输入外群序列位置回车Y 回车生成“outfile文件”改名为:5“outtree文件”改名为:65、Proml (应用程序)输入开始的.phy文件改U:直接输入U,回车,应显示NO,use use trees in input file;O改外群。
输出的文件名改为7,intree。
基因DNA序列BLAST及系统发育树构建操作步骤
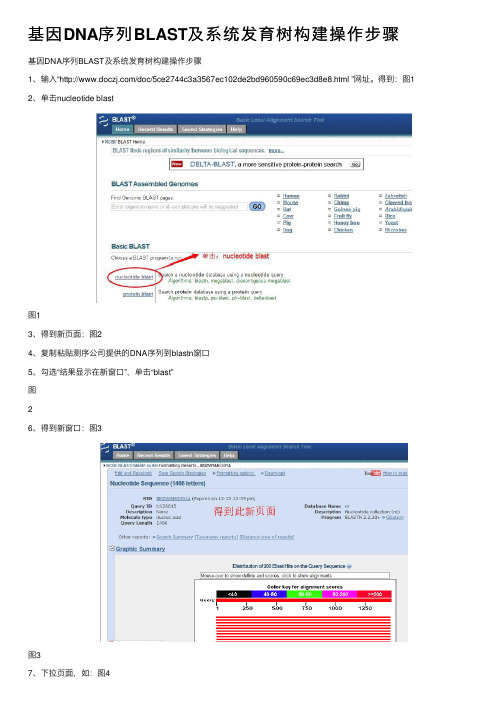
基因DNA序列BLAST及系统发育树构建操作步骤基因DNA序列BLAST及系统发育树构建操作步骤1、输⼊“/doc/5ce2744c3a3567ec102de2bd960590c69ec3d8e8.html ”⽹址。
得到:图12、单击nucleotide blast图13、得到新页⾯:图24、复制粘贴测序公司提供的DNA序列到blastn窗⼝5、勾选“结果显⽰在新窗⼝”,单击“blast”图26、得到新窗⼝:图3图37、下拉页⾯,如:图48、选择Accession下的基因编码。
⼊选第⼀个“HQ711983.1”图49、得到新窗⼝如:图510、单击FASTA图511、得到如:图612、复制菌株种名跟16SrDNA序列,并粘贴到“.txt”⽂件中图613、新建“.txt”⽂件,如:图714、把测序菌株16SrDNA序列和选取blast所得的匹配度⾼的菌株16SrDNA序列粘贴在“.txt”⽂件下,在菌株种名前加“>”,每个序列后空⼀⾏图715、复制“.txt”⽂件,改“.txt”⽂件为“.fasta”⽂件,如:图8图816、⽤MEGA 4.0.2软件打开“.fasta”⽂件,如:图917、单击Alignment⾥的Alinment Explorel/CLUSAC,如:图1018、单击Alignment⾥的Align by clustalW,如:图1119、在弹出窗⼝中单击OK,如:图12图12 19、在弹出窗⼝中单击OK,如:图13图13 20、等待数据运算,如:图14图1421、运算结束,关闭当前窗⼝。
如:图1522、在弹出的第⼀个窗⼝单击NO图15 23、在弹出的第⼀个窗⼝单击YES,如:图16图1624、将⽂件以“.meg”格式保存,如:图17图1725、关闭其余窗⼝,打开刚刚保存的“.meg”⽂件,如:图18图1826、选择phyloeny下的Bootsteap Test of phylogeng下的Neighbor-Joining...,如:图19图1927、在弹出窗⼝单击Compulte,如:图20图2028、单击树状图,得到系统发育树,建议截图保存,如:图21图21。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
系统发育树的编辑与图形输出* 可用于系统发育树编辑与图形输出的软件很多,可根据研究目的和使用习惯选择其中的一种或几种使用。
与系统发育分析软件一样,各种编辑系统发育树的软件也可在一下网址下载:/phylip/software.html#Plottinghttp://bioinfo.unice.fr/biodiv/Tree_editors.htmlDendroscope的使用* 该软件可免费下载,地址是:rmatik.uni-tuebingen.de/software/dendroscope* 该软件安装后必须注册,否则部分功能无法解锁,注册密码可在下载软件的网页内的“Dowload”条目下点击“Obtain a license key online”[Use of the program requires a license. Academic licenses are freely available to all academic users. Usage in non-academic settings requires a free commerical license. All uses of the program must be acknowledged by citing our paper on Dendroscope.Obtain a license key online.]Your personal licence-key is:fuiceGUANGXI NORMALfuicewww@11590779一、树的导入和选择运行“Dendroscope”,点击“Fil e→Open”,在弹出的“打开”对话框中指定要打开的树文件,点击“确定”按钮即可导入树文件。
如果一个文件中包含多棵树,可用“Tre e→Next Tree”和“Tre e→Previous Tree”菜单命令选择。
用“Fil e→Recent Files”可直接打开最近编辑过的树文件。
用“Window s→Enter Tree”菜单命令可以在弹出的“Input”小窗口中直接用键盘输入一棵Newick 格式的树。
二、树的编辑1. 分支的选择:用“Selec t→……”菜单命令可根据需要选择部分或全部分支。
“Selec t→Select All”:选择全部分支和节点。
用“Selec t→Deselect All”或将鼠标移到非树区域点击左键可取消全部选择。
“Selec t→Select Nodes”:选择全部分支节点,包括末端分类单元;按住“Shift”键点击鼠标左键可选定任意数量的分支节点。
用“Selec t→Deselect Nodes”可取消对节点的选择。
“Selec t→Select Edges”:选择全部分支;按住“Shift”键点击鼠标左键可选定任意数量的分支。
用“Selec t→Deselect Edges”可取消对分支的选择。
“Selec t→Select Labeled Nodes”:选择有标签的节点;按住“Shift”键点击鼠标左键可选定任意数量的分支节点。
用“Selec t→Deselect Nodes”可取消对这些节点的选择。
“Selec t→Select Labeled Leaves”:选择树的分支末端(即树叶leaves),按住“Shift”键点击鼠标左键可选定任意数量的分支末端。
用“Selec t→Deselect Nodes”可取消对这些分支末端的选择。
“Selec t→Select Root”:选择树根节点,用“Selec t→Deselect Nodes”可取消对树根节点的选择。
“Selec t→Select Non-terminal”:选择不与分支末端相连的分支,按住“Shift”键点击鼠标左键可选定任意数量的非末端分支。
用“Selec t→Deselect Edges”可取消对这些分支的选择。
“Selec t→Invert Selection”:可选定与刚才的选定的对象相反的对象。
例如,先用“Selec t→Select Nodes”命令选定分支节点,则用“Selec t→Invert Selection”命令可转而选定所有分支;先用“Selec t →Select Non-terminal”命令选定了不与分支末端相连的分支,则用“Selec t→Invert Selection”命令可转而选定所有的与分支末端相连的分支;先用“Selec t→Select Root”选定了树根节点,则用“Selec t →Invert Selection”命令可转而选定所有的非根节点和分支;其余类推。
2. 分支塌陷、提取子树及导入分类单元图像“Optio n→Collapse”:[Collapse selected nodes]可以使选定节点以后的分支塌陷,合并为一个关系不确定的分支群。
用“Optio n→Uncollapse”命令可以指定恢复塌陷的分支;用“Optio n→Uncollapse Subtree”命令可以恢复选定节点以下的所有塌陷的子树。
“Optio n→Collapse Complement”:[Collapse the complement of currently selected nodes]使当前选定节点的互补分支(即与选定节点构成单系的分支?)塌陷。
用“Optio n→Uncollapse”命令可以指定恢复塌陷的分支;用“Optio n→Uncollapse Subtree”命令可以恢复选定节点以下的所有塌陷的子树。
“Optio n→Collapse At Level”:[Collapse all nodes at given depth in tree]在指定的分支水平上使分支塌陷;分支水平的计算方法是从树根算起,第1个不与末端分支直接连接的分支为1级分支,1级分支之后的不与末端分支直接连接的分支为2级分支,其余类推。
用“Optio n→Uncollapse”命令可以指定恢复塌陷的分支;用“Optio n→Uncollapse Subtree”命令可以恢复选定节点以下的所有塌陷的子树。
“Optio n→Extract Subtree”:[Extracts the subtree induced by the set of selected nodes]提取由选定的节点集形成的子树。
“Optio n→Load Taxon Images”:[Load taxon images from a directory],从指定路径导入分类单元图像(导入后若想删除,应该如何操作?)。
“Optio n→Set Image Size”:可设置导入的分类单元图像的高度。
“Optio n→Image Position”:[Set layout of images to the given position of label]调节分类单元图像在与分支末端标签(分类单元名称)的相对位置。
* 在“Optio n→……”菜单命令下还可生成不同要求的合一树,但一般用户不会使用该功能。
3. 线条及分支末端标签(分类单元名称)的编辑(1)一般特性的编辑选定分支或分支末端标签,点击“Edi t→Format”菜单命令(或将鼠标移到选定的对象上单击右键,在弹出菜单中选择“Edit Edge Label”选项),在弹出来的“Format”对话框中可编辑线条粗细(Edge width)、线条形状(Edge style)、节点大小(Node size)、节点形状(Node shape)、线条和末端标签的颜色(在调色板Swatches中调节,要选中“Line color”和“Label color”才能调节,“Recent”小调色板记录了最近使用过的颜色,因此,只要小调色板中有需要的颜色,也可以从小调色板中选择调整)、标签的字体(Font)/字号(Size)/粗体(Bold)/斜体(Italic)甚至旋转标签(“rotate labe left”和“rotate label right”图标,要使标签恢复原来的位置,点击“Vie w→Reposition labels”菜单命令)。
(2)末端分支标签的查找与替换用“Edi t→Find/Replace”菜单命令可以查找指定的末端分支标签并用新的标签替换。
选定末端分支标签后单击右键,在弹出菜单中点击“Edit Node Label”选项将弹出“Input”对话框,在“Edit Node Label”栏内输入新的标签名称,点击“OK”按钮即可替换原来的标签。
4. 系统发育树的图形布局编辑使用“Tre e→……”菜单命令可以选择自己喜欢的或符合需要的图形布局。
5. 图形的缩放与节点/线条标签的显示及隐藏。
(1)图形的放大与缩小:点击“Vie w→Fully Contract ”或“Vie w→Zoom to Fit”菜单命令可以使树紧缩而在屏幕上显示整棵树的轮廓;点击“Vie w→Fully Expand ”菜单命令可以使树的分类单元紧密排列但又可以清楚浏览(程序默认的最佳显示比例);将鼠标移到树的区域内的任何位置,滚动鼠标滚轮可调节树的分类单元之间的间隔(即紧缩或扩展程度),以便浏览;点击“Vie w→Use Magnifier”菜单命令可以显示放大镜用于放大和缩小图的显示比例(放大镜以鱼眼方式显示树的分类单元之间的间隔,即屏幕中间的分类单元之间间隔最宽,两侧依次紧缩,一般情况下不必使用),再次点击该命令可以隐藏放大镜。
将鼠标移到窗口右侧的滚动条区域,滚动鼠标滚轮可以上下滚动浏览树的分支,也可直接用鼠标拖动滚动条或点击滚动条上下两端的“▲”和“▼”图标来滚动浏览。
(2)节点/线条标签的显示、隐藏及查找/替换用“Vie w→Show Node Labels”菜单命令可以显示节点标签,用“Vie w→Hide Node Labels”可以隐藏节点标签。
用“Vie w→Show Edge Weights”或“Vie w→Show Edge Labels”菜单命令可以显示线条标签,“Vie w →Hide Edge Labels”可以隐藏线条标签。
“Vie w→Sparse Labels”:Show sparse non-overlapping subset of all labels.“Vie w→Radial Labels”:Rotate labels to match directions of edges.“Vie w→Reposition Labels”:Reset the positions of all labels.三、树的输出1. 以文本形式保存树文件经过编辑后的树文件若想以文本形式保存,最好使用“Fil e→Save As…”另存,以便保留系统发育分析时生成的原始树文件供不同分析时使用。