同源建模
蛋白质结构预测方法

蛋白质结构预测方法随着生物科技和计算机技术的快速发展,蛋白质结构预测方法已经成为当今生物学中的热门话题。
蛋白质是生命体中最基本的一种生物大分子,对于许多生命活动和疾病的研究都具有重要的作用。
然而,了解蛋白质的结构对于研究其功能和相互作用至关重要。
本文将介绍一些常见的蛋白质结构预测方法。
一、亚氨酸序列分析法亚氨酸序列分析法是一种基于蛋白质多肽链上各个氨基酸的组成及其排列顺序来预测蛋白质空间结构的方法。
这种方法在理论上已经被证明是可行和准确的。
然而,由于该方法在预测过程中可能会受到亚氨酸序列中缺失信息的影响,因此需要借助其他方法进行补充。
二、同源建模法同源建模法是一种比较广泛使用的蛋白质结构预测方法。
该方法依据细胞中已知结构的蛋白质对于待预测蛋白质的模板效应进行预测,从而得到待预测蛋白质的结构。
该方法的优点在于它能够对大量的蛋白质进行预测,并且往往能获得高质量的结构预测结果。
然而,该方法的主要缺点是仅适用于那些与已知结构相似的蛋白质。
三、Ab initio方法Ab initio方法是一种从头开始预测蛋白质结构的方法,它不依赖于与已知结构相似的蛋白质。
这种方法基于物理力学和统计学知识进行计算,尝试预测分子的基本构筑原理。
这种方法在处理具有折叠密码学特性的蛋白质时比较准确,但是在面对大分子的复杂蛋白质时常常出现预测的误差。
四、网络方法网络方法是一种将蛋白质折叠预测看作一个大型优化问题的方法,它通过构建各种相互作用网络来预测蛋白质的结构。
这种方法在处理大分子蛋白质的折叠过程中具有较好的表现,也是目前研究中的热门和前沿方向之一。
五、机器学习方法机器学习方法是一种基于人工智能理论和算法的蛋白质结构预测方法。
该方法可以构建出一个有效的预测模型,然后通过灵活的机器学习算法对蛋白质信息进行分析来预测蛋白质的结构。
该方法在处理大分子的复杂蛋白质时常常具有很好的预测效果,但是它的缺点在于需要大量的已知数据用于训练模型。
同源建模的可行性研究报告

同源建模的可行性研究报告一、引言同源建模是一种新兴的建模方法,旨在通过对不同数据源中的同源数据进行整合和建模,实现更加综合和全面的数据分析。
在信息化浪潮的推动下,各行各业的数据呈现出多样化和复杂化的特点,传统的数据建模方法已经难以满足日益增长的数据需求。
因此,同源建模作为一种新的数据建模方法,具有很大的发展潜力和价值。
本报告旨在对同源建模的可行性进行研究和分析,为同源建模的应用和推广提供理论支持和实践指导。
二、同源建模的概念和原理1. 同源建模的概念同源建模是指基于不同数据源中的同源数据进行数据整合和建模的方法。
所谓同源数据,是指来自不同数据源但具有相同含义和结构的数据。
同源建模旨在通过对同源数据的整合和建模,实现更加综合和全面的数据分析,从而为决策提供更加丰富和全面的信息支持。
2. 同源建模的原理同源建模的原理主要包括数据整合、特征提取和模型建立。
首先,需要对不同数据源中的同源数据进行整合,将其统一到一个数据源中。
然后,通过特征提取,将同源数据中的特征进行提取和筛选,以便进行后续的建模分析。
最后,基于整合后的同源数据,建立数据模型,进行数据分析和决策支持。
三、同源建模的应用场景同源建模的应用场景主要包括金融领域、医疗领域、互联网领域等。
具体应用场景如下:1. 金融领域:银行、证券、保险等金融机构需要对客户的各种数据进行分析和预测,以便进行风险评估和决策支持。
同源建模可以帮助金融机构对客户的不同数据进行整合和建模,实现更加全面和准确的风险评估。
2. 医疗领域:医疗机构需要对患者的临床数据、影像数据、基因数据等进行分析和诊断,以支持临床决策和治疗方案选择。
同源建模可以帮助医疗机构对不同数据源中的同源数据进行整合和建模,实现更加全面和准确的诊断和治疗支持。
3. 互联网领域:互联网企业需要对用户的行为数据、社交数据、消费数据等进行分析和预测,以支持产品设计和营销决策。
同源建模可以帮助互联网企业对不同数据源中的同源数据进行整合和建模,实现更加全面和准确的用户画像和预测分析。
同源建模工具easymodeller4.0操作说明

同源建模工具easymodeller4.0操作说明easymodeller是modeller的图形操作界面,方便同源建模EasyModeller 4.0 使用说明----By Xm 中科院昆明动物研究所20XX 年.3.11easymodeller是modeller的图形操作界面,方便同源建模Before using需要安装的软件:Python(2.5,2.6,2.7系列均可,3系列不可)Modeller() Easymodeller 4.0 RasMol (windows版) Office Excel*以上软件都能在官网或网站上下载,软件需要安装在C盘默认路径下,easymodeller 需安装在modeller目录下,所有安装路径中都不要出现中文!!easymodeller是modeller的图形操作界面,方便同源建模同源建模的基本步骤1、模板蛋白搜索:PDB数据库、BLAST(或PSI-BLAST) 、获取模板(一个或多个)2、比对结果的校正3、主链生成4、环区建模5、模型优化6、合理性检测easymodeller是modeller的图形操作界面,方便同源建模模板.pdb文件准备选择align结果中的蛋白,就可以找到相应的蛋白3D 结构,下载pdb文件即可同源度,30% 时方可用与目标蛋白序列同源的蛋白easymodeller是modeller的图形操作界面,方便同源建模Start using右击,win7或vista系统下选择“以管理员身份运行”出来的界面easymodeller是modeller的图形操作界面,方便同源建模①目标氨基酸序列,无空格,需全部大写②导入模板PDB文件③勾选需要比对的模板④点击开始比对选中的模板easymodeller是modeller的图形操作界面,方便同源建模⑤点击,align模板easymodeller是modeller的图形操作界面,方便同源建模⑥点击,align模板和目标序列easymodeller是modeller的图形操作界面,方便同源建模⑦点击,生成模型easymodeller是modeller的图形操作界面,方便同源建模⑧选中生成的模型,进行预览⑨对模型显示进行设置easymodeller是modeller的图形操作界面,方便同源建模点击,选择(可多选)easymodeller是modeller的图形操作界面,方便同源建模。
PpKO的同源建模101009

梨贝壳杉烯氧化酶(PpKO)三维结构的同源模建及功能预测摘要:以本试验室已克隆获得的梨贝壳杉烯氧化酶PpKO为试材,以人的微粒体细胞色素P450的A链的晶体结构为模板,综合运用生物信息学方法及分子模拟手段,构建了与其高度同源、催化功能相似的晶体结构。
通过Structure assessment和SA VS对模建结构的合理性进行了分析,结果表明利用Swiss-Model同源模建结果较好。
在此基础上,运用细胞色素P450酶系催化循环酶促动力学对PpKO的催化机理进行了初步分析,这为揭示贝壳杉烯氧化酶(KO)的生物学功能及抑制剂药物分子设计提供了有益的信息。
关键词:贝壳杉烯氧化酶;同源模建;蛋白质三维结构贝壳杉烯氧化酶(KO)是赤霉素合成途径中的关键酶(Hedden,1997),位于内质网且与叶绿体膜结合的依赖细胞色素P450(CYP450)和NADPH的单加氧酶。
通过三步连续的羟基化作用,将内根-贝壳杉烯的C-19甲基氧化,分别形成内根-贝壳杉烯醇、内根-贝壳杉烯醛和内根-贝壳杉烯酸(Helliwell,1999),该过程使6碳的B环转化为5碳的C环,从而形成赤霉素的母环结构(Lange,1998)。
NCBI保守域分析表明PpKO属于细胞色素超家族P450系。
细胞色素P450家族成员之间的一级结构差异较大,但空间结构却有着较大的相似性,含有由含亚铁血红素和半胱氨酸组成的活性中心(Rupasinghe,2006)。
目前数据库中已有经实验获得的晶体结构,较易获得模建所需的模板。
不过,我们在PDB数据库中搜索发现,目前尚无KO X光晶体衍射的晶体结构,只有拟南芥(UniProt:Q93ZB2)和赤霉菌(UniProt:A9P5P8)的同源模建晶体结构。
KO三维结构详细信息的缺失必然阻碍对其功能的理解和药物开发的滞后。
同源模拟法是根据同源蛋白质三级结构的保守性超过蛋白质序列的理论,通常在蛋白质序列上稍微的改变其三级结构亦改变很小。
蛋白质结构的预测和设计

蛋白质结构的预测和设计蛋白质是生命体内最基本的分子机器,它们完成了生命中几乎全部的功能。
然而,在理解蛋白质功能的基础上,我们面临的一个重要挑战是如何探究蛋白质的三维立体结构。
在人类基因组计划完成后,我们已经识别出了大约20,000种编码人类蛋白质的基因,但是仅有不到一半的蛋白质结构被解析。
因此,预测和设计蛋白质结构是当前生命科学领域内非常重要的研究方向。
一、蛋白质的结构预测蛋白质结构预测是通过计算机模拟来预测蛋白质的三维立体结构的过程。
它有助于了解蛋白质的功能和相互作用,并在药物开发、基因工程和分子演化等领域中发挥重要作用。
目前,主要有以下几种方法进行蛋白质结构预测。
1.同源建模同源建模是通过在已经解析的蛋白质结构库中搜索和比较已知的蛋白质结构进行预测的一种方法。
这种方法主要适用于已知序列与已知结构高度相似的蛋白质。
2.蒙特卡罗模拟蒙特卡罗模拟是通过在空间内采用随机数的方法来模拟蛋白质的折叠过程,以得到最稳定的蛋白质三维结构的一种方法。
3.基于物理性质的模拟基于物理性质的模拟是通过计算机模拟蛋白质原子在环境中的物理相互作用和动力学性质来预测蛋白质三维结构的一种方法。
这种方法通过物理模型计算蛋白质的能量最小值,以预测出最稳定的结构。
除了以上方法,还有一些基于机器学习算法、分子动力学模拟等技术的方法也被用于蛋白质结构预测。
但是,所有这些方法都存在重大局限性,如预测结果不准确、模型偏差过大等问题,限制了其在实际应用中的应用。
二、蛋白质的结构设计蛋白质结构设计是通过设计人工序列来实现有特定功能的蛋白质结构的过程。
它可以用于开发新型药物、分子传感器、嫩肤等领域。
经过近年来的探索和研究,已经发现了一些有效的蛋白质结构设计方法。
1.方法一该方法是将构建的蛋白质折叠成模型后,通过改变其氨基酸序列,以优化其折叠或使其保持特定的折叠状态。
重要的是,最好根据确定的目标蛋白质结构,为蛋白质设计完整的折叠拓扑和氨基酸序列。
swiss-model的使用

预测的三维结构
SWISS-MODEL网页界面
我们再选择联配模式
接着会出现下面的界面
出现的结果
谢谢聆听
Thank You
下边为大家演示一下简捷模式的操作
SWISS-MODEL网页界面
我们选择简捷模式
填写正确的邮箱,会有邮件
输入蛋白质序列
提交以后会出现
在线显示的有的时候速度有点慢,耐心等待
Ps:珍惜劳动成果。这可是等了一个小时的成果
反馈的结果也可以在 myWorkspace 中查看,自己的邮箱 是用户名,邮件里边有password
可以把结果保 存下来
SWISS-MODEL同源建模简介
SWISS-MODEL 是一个自动化的蛋白质比较建模服务器, 该服务器提供用户三种模式可选择: Automatic mode(简捷模式): 用于建模的氨基酸序列或是 Swiss-Prot/TrEMBL (/sprot )编目号 (accession)可以直接通过web界面提交。服务器会完全自 动地为目标序列建立模型。 用户可以选择指定模板结构,模 板可以来自由PDB数据库( )抽取得到 的内建模板库,也可以上传PDB格式的坐标文件。 Alignment mode(联配模式): 这个模式需要多序列联配的 结果,序列中至少包括目标序列和模板(最多可输入5条序 列)。服务器会基于比对结果建模。用户需要指明哪一条序 列作为目标序列,哪一条又作为模板。 Project mode(项目模式): 这种模式允许用户提交经过手工优 化的请求给服务器。DeepView被用来建立一个项目文件, 它包含了模板结构,以及目标序列与模板的联配结果。这个 结果也要上传到服务器。这种方式提供对建模过程中细
discovery 同源建模使用方法

4 打开选定的结构 右键选择1n5u,点击load 右键选择1n5u,点击load selected structure。观察Hsa结构。 structure。观察Hsa结构。
点击 Sequence Window,然后点击右键 Window,然后点击右键 选择 Insert Sequence | From Windows…. 在对话框中,选择 1n5u_A,然后按OK 1n5u_A,然后按OK 。这样就成功地讲1n5u序列添加到 。这样就成功地讲1n5u序列添加到 Sequence window中。 window中。
说明: 途中红色部分为较为严重的过近接触 atom clashes 绿色的为不合理的二面角 紫色的球为不合理的Cb 紫色的球为不合理的Cb 橘黄色的残基为不合理的rotamer 橘黄色的残基为不合理的rotamer
6使用 MODELER 建立一个 3D 结构
在 Protocol里,展开 Protein Modeling Protocol里,展开 文件夹,打开 Homology Building protocol。 protocol。 然后在 Parameters里,Input Sequence Parameters里,Input Alignment 选择 Sequnce Window(1)。 Window(1)。 展开 Input Sequence Alignment, Alignment, Input Model Sequence 设置为 bsa。 bsa。 在 Input Template Structure 选择 1n5u。根据需要选择 1n5u。根据需要选择 Optimization Level。 Level。 最后运行 protocol 即可。
步骤一:上传文件 步骤一:上传文件
同源建模详细讲解-整理版

05
同源建模的案例分析
案例一:自然语言处理领域的应用
总结词
自然语言处理领域是同源建模的重要应用场景之一, 通过同源建模技术,可以更好地理解和处理自然语言 数据。
详细描述
在自然语言处理领域,同源建模技术被广泛应用于文本 分类、情感分析、机器翻译等方面。通过构建同源模型 ,可以更好地捕捉文本中的语义信息和上下文信息,提 高处理效率和准确性。例如,在文本分类中,同源建模 技术可以帮助我们更好地理解文本的主题和分类;在情 感分析中,同源建模技术可以更准确地判断文本的情感 倾向;在机器翻译中,同源建模技术可以提高翻译的准 确性和流畅性。
同源建模详细讲解整理版
04
同源建模的优缺点分析
目录
• 同源建模概述 • 同源建模的关键技术 • 同源建模的实现步骤 • 同源建模的优缺点分析 • 同源建模的案例分析 • 同源建模的未来发展与展望
优点分析
高效性
同源建模基于相似性原理,能够快速建立 模型,提高建模效率。
灵活性
同源建模适用于多种数据类型和场景,具 有较好的灵活性。
技术发展趋势
智能化
同源建模技术将进一步融合人工智能和机器学习算法,实 现建模过程的自动化和智能化,提高建模效率和精度。
精细化
随着计算能力和数据量的提升,同源建模将向更精细化的 方向发展,能够处理更复杂的模型和更高精度的数据。
多学科交叉
同源建模将进一步与物理学、数学、工程学等多个学科交 叉融合,拓展其应用领域和解决复杂问题的能力。
05
同源建模的案例分析
同源建模的基本原理
1 2 3
基于共同祖先的遗传信息
同源建模利用具有共同祖先的物种之间的遗传信 息,通过比较它们的基因和蛋白质序列,找出相 似性和差异性。
5.5计算方法预测三级结构-02-同源建模法SWISS-MODEL

《生物信息学》第五章:蛋白质结构预测与分析(第二部分) 计算方法预测三级结构:同源建模法SWISS-MODEL预测蛋白质三级结构的首选方法是同源建模法(homolog modeling)。
该方法基于原理:相似的氨基酸序列对应着相似的蛋白质结构。
比如三个蛋白质,它们在序列水平上十分相似,解析出的结构也十分相似。
第四个蛋白质的序列和前面三个也高度相似,那么就可以比着前三个结构的样子“画”出第四个的样子。
所以同源建模法的关键就是找到一个好的模板。
好的模板要求,在序列水平上模板(template)要与目标(target)蛋白质具有超过30%的一致度。
同源建模法操作流程如下(图1):图1. 同源建模法操作流程1. 确定模板:找到与目标蛋白质同源的已知蛋白质结构作为模版(目标序列与模版序列间的一致度要≥30%)。
2. 序列比对:为目标序列与模板序列创建序列对比。
模板可以选取多个,通过做多序列比对,各取所长,让模板序列中与目标序列相似的片段尽可能多的覆盖整个目标序列,同时要尽量避免没有模板参考的断口。
3. 计算模型:通过序列比对,将目标序列里的氨基酸替换到模板结构里对应的氨基酸所在的空间位置上。
这一步通过同源建模软件来实现。
4.换模板或修正序列比对,重新构建模型,再次评估。
SWISS-MODEL()它能帮助完成上述步骤中从模板选取到创建序列比对,再到计算模型,以及最后的质量评估的全部过程。
你需要做的只是:输入目标序列,点Build Model(创建模型)(图2左)。
大约三到五分钟之后就会返回结果。
如果这种自动挡模式不能满足你的要求,可以通过点击Search For Templates切换成手动挡,以便指定模板。
也可以直接把做好的目标序列与模板序列之间的序列比对按照指定格式黏贴到输入框里,再点击Build Model(创建模型)(图2右)。
这时,SWISS-MODEL会根据输入的特定格式的序列比对,识别出哪个是目标序列,哪个是模板,并自动从PDB数据库下载模板结构,最后根据输入的比对计算结构模型。
同源建模

确定模板
构建三维模型
找出结构保守性的主链结构片段,进行主链结 构建模 找出建模变化区域,一般为连接二级结构片段 变化的区域,进行环区建模 侧链建模
三维模型准确 性检验
用 V erify 一3D 程序对构建的模型进行评估得出, 所构建的模型三维结构同血凝素氨基酸序列相容 性良好,构建的模型是可信的。
人感染甲型 H 7 N 9 亚型禽流感病毒血凝 素蛋白同源建模
模板选择和序列比对
在PDB数据库中搜索模板 以 12 条人感染 H 7N 9 亚型流感 病毒血凝素作全 序列,分别 构建两组参 比序列数 据集 。第一组 参 与比序列数 据集是 用 BL A ST 程序 在G enBank 数据库中搜索到的同新发 H 7N 9 亚 型毒株血凝 素接近 的序列。第二组参比序列 是在 Gen—Bank 数据库中采用分层抽样的方法 ,在 H 1~H 16亚 型中每个亚型随机抽取 2 或 3 条序列。用 M U SC L E 软件 对上述数据集进行多重序列比对。 确定4DJ6为模板( 因为4DJ6同 A G L44438 序列算机硬件和预测方法的发展,同源建模将会在蛋白质设 计和计算机辅助药物设计等方面应用广泛。在后基因组时代,蛋 白质结构同源建模将起着关键桥梁作用,是基因组、蛋白质组等 信息应用于生物工程中。
Thank You!
同源建模
同源建模概述及原理
使用这种方法有两个前提条件:
首先,目标序列的同源蛋白质中某一个或者几个结构已经被解析(x射线或者 NMR结构均可); 其次,目标序列与该蛋白质的同源性足够高,一般50%以上的同源性能够得 到比较合理的构象。
原理是序列相似则结构相似,即存在同源关系的两条序列具有相似的结构。 特点同源建模是目前从序列预测结构的最切实可行、应用最广泛的一种方
蛋白质三级结构预测(swiss-model同源建模)
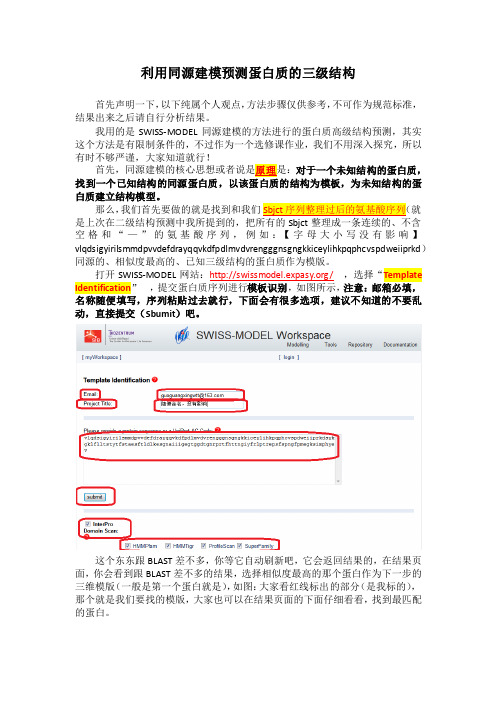
利用同源建模预测蛋白质的三级结构首先声明一下,以下纯属个人观点,方法步骤仅供参考,不可作为规范标准,结果出来之后请自行分析结果。
我用的是SWISS-MODEL同源建模的方法进行的蛋白质高级结构预测,其实这个方法是有限制条件的,不过作为一个选修课作业,我们不用深入探究,所以有时不够严谨,大家知道就行!对于一个未知结构的蛋白质,白质建立结构模型。
那么,我们首先要做的就是找到和我们空格和“—”的氨基酸序列,例如:【字母大小写没有影响】vlqdsigyirilsmmdpvvdefdrayqqvkdfpdlmvdvrengggnsgngkkiceylihkpqphcvspdweiiprkd)同源的、相似度最高的、已知三级结构的蛋白质作为模版。
打开SWISS-MODEL网站:/,选择“Template Identification,提交蛋白质序列进行模板识别,如图所示,注意:邮箱必填,名称随便填写,序列粘贴过去就行,下面会有很多选项,建议不知道的不要乱动,直接提交(Sbumit)吧。
这个东东跟BLAST差不多,你等它自动刷新吧,它会返回结果的,在结果页面,你会看到跟BLAST差不多的结果,选择相似度最高的那个蛋白作为下一步的三维模版(一般是第一个蛋白就是),如图:大家看红线标出的部分(是我标的),那个就是我们要找的模版,大家也可以在结果页面的下面仔细看看,找到最匹配的蛋白。
这里还有一点要作说明,就是上图标出的代码是PDB编号,前四个表示PDB- Code,最后一位表示Chain-ID,具体什么意思,大家有兴趣就去了解一些吧。
接下来,去NCBI串串门吧,在NCBI中搜索上面查到的蛋白的PDB号,一般输入前四位就行啦,注意:搜索蛋白库(Protein)。
找到以后,以FASTA格式显示。
接下来,我们再回到SWISS-MODEL,接下来就是重点和难点啦,在线提交序列进行同源建模分析,这个在线提交不是大家想象的那么容易,这个耗费了我大部分的时间,说到这里我就想画个圈圈诅咒它,大家注意啦~~~~~~~~~~~SWISS-MODEL 是一个自动化的蛋白质比较建模服务器,该服务器提供用户三种模式可选择:Automatic mode(简捷模式): 用于建模的氨基酸序列或是Swiss-Prot/TrEMBL (/sprot )编目号(accession)可以直接通过web界面提交。
同源建模_精品文档

Verify3D
比较模型和氨基酸 一级结构的关系
人感染甲型 H 7 N 9 亚型禽流感病毒血凝 素蛋白同源建模
模板选择和序列比对
在PDB数据库中搜索模板 以 12 条人感染 H 7N 9 亚型流感 病毒血凝素作全
序列, 分别 构建两组参 比序列数 据集 。第一组 参 与比序列数 据集是 用 BL A ST 程序 在G enBank 数据库中搜索到的同新发 H 7N 9 亚 型毒株血凝 素接近 的序列。第二组参比序列 是在 Gen—Bank 数据库中采用分层抽样的方法 , 在 H 1~H 16亚型 中每个亚型随机抽取 2 或 3 条序列。用 M U SC L E 软件 对上述数据集进行多重序列比对。
确定模板
确定4DJ6为模板( 因为4DJ6同 A G L44438 序列 相似度最高, 为97%)
构建三维模型
找出结构保守性的主链结构片段, 进行主链结 构建模
找出建模变化区域, 一般为连接二级结构片段 变化的区域, 进行环区建模
侧链建模
三维模型准确 性检验
用 V erify 一3D 程序对构建的模型进行评估得出, 所构建的模型三维结构同血凝素氨基酸序列相容 性良好, 构建的模型是可信的。
随着计算机硬件和预测方法的发展, 同源建模将会在蛋白质设 计和计算机辅助药物设计等方面应用广泛。在后基因组时代, 蛋 白质结构同源建模将起着关键桥梁作用, 是基因组、蛋白质组等 信息应用于生物工程中。
Thank You!
开始
同源性对比
选择模板
靶序列与模板结构 进行比对 使用模板结构的信 息建立靶序列的模型
评价模型
模型 满意?
结束
同源建模流程图
同源建模的评价
RMSD
同源建模法的步骤

同源建模法的步骤同源建模法是一种在系统工程中常用的建模方法,用于描述和分析不同层次、不同粒度、不同领域之间的关系和交互。
它可以帮助我们更好地理解和把握系统的整体结构和行为,从而指导系统设计和优化。
本文将介绍同源建模法的步骤,并提供一些实例来帮助读者更好地理解该方法。
步骤一:确定研究对象在使用同源建模法之前,首先需要明确研究对象是什么。
研究对象可以是一个产品、一个系统、一个流程等等。
确定研究对象的范围和边界非常重要,它决定了后续建模的深度和广度。
步骤二:收集相关信息在进行建模之前,需要收集与研究对象相关的信息。
这些信息可以来自于文档、实验、调查等多种渠道。
收集到的信息应该包括系统的功能、性能、约束条件等方面的内容。
步骤三:确定系统组成部分根据收集到的信息,我们可以确定系统由哪些组成部分构成。
这些组成部分可以是物理实体(如设备、零件等),也可以是逻辑模块(如子系统、模块等)。
确定系统组成部分有助于我们理解系统的结构和功能。
步骤四:建立元模型元模型是对系统组成部分进行抽象和概括的模型。
它描述了系统组成部分之间的关系和交互。
建立元模型的过程可以采用层次化、分类化的方法,将系统组成部分按照一定的规则进行划分和组织。
我们要研究一个汽车的行驶过程,可以将其划分为发动机、变速器、车轮等几个组成部分。
我们可以建立一个元模型,描述这些组成部分之间的关系和交互。
步骤五:建立同源关系同源关系是指不同层次、不同粒度、不同领域之间具有相似结构或功能的元素之间的关系。
通过建立同源关系,我们可以更好地理解和描述系统的整体结构和行为。
在汽车的例子中,发动机、变速器和车轮都是与汽车行驶相关的元素。
它们之间具有相似的结构和功能,因此可以建立同源关系。
步骤六:建立行为模型行为模型是对系统行为进行描述的模型。
它可以包括系统的状态变化、事件触发、动作执行等方面的内容。
建立行为模型可以帮助我们更好地理解和分析系统的行为特征。
在汽车的例子中,我们可以建立一个行为模型,描述汽车启动、加速、减速等过程中发动机、变速器和车轮之间的相互作用。
分子动力学优化同源建模得模型

分子动力学优化同源建模的模型主要包括以下步骤:
构建同源模型:首先,通过序列相似性搜索工具为查询序列搜索序列相似程度较高的已知结构蛋白作为候选模版。
然后,比对对齐后,那些与模版序列完全相同的残基所有原子座标完全拷贝自模版,而那些相似残基仅有骨架原子取自模版——侧链座标另行构建,那些缺失序列则通过查找一个Loop 数据库来进行补缺。
最后,这个虚拟的预测模型在特定的势场参数下进行能量优化,得到初始同源模型。
分子动力学模拟:在建模蛋白质结构基础之上,利用动力学软件进行分子动力学模拟,以得到其最优结构模式。
通过分子动力学优化同源建模的方法,可以更加精确地预测蛋白质的三维结构,为后续的药物设计等工作提供基础。
以上内容仅供参考,建议咨询生物学专家或查阅相关文献资料获取更详细的信息。
同源建模的实验报告(3篇)

第1篇一、实验目的1. 熟悉同源建模的基本原理和方法;2. 掌握同源建模的实验步骤和操作技巧;3. 通过同源建模预测蛋白质的三级结构,并验证预测结果的准确性。
二、实验原理同源建模是一种基于生物信息学的方法,利用已知蛋白质的三维结构作为模板,通过计算机模拟和计算,预测未知蛋白质的三维结构。
该方法基于以下两个原理:1. 蛋白质的结构由其氨基酸序列唯一决定,知道其一级序列,在理论上就可以获取其二级结构以及三级结构;2. 蛋白质的三级结构在进化中更稳定或者说更保守。
如果两个蛋白质的氨基酸序列有50%相同,那么约有90%的α-碳原子的位置偏差不超过3,这是同源模型化方法在结构预测方面成功的保证。
三、实验材料与仪器1. 实验材料:蛋白质序列、NCBI数据库、SwissModel在线平台、MOE软件;2. 实验仪器:计算机、网络连接。
四、实验步骤1. 收集蛋白质序列:在NCBI数据库中搜索目标蛋白质的序列,获取其一级结构信息。
2. 模板搜寻:使用SwissModel在线平台进行模板搜寻,寻找与目标蛋白质序列具有较高同源性的已知蛋白质结构作为模板。
3. 模型构建:根据模板蛋白质的三维结构,使用SwissModel在线平台构建目标蛋白质的三维结构模型。
4. 模型优化:使用MOE软件对模型进行优化,包括分子力学优化、能量最小化等。
5. 模型验证:通过比对实验结果和预测结构,验证同源建模的准确性。
6. 结果分析:分析预测结构的合理性,解释蛋白质的功能和作用机理。
五、实验结果与分析1. 模板搜寻:在SwissModel在线平台中,成功找到与目标蛋白质序列具有较高同源性的模板蛋白质。
2. 模型构建:根据模板蛋白质的三维结构,成功构建了目标蛋白质的三维结构模型。
3. 模型优化:使用MOE软件对模型进行优化,优化后的模型具有更低的能量。
4. 模型验证:通过比对实验结果和预测结构,验证同源建模的准确性。
预测结构在关键区域与实验结果基本一致。
同源建模详细讲解-整理版

同源建模的基本步骤
• • • • • •
1、模板蛋白搜索
PDB数据库、BLAST(或PSI-BLAST) 、获取模板(一个或多个)
2、比对结果的校正
3、主链生成
4、环区建模
5、模型优化 6、合理性检测
同源建模在线服务器
Swiss-model
I-TASSER
ESyPred3D
HOMCOS 1.0
同源建模软件
• • •
Automated mode:自动模式,可以称为是最傻瓜的方式
提交自己的氨基酸序列+邮箱即可
适用:一致性较高时
邮箱 模型命名
氨基酸序列
Swiss-port/TrEMBL登录号
Alignment mode
• •
•
Alignment mode: 比对模式
提交目标蛋白与模板蛋白的序列比对结果(FASTA,MSF,ClustalW等 格式) 适用:1、较高的相似性 2、利用Auto模式未必能找到最合适模板的情况 3、使用者有目的的使用特定的模板蛋白
Chiron进行优化
•
•
我们考虑采用计算量较少的Chiron服务器对模建结构的clash进行 处理。
swiss-model的使用

SWISS-MODEL网页界面
我们选择简捷模式
精选课件
3
填写正确的邮箱,会有邮件
精选课件
输入蛋白质序列
4
提交以后会出现
在线显示的有的时候速度有点慢,耐心等待
Ps:珍惜劳动成果。这可是等了一个小时的成果
精选课件
5
反馈的结果也可以在 myWorkspace 中查看,自己的邮箱
是用户名,邮件里边有password
精选课件
6
可以把结果保 存下来
精选课件
7
精选课件
8
精选课件
9
预测的三维结构
精选课件
10
SWISS-MODEL网页界面
我们再选择联配模式
精选课件
11
精选课件
12
精选课件
13
接着会出现下面的界面
精选课件
14
出ank You
下边为大家演示一下简捷模式的操作
❖ Alignment mode(联配模式): 这个模式需要多序列联配的 结果,序列中至少包括目标序列和模板(最多可输入5条序 列)。服务器会基于比对结果建模。用户需要指明哪一条序 列作为目标序列,哪一条又作为模板。
❖ Project mode(项目模式): 这种模式允许用户提交经过手工优 化的请求给服务器。DeepView被用来建立一个项目文件, 它包含了模板结构,以及目标序列与模板的联配结果。这个 结果也要上传到服务器。这精选种课方件 式提供对建模过程中细 2
精选课件
16
SWISS-MODEL 同源建模的方法
精选课件
1
SWISS-MODEL同源建模简介
❖ SWISS-MODEL 是一个自动化的蛋白质比较建模服务器, 该服务器提供用户三种模式可选择:
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
同源建模
定义:同源建模又称为比较建模,其原理是序列相似则结构相似,即存在同源关系的两条序列具有相似的结构。
当两条序列的同源性大于30%时,序列的同源性能够暗示两者结构相似,序列的同源性越高则结构模型的准确性越高。
步骤:1)寻找已知结构的蛋白质作为模板;
(2)目标和模板的序列比对;
(3)以模板结构为原型,构建目标的主链结构模型;
(4)在目标模板比对中形成空位的区域,使用环区建模得到完整的主链结构模型;
(5)构建并优化模型的侧链;
(6)对整个结构模型进行优化。