蛋白质同源建模评价
GMQE和QMEAN:评价蛋白质三维结构预测模型的质量的两种方法

GMQE和QMEAN:评价蛋白质三维结构预测模型的质量的两种方法蛋白质是生命的基本组成单位,它们的三维结构决定了它们的功能和特性。
因此,预测蛋白质的三维结构对于生物学研究和药物设计非常重要。
然而,蛋白质的三维结构往往难以通过实验方法获得,所以需要借助计算方法来进行预测。
蛋白质三维结构预测是一种根据蛋白质的氨基酸序列,推测其空间结构的技术。
蛋白质三维结构预测的方法有很多,其中最常用的一种是同源建模法(Homology Modeling)。
同源建模法是一种基于序列相似性的方法,它是根据已知结构的蛋白质(模板蛋白质)来构建未知结构的蛋白质(目标蛋白质)的结构模型。
同源建模法的基本假设是,如果两个蛋白质的序列相似度很高,那么它们的结构也很可能相似。
同源建模法的步骤包括:(1)根据目标蛋白质的序列,在数据库中搜索合适的模板蛋白质;(2)根据目标蛋白质和模板蛋白质的序列,进行序列比对,得到对齐方式;(3)根据对齐方式,将模板蛋白质的结构信息转移给目标蛋白质,得到初始的结构模型;(4)对初始的结构模型进行优化和修正,得到最终的结构模型。
同源建模法的优点是,它可以利用已有的结构信息,快速地生成结构模型。
同源建模法的缺点是,它依赖于模板蛋白质的质量和数量,以及序列比对的准确性。
如果目标蛋白质和模板蛋白质的序列相似度很低,或者没有合适的模板蛋白质,或者序列比对有误,那么同源建模法的结果就可能有很大的误差。
因此,评价同源建模法生成的结构模型的质量是非常重要的。
评价结构模型的质量的方法有很多,其中两种比较常用的方法是GMQE和QMEAN。
GMQE和QMEAN都是综合评估结构模型的质量的方法,但是它们有不同的依据和计算方式。
GMQE是全球模型质量估计(Global Model Quality Estimation)的缩写,它是一种基于模板的质量评估方法,它主要考虑了目标蛋白质和模板蛋白质之间的关系。
GMQE的分数是一个0到1之间的数字,表示了模型的预期准确性和目标蛋白质的覆盖范围。
人SEPT9蛋白同源建模和活性位点分析_王俊生

第42卷 第2期2014年3月河南师范大学学报(自然科学版)Journal of Henan Normal University(Natural Science Edition) Vol.42 No.2 Mar.2014 文章编号:1000-2367(2014)02-0107-07人SEPT 9蛋白同源建模和活性位点分析王俊生,阮先乐,范小芳,王永立,陈 龙(周口师范学院生命科学与农学学院,河南周口466001)摘 要:利用生物信息学在线软件预测了人SETP 9蛋白质的二级结构和模体信息,同时对其三级结构进行同源建模和模建结果质量评价,其次预测了该蛋白质的活性位点信息,旨在从蛋白质序列特征和分子结构水平理解其在人类生理病理过程中的作用.结果表明,模建的SEPT 9蛋白结构品质较高,具有7段α-螺旋和2组β-折叠结构,是一个典型的α/β类蛋白,表面呈弱正电势分布;人SEPT 9蛋白具有8个不同模体,可能参与不同生化反应或执行不同的功能.搜寻获得了人SEPT 9蛋白配基结合位点有10个,其中位点1可能是该蛋白的活性位点.这些研究结果对理解人SEPT 9蛋白功能以及配基结合位点定位非常重要,也为针对SEPT 9蛋白的分子对接和药物从头设计提供了理论基础.关键词:人SEPT 9蛋白;同源模建;活性位点中图分类号:Q518.2文献标志码:ASeptin广泛存在于真菌[1]、线虫[2]、果蝇[3-4]、爪蟾[5]及哺乳动物[6]等绝大多数真核生物中.研究[7]表明,人基因组中存在多达13种Septin基因亚家族,并且与细胞的重要生物功能密切相关,从血管生成到小泡运输,从癌症到神经系统一系列疾病等[8-10].SEPT 9蛋白最早是在白血病中作为粒细胞/淋巴细胞/白血病的伴随蛋白得到鉴定的[11],并且是从胸癌和卵巢癌的等位基因失衡区中克隆获得的[12,13].SEPT9基因位点复杂,至少编码5种不同的蛋白质单体,能够和其他SEPT蛋白形成低聚体复合物[14,15],不同的SEPT 9蛋白单体均具有一个GTPase保守结构域,该结构域含有核定位信号,他们的差异主要表现在氨基酸组成上[7-8].不同的SEPT9变异剪接本以及SEPT 9蛋白表达的改变被认为与前列腺癌[2]、卵巢癌[9,16]、胸癌[17]、结肠癌[18]、头颈部癌[19-20]等疾病密切相关,也与遗传性神经痛性肌萎缩[21]密切相关.随着对SEPT蛋白的研究日益深入,人类SEPT蛋白之间的互作以及功能不断被证实,如SEPT 9蛋白与SEPT 2和SEPT 7蛋白互作[22],可能参与了细胞骨架的组装完成;SEPT 2、6和7之间存在相互作用[23],也有研究[24]表明,该SEPT9与AHNAK,eIF4E和S100A11基因共同在伪足突起、癌症细胞的迁移和侵袭中表现出很重要的作用.然而,关于蛋白的三级结构信息以及基于三级结构的活性位点信息尚未见报道,因此本研究基于SEPT 9蛋白突变剪接体(AAH54004.1)氨基酸序列,利用生物信息学方法预测其二级结构、模体类别和数量,并利用同源建模方法模建了该蛋白质的空间结构,分析和平估了模建的质量,最后预测了该蛋白的活性位点信息,为治疗该基因突变引起的人类疾病进行药物设计奠定理论基础.1 材料和方法1.1 序列来源在美国国立信息技术中心NCBI数据库中查询下载人SEPT 9蛋白序列,序列获取号为AAH54004.1,由美国国立卫生研究院、哺乳动物基因中心、国家癌症研究院对人眼部癌变组织的测序项目提供[25],该蛋白由422个氨基酸组成,定位在人体第17号人色体上(17q25.2-q25.3),与卵巢癌、遗传性神经痛、肌萎缩、白血病等多种人体病症相关.收稿日期:2013-11-26基金项目:河南省教育厅项目(14B210022);河南省高校博士科研启动经费(ZKSYBSCX201110)作者简介:王俊生(1970-),男,陕西蒲城人,周口师范学院副教授,博士,主要从事生物信息数据挖掘研究.1.2 二级结构和蛋白质模体预测利用NPS(Network Protein Sequence analysis,http://npsa-pbil.ibcp.fr)提供的Proscan,对人SEPT9蛋白序列进行模体预测;使用在线网站Expasy的Coils程序预测卷曲螺旋区域,用Multicoil程序预测二聚体和三聚体.1.3 模建、蛋白质表面静电构象和模建结果评价分析方法利用CPHmodels 3.2在线服务器(http://www.cbs.dtu.dk/services/CPHmodels/)对人SEPT 9蛋白序列(gb|AAH54004.1)进行同源建模,获取其三级结构信息,并且用Rasmol视图软件可视化.模建结果检测评价采用NIH MBI实验室提供的结构基因组学和蛋白质确证分析服务器(http://nih-server.mbi.ucla.edu/SAVES/)中提供的不同软件,从拉氏散点图、3D-1D符合度两方面进行评价.蛋白质表面静电构象通过计算SEPT 9蛋白(AAH54004.1)各氨基酸残基的静电,使用软件Swiss-PdbViewer4.0.1模建蛋白质表面静电构象.1.4 蛋白质活性位点分析活性位点分析采用在线软件Q-SiteFinder(http://www.modelling.leeds.ac.uk/qsitefinder/)[26].均采用软件默认设置参数.2 结果与分析2.1 人SEPT9蛋白的二级结构预测利用SPOMA在线软件对人SEPT 9蛋白二级结构预测结果表明(如图4),该蛋白质序列中α螺旋占29.62%,β-延伸链占12.32%,β转角占3.32%,无规则卷曲占54.74%,而利用GOR4软件对该蛋白二级结构预测表明,该蛋白质序列中α螺旋占34.36%,延伸链占15.64%,无规则卷曲占50.00%,无β转角.与SOPMA软件预测结果基本一致.从SOPMA预测的β转角结构来看,所有6个β转角结构均不超过4个氨基酸残基(5个由2个残基构成,1个由3个残基构成),因此该蛋白的二级结构中β转角不会形成环(Loop)结构.2.2 人SEPT9蛋白的特殊卷曲螺旋预测对人SEPT 9蛋白二级结构卷曲螺旋区域进行预测,结果(图2-A)表明,在蛋白质序列上分布着几段可能的卷曲螺旋区域,其中215-242区段形成卷曲螺旋的可能性较大,但预测得分没有超过0.4,其他区段得分更小.使用在线网站Expasy的Multicoil程序预测该蛋白质的二聚体、三聚体卷曲螺旋(使用PairCoil算法),蓝线表示二聚体卷曲螺旋预测分值,红线表示三聚体卷曲螺旋预测分值,预测的分值皆为0.因此,该蛋白质难以形成稳定二聚体、三聚体卷曲螺旋(图2-B).2.3 人SEPT9蛋白的模体预测对人SEPT 9蛋白序列进行模体预测,结果(表1)表明,该蛋白包含1个N-糖基化位点、1个cAMP-和801河南师范大学学报(自然科学版) 2014年cGMP依赖的蛋白激酶磷酸化位点、4个蛋白激酶C磷酸化位点、9个酪蛋白激酶II磷酸化位点、1个酪氨酸激酶磷酸化位点、1个N段肉豆蔻酰基化位点、2个酰胺化位点、1个ATP/GTP结合位点,模体类型和氨基酸位置详细信息见下表.表1 人SEPT 9蛋白的模体预测模体名称序列号模式类型氨基酸序列随机概率N-糖基化位点PS00001N-{P}-[ST]-{P}363-366:NTTH 5.138e-03依赖cAMP和cGMP的蛋白激酶磷酸化位点PS00004[RK](2)-x-[ST]267-270:KRLS 1.572e-03蛋白激酶C磷酸化位点PS00005[ST]-x-[RK]71到73:TPR;91到93:TPR;160到162:SRK;257到259:SLR1.423e-02酪蛋白激酶II磷酸化位点PS00006[ST]-x(2)-[DE]57到60:SRLE;91到94:TPRD;103到106SRNE;119到122:SILE;167到170:TSEE;195到198:TVID;286到289:TLEE;338到341:SDHE;365到368:THCE1.482e-02酪氨酸激酶磷酸化位点PS00007[RK]-x(2,3)-[DE]-x(2,3)-y107到114:KAPVDFG4.074e-044.083e-04N-肉豆蔻酰基化位点PS00008G-{EDRKHPFYW}-x(2)-[STAGCN]{P}144到149:GLGKS 1.397e-02酰胺化位点PS00009x-G-[RK]-[RK]345到348:NGKR;350到353:LGRKV8.636e-04ATP/GTP结合位点PS00017[AG]-x(4)-G-K-[ST]141到148:GQSGLGKS 7.781e-052.4 利用CHP同源建模法的建模结果与评价以人Septin 3蛋白的GTPase域的三维结构(序列号为3SOP.B,与待建模序列相似性为69.6%,显著性水平为1e-106,模板长度270个残基,覆盖待建模板的63.7%)为模板,利用CPHmodels同源建模法进行建模,结果(见图3-A)表明,人SEPT9蛋白三级结构中α-螺旋、B-折叠、B-转角,无规则卷曲(白色)结构清晰可见,由7段α-螺旋和2组β-折叠构成,其中1组平行β-折叠片层结构由6个β折叠单链组成,位于蛋白质内部,另一组β-折叠片层结构由3个较小的β折叠单链组成,靠近蛋白质外部,无规则卷曲(包括β-转角)起着连接α-螺旋或β-折叠的作用.因此该蛋白是一个典型的α/β类蛋白.利用What_check程序获得模建结果的拉氏图(Ramachandran plot)(图3-B),结果表明,拉氏图中最合理区(图3B中的A,B,L区)和一般允许区(图3B中a,b,p)以及免强允许区(图3B中-a,-b和-p区)的残基数占比分别为87.8%、11.4%和0%,不允许区(白色区)为0.8%,在允许区内总计占99.2%,因此,模建结果比较理想;进一步利用verfy3D-1D软件检测模建结果的原子结构(3D)与序列(1D)的符合度,结果901第2期 王俊生,等:人SEPT 9蛋白同源建模和活性位点分析(图4)表明,最大得分0.68,最小得分-0.03,并且平均得分大于0.2的残基数占建模总残基数(270个氨基酸位置,包含一个空位)的84.7%,出于可接受的水平.因此,利用CHPmodel同源建模法获得的人SEPT 9蛋白的三级结构符合蛋白质立体化学品质规则.2.5 人SEPT9蛋白表面电位预测通过计算SEPT 9蛋白(AAH54004.1)各氨基酸残基的静电,使用软件Swiss-PdbViewer4.0.1模建蛋白质表面静电构象(图5),正电势与负电势共存于蛋白质表面,其中表面正电集团成族分布,占据较多的位置,表面整体呈正弱电势,其中负电势的残基成较小的簇状分布,负电残基分布在169、170、177、184、186、187、198、204、209、221、224、229-230、241、262、264、283、288-289、300、307、313、315-317、319-320、325、329、339、341、360、362、368、374、386和394位;其中288~289位的Glu呈较强负电势分布在表面,300、307、315、317和320位的Asp以及31,316,319位的Glu形成一个较大的负电集团,也分布在蛋白质表面,这些部位可能会影响到分子间的识别和结合,其余负电散布在蛋白质内部.蛋白内部正负电荷分布相对较均匀,整体偏中性.2.6 活性位点分析活性位点是指蛋白质立体结构中形成的能够结合配基的活性腔(口袋),利用Q-SiteFinder软件对人SEPT 9蛋白的配基结合位点进行预测,结果(图6)表明,该蛋白总体积为2.666 3E-26m3,含有10个可能的配基结合位点,大多数靠近蛋白质结构表面;各位点空腔体积(见表2)有明显差异,体积最大的位点1和最小的位点9的体积分别为2.16和1.26E-28m3.表2表明,组成各配基结合位点的氨基酸残基种类和数目具有较明显差异,可能影响了不同位点的空腔大小.011河南师范大学学报(自然科学版) 2014年表2 人SEPT9蛋白配基结合位点信息活性位点序号位点体积/(1E-30m3)活性位点氨基酸残基及其位置编号1 216Ser148,Ile151,Asn152,Ser168,Ile172,Pro173,Lys174,Thr175,Ile176,Glu177,Ile178,Lys179,Ile181,Asp1982 200Tyr309,pro310,Gln311,phe314,Asp315,Asp320,Asn324,Asn383,Asp386,Ile387,Ser390,Ile3913 163Ser143,Gly144,Leu145,Gly146,Lys147,Ser148,Thr149,Ile172,pro173,Lys174,thr175,4 200Asn152,Lys156,lys162,pro166,Thr167,Ser168,Glu169,Glu170,Asp339,His340,Glu341,5 152The298,Leu301,L eu302,Ile306,Asp307,Val308,Pro310,Gln311,Lys312,Arg328,Ile3316 170Met266,Lys267,Arg268,Ser270,Lys271,Val272,Asn304,Gly306,Ile307,Asp311,Gln313,Phe314,Ile387,Ile391,His3927 134Gln228,GLu229,Glu230,Val231,Asn232,Ile233,Asn234,Lys236,Ile239,Pro2408 153Glu134,Asn136,Ser180,Thr182,Lys193,Thr195,Ile197,Phe218,Gln222,Asp241,Thr242,Arg243,Val2449 126Asn220,Tyr223,Lys271,Val272,Val273,Ile391,His392,Phe393,Ala395,Tyr396,Lys39910 133Ile178,Thr199,Pro200,Gly201,Phe202,Gly203,His205,Cys211,Pro214,Ile215注:人SEPT 9蛋白体积为2.6663E-2.6m33 讨论和结论本研究预测了人SEPT 9蛋白的二级结构,并以人SEPT 3蛋白结构为模板,采用同源建模方法首次获得了人SEPT 9蛋白的三维结构,模建的原子空间分布质量经拉氏图检验,蛋白质主链上大多数氨基酸的Φ角和Ψ角分布在合理的区域,并且原子空间结构(3D结构)和氨基酸序列(1D结构)的兼容性或适配性较好.通过对模建的三级结构可视化,清晰地表明,该蛋白具有7段α-螺旋和2组β-折叠结构,其中1组平行β-折叠片层结构由6个β折叠单链组成,位于蛋白质内部,另一组β-折叠片层结构由3个较小的β折叠单链组成,靠近蛋白质外部,无规则卷曲(包括β-转角)起着连接α-螺旋或β-折叠的作用,这是一个典型的α/β类蛋白;计算其表面电荷分布,表明该蛋白质表面呈弱正电势分布,有2个相对集中的负电集团,内部正负电荷分111第2期 王俊生,等:人SEPT 9蛋白同源建模和活性位点分析布较均匀.Q-SiteFinder通过聚类蛋白质表面上范德华力(甲基)探针有利的区域来定位配体,该算法使用一个精度为基础的阈值来验证成功与否,精度定义为与配体距离1.6埃以内的探针在一个集群内的百分比,并且以25%的精确度阈值来定义一个成功的预测[26].该算法已被证明在前3名有正确预测的情况下,对于Nissink等描述的GOLD对接测试集(134个蛋白质-配体复合体)取得了90%的成功预测[27].Q SiteFinder预测的第一个位点作为结合位点平均准确率达到了68%[26].本研究预测获得了人SEPT 9蛋白可能的配基结合位点有10个,空腔体积大小不等,而这些位点体积大小与实际可能结合的配基体积大小差异较小,并具有较高的精确性,这一点在后续的分子对接、从头药物设计或结构鉴定或功能位点比较等研究中具有非常重要的作用[25].另外我们也利用基于口袋侦测算法的CASTp软件(http://sts.bioengr.uic.edu/castp/view/viewer.php)对该蛋白的配基结合位点进行了预测,结果预测的结合位点中口袋体积最大的位点1与Q SiteFinder预测的位点1在氨基酸组成上仅2个氨基酸差异,即多了Thr167,但缺少了Ile178,基本可以肯定两者是同一个活性位点.在一般情况下,利用CASTp进行配基结合位点预测,有74%的蛋白配基结合位点被确定位于最大的口袋[28],因此基于两种不同算法获得较为一致的预测结果,可以推测位点1应该是SEPT 9蛋白的真正活性位点,可以考虑把活性位点1作为后续研究重点,用于理论药物设计研究.当然其他配基结合位点能否成为真正的活性位点还需要通过其他技术方法进行进一步的研究.以上研究结果为进一步研究人SEPT 9蛋白结构与功能关系提供了理论依据,同时也为设计新型的人SEPT 9蛋白结构制剂,寻找神经性退行病、癌症和肿瘤缓解药物奠定了基础.参 考 文 献[1] Adam J C,Pringle J R,Peifer M.Evidence for functional differentiation among drosophila septins in cytokinesis and cellularization[J].Molecular Biology of the Cell(Print),2000,11(9):3123-3135.[2] Sharon A,Wang R,Matzkin H,et al.Msf-a interacts with hypoxia-inducible factor-1and augments hypoxia-inducible factor transcrip-tional activation to affect tumorigenicity and angiogenesis[J].Cancer Research,2006(66):856-866.[3] Barral Y,Mermall V,Mooseker M S,et al.Compartmentalization of the cell cortex by septins is required for maintenance of cell polarityin yeast[J].Molecular Cell,2000,5(5):841-851.[4] Beites C L,Xie H,Bowser R,et al.The septin CDCrel-1binds syntaxin and inhibits exocytosis[J].Nature Neuroscience,1999,2(5):434-439.[5] Berlin A,Paoletti A,Chang F.Mid2p stabilizes septin rings during cytokinesis in fission yeast[J].The Journal of cell biology,2003,160(7):1083-1092.[6] Bourne H R,Sanders D A,Mccormick F.The GTPase superfamily:conserved structure and molecular mechanism[J].Nature,1991,349(6305):117-127.[7] Russell S E,Hall P A.Septin genomics:a road less travelled[J].Biological Chemistry,2011,392(8/9):763-767.[8] Peterson E A,Petty E M.Conquering the complex world of human septins:implications for health and disease[J].Clinical Genetics,2010,77(6):511-524.[9] Connolly D,Abdesselam I,Verdier-Pinard P,et al.Septin roles in tumorigenesis[J].Biological Chemistry,2011,392(8/9):725-738.[10] Roeseler S,Sandrock K,Bartsch I,et al.Septins,a novel group of GTP-binding proteins:relevance in hemostasis,neuropathology andoncogenesis.[J].Klinische Paediatrie,2009,221(3):150-155.[11] Osaka M,Rowley J D,Zeleznik-le N J.MSF(MLL septin-like fusion),a fusion partner gene of MLL,in a therapy-related acute mye-loid leukemia with at(11;17)(q23;q25)[J].Proceedings of the National Academy of Sciences,1999,96(11):6428-6433.[12] Kalikin L M,Sims H L,Petty E M.Genomic and expression analyses of alternatively spliced transcripts of the MLL septin-like fusiongene(MSF)that map to a 17q25region of loss in breast and ovarian tumors[J].Genomics,2000,63(2):165-172.[13] Russell S E,Mcilhatton M A,Burrows J F,et al.Isolation and mapping of a human septin gene to a region on chromosome 17q,com-monly deleted in sporadic epithelial ovarian tumors.[J].Cancer Research,2000,60(17):4729-4734.[14] Sandrock K,Bartsch I,Bl ser S,et al.Characterization of human septin interactions.[J].Biological Chemistry,2011,392(8/9):751-761.[15] Peterson E A,Kalikin L M,Steels J D,et al.Characterization of a sept9interacting protein,sept14,a novel testis-specific septin[J].Mammalian Genome,2007,18(11):796-807.211河南师范大学学报(自然科学版) 2014年[16] 吕讷男.Septin-9和Clusterin在卵巢上皮性癌患者外周血和肿瘤组织中的蛋白水平及其临床意义[D].北京:北京协和医学院,2011.[17] Gonzalez M E,Peterson E A,Privette L M,et al.High SEPT9_v1expression in human breast cancer cells is associated with oncogenicphenotypes.[J].Cancer Research,2007,67(18):8554-8564.[18] Tóth K,Galamb O,Spisák S,et al.The influence of methylated septin 9gene on RNA and protein level in colorectal cancer.[J].Pa-thology and Oncology Research,2011,17(3):503-950.[19] Stanbery L,D'silva N J,Lee J S,et al.High SEPT9_v1expression is associated with poor clinical outcomes in head and neck squamouscell carcinoma.[J].Translational oncology,2010,3(4):239-245.[20] Bennett K L,Romigh T,Eng C.Disruption of transforming growth factor-beta signaling by five frequently ethylated genes leads to headand neck squamous cell carcinoma pathogenesis.[J].Cancer Research,2009,69(24):9301-9306.[21] Hannibal M C,Ruzzo E K,Miller L R,et al.Sept9gene sequencing analysis reveals recurrent mutations in hereditary neuralgic amyot-rophy[J].Neurology,2009,72(20):1755-1759.[22] 郭 佳.酿酒酵母轴向出芽地标蛋白Bud3p与septin细胞骨架相互作用的研究[D].武汉:武汉大学,2011.[23] Ito M,Okui H,Nakagawa H,et al.Synthesis and insecticidal activity of novel dihydropyrrole derivatives with n-sulfanyl,sulfinyl,andsulfonyl moieties[J].Bioorganic &Medicinal Chemistry,2003,11(4):489-494.[24] Shankar J,Messenberg A,Chan J,et al.Pseudopodial actin dynamics control epithelial-mesenchymal transition in metastatic cancercells[J].Cancer Research,2010,70(9):3780-3790.[25] Strausberg R L,Feingold E A,Grouse L H,et al.Generation and initial analysis of more than 15,000full-length human and mouse cD-NA sequences[J].Proceedings of the National Academy of Sciences of the United States of America,2002,99(26):16899-16903.[26] Laurie A T,Jackson R M.Q-sitefinder:an energy-based method for the prediction of protein-ligand binding sites[J].Bioinformatics,2005,9(21):1908-1916.[27] Nissink J W,Murray C,Hartshorn M,et al.A new test set for validating predictions of protein-ligand interaction[J].Proteins,2002,49(4):457-471.[28] Dundas J,Ouyang Z,Tseng J,et al.CASTp:computed at as of surface topography of proteins with structural and topographical map-ping of functionally annotated residues[J].Nucleic Acids Research,2006,34:116-118.Homology Modeling of Human SEPT9 Proteinand Analysis of Its Active SiteWANG Junsheng,RUAN Xianle,FAN Xiaofang,WANG Yongli,CHEN Long(College of Life Sciences and Agriculture,Zhoukou Normal University,Zhoukou 466001,China)Abstract:To reveal the function of human SEPT 9protein in physiological and pathological process through protein se-quence characteristics and molecule structure,its secondary structure,tertiary structures and motif were predicted by onlinewebsite.And the structural quality of atomic model(3D)were assessed by Ramachandran graph and compatibility test of 3Dvs1Dstructure,then its active sites were predicted.The results showed that the tertiary structures of human SEPT 9proteinwith higher quality was obtained,it comprised sevenα-helix and two types ofβ-extended strand,belonged toα/βprotein,thesurface in tertiary structure appeared weak positive charge.Eight different motif patters were found in amino acid sequence,which means that it may be involved in different biochemical reaction or perform different functions.At last,10different lig-and-bing sites in its tertiary structures were found,and the first one may be its real active site.The results were very importantto understand the function and to identify the location of ligand-binding sites for human SEPT 9protein,and also provided abasic information for molecular docking and de novo drug design.Key words:human being;SEPT 9protein;homology modeling;active site311第2期 王俊生,等:人SEPT 9蛋白同源建模和活性位点分析。
同源模型化法预测蛋白质结构的具体方法

同源模型化法预测蛋白质结构的具体方法同源模型化法是一种基于已知蛋白质结构来预测未知蛋白质结构的方法。
它是最早开发的蛋白质结构预测方法之一,也是最为成功和常用的方法之一。
下面将详细介绍关于同源模型化法预测蛋白质结构的具体方法。
1. 数据库选择和比对同源模型化法的第一个步骤是选择相关的结构数据库,并将待预测的蛋白质序列与这些数据库中已解析的结构进行比对。
目前,常用的数据库包括PDB、SWISS-MODEL和PHYRE 等。
比对通常使用BLAST或PSI-BLAST等算法,以寻找已知结构与待预测蛋白质的序列相似度较高的同源蛋白质。
2. 模板选择从比对结果中,选择相似度最高的已解析蛋白质结构作为模板。
模板选择的质量和准确性对于蛋白质结构预测的质量具有至关重要的影响。
3. 序列比对和合并将待预测蛋白质的序列与选定的模板序列进行全局比对并合并,以确定两者之间的结构异构体差异和相似性。
这一步骤通常使用多序列比对算法,并且需要手动校对和手动调整,以保障序列和结构的一致性和可靠性。
4. 结构拟合根据序列比对的结果,在已有模板结构的基础上对待预测蛋白质进行拟合,以得到一个初步的结构模型。
结构拟合通常使用最小二乘法进行优化,以便使拟合后的蛋白质结构与模板结构尽量重合。
5. 质量评估对预测的蛋白质结构进行质量评估,以确定预测模型的可靠性和准确度。
质量评估包括一系列的评估指标,如Ramachandran图、能量值、概率评估和实验验证等。
6. 侧链构象优化在蛋白质结构预测过程中,侧链的位置通常依赖于主链的位置。
需要对侧链进行构象优化,以提高预测结构的可靠性和准确性。
7. 模型重构和优化在很多情况下,预测的蛋白质结构可能存在结构上的错误或不合理之处。
需要对模型进行重构和优化,以提高预测结构的可靠性和准确性。
8. 能量最小化能量最小化是为了使蛋白质结构达到最佳的稳定状态。
在这一步中,使用能量函数对预测结构进行优化,并进行优化和调整,以使结构的能量尽可能小。
基于同源建模的蛋白质结构预测方法的研究

基于同源建模的蛋白质结构预测方法的研究
同源建模是一种预测蛋白质结构的方法,它利用已知结构的蛋白质来推断目标蛋白质的结构。
这种方法的核心思想是:如果两个蛋白质的序列相似度很高,它们的结构也很可能相似。
因此,如果已知一个与目标蛋白质序列相似的蛋白质的结构,我们就可以通过同源建模来预测目标蛋白质的结构。
同源建模的具体步骤包括:选择已知结构与目标蛋白质序列相似度较高的蛋白质作为模板;将目标蛋白质序列与模板序列进行比对,确定相应的残基对应关系;利用这些对应关系,建立目标蛋白质的初始结构;通过模拟退火、分子动力学等算法对初始结构进行优化,得到最终的蛋白质结构。
同源建模的优点是能够高效地预测目标蛋白质的结构,尤其是在没有实验测定结构的情况下。
同时,同源建模还可以帮助研究人员理解蛋白质的结构与功能之间的关系,为药物设计等方面提供有价值的信息。
同源建模也存在一些限制,例如模板蛋白质与目标蛋白质的序列相似度必须较高,否则预测结果可能不准确;此外,同源建模无法预测具有新型结构或结构中存在大量不规则构象的蛋白质的结构。
总之,同源建模是一种重要的蛋白质结构预测方法,其在生物学、药物设计等领域具有广泛的应用前景。
蛋白质结构预测方法及其应用前景

蛋白质结构预测方法及其应用前景引言:蛋白质是构成生物体的重要组成部分,它们在细胞内发挥着关键的功能。
了解蛋白质的结构对于理解其功能和参与药物开发具有重要意义。
然而,实验手段通常耗时、昂贵且可能有限,因此研究人员一直在寻求一种准确、高效的方法来预测蛋白质的结构。
本文将介绍几种常见的蛋白质结构预测方法,并探讨其在生物医学领域中的应用前景。
一、同源建模方法同源建模是一种基于相似蛋白质序列的结构预测方法。
它假设具有相似序列的蛋白质可能具有相似的结构,因此通过与已解析的蛋白质结构进行比较,可以推断出目标蛋白质的结构。
这种方法的优势在于准确性高、时间效率高,已被广泛应用于生物医学研究中。
例如,同源建模方法可以用于预测蛋白质-蛋白质相互作用,帮助研究人员理解蛋白质间的相互作用机制,有助于药物研发和疾病治疗。
二、蛋白质折叠动力学模拟蛋白质折叠动力学模拟是一种基于物理力学原理的结构预测方法。
它通过模拟蛋白质分子的运动过程,推测出最稳定的蛋白质结构。
这种方法的优势在于可以考虑蛋白质分子的动态过程,从而更好地预测其结构。
蛋白质折叠动力学模拟在酶的催化机制研究、蛋白质结构稳定性预测等方面具有广泛的应用前景。
三、基于机器学习的方法随着机器学习的快速发展,越来越多的研究人员开始将其应用于蛋白质结构预测中。
例如,基于深度学习的神经网络模型可以通过学习大量已知蛋白质结构的数据,来预测未知蛋白质的结构。
这种方法的优势在于可以自动从大量数据中提取特征,并学习蛋白质的结构模式。
基于机器学习的方法在蛋白质药物设计、蛋白质功能预测等领域有着广阔的应用前景。
四、蛋白质结构预测的应用前景蛋白质结构预测方法的不断发展,为生物医学领域带来了广泛的应用前景。
首先,结构预测可以帮助揭示蛋白质的功能机制,从而推动药物研发和疾病治疗。
其次,结构预测可以用于预测蛋白质-蛋白质或蛋白质-小分子/药物的相互作用,为药物设计和药效评估提供重要信息。
再者,结构预测还可以用于研究蛋白质折叠与稳定性,有助于理解蛋白质的功能和突变对其结构和功能的影响。
蛋白质结构预测与设计方法

蛋白质结构预测与设计方法随着生物信息学和基因工程技术的发展,蛋白质结构预测和设计成为了重要的研究领域。
蛋白质是生命体中极其重要的一类分子,它不仅是生物体内激素、抗体和酶等生物功能大分子,同时还是生物化学研究中的重要探针。
因此,研究蛋白质结构预测与设计方法,对于理解生物化学和生物医学等领域具有十分重要的意义。
本文将从蛋白质结构与功能、蛋白质结构预测与设计原理、常用的蛋白质结构预测与设计方法等方面进行论述。
一、蛋白质结构与功能从化学结构上来看,蛋白质是一类高分子碳水化合物,是由氨基酸以肽键相连形成的长链分子。
其化学结构的特点是: 在分子中存在大量的氢键、离散键、与烷基相互作用等影响蛋白质结构的重要因素,这些因素使得蛋白质结构非常复杂,蛋白质的空间构象很有规律但也非常灵活。
蛋白质能够完成生命体内的各种功能是因为它的结构对相关生物分子的识别和互作起着至关重要的作用。
蛋白质的功能直接取决于它的三维结构:在结构基础上,蛋白质能够稳定地与其他生物分子发生相互作用。
这种作用在生物体内具有不可替代的作用,如酶的催化过程、神经传导、免疫反应等,这些生物学过程的承担者往往就是各种蛋白质。
二、蛋白质结构预测与设计原理生物中的蛋白质结构具有很高的多样性,但却难以被实验记录下来。
从这个意义上来看,蛋白质结构预测和设计的意义就显得尤为重要了。
蛋白质结构预测和设计的目的在于确定蛋白质的三维结构,并根据结构和功能间的关系进行分析和设计。
与传统的研究方法不同的是,蛋白质结构预测和设计方法是通过计算模拟分析确定蛋白质的可能结构和功能。
蛋白质结构预测和设计的方式是根据比较理性的原理进行的,即参照已知的蛋白质结构确定未知蛋白质的结构。
在比较的过程中,应选择最相似的结构模板,使模板结构适应于预测的蛋白质序列。
这样,就可以用已知结构的物质为通准存储器,在此基础下为预测或设计目标型蛋白质求数学解,进而为找到合适的解答方案提供重要的基础。
三、常用的蛋白质结构预测与设计方法(1)同源建模法同源建模法是最常用的蛋白质结构预测方法,在大量已知蛋白质结构模板中,通过序列比较找到最相似的结构模板,对预测蛋白质的三维结构进行模拟并预测其结构。
蛋白同源建模及分子对接知识讲解

蛋白模型 局部(每个氨基酸)Z-score
Z-score在pdb数据库中所有蛋白中的分布
接近91% >80% -0.10 ~ 0.10 <1.0
Autodock 4.0分子对接
• 受体:以Swiss-model构建的CueO模型为例, 未经优化。
• 配体:文献中所给出的CueO的底物之一— —二乙醇胺(Diethanolamine)
Diethan olamine
准备受体和配体 CueO
Grid box参数设置
分子对接结果展示
待解决的问题
• 1. 蛋白模型的评估还需完善。 • 2.蛋白模型的优化:因为在线网站Chiron
的优化效果并不好,所以在查阅文献后, 拟用本地软件olex2进行局部优化。 • 3.分子对接的评价。 • 4.为确定酶底物,最好补充一个虚拟底物 筛选试验,拟用Autodock Vina软件完成。
中的区域。可能是因为预测模型中Cu离子的缺失,导致 对活性周围的残基电子云分布,肽键角度,二级结构等 造成了影响。
蛋白模型的优化
Chiron网站界面
Chiron优化前后分子能量对比
Save检测优化后的模型
项目 Procheck Ramachandran plot
ERRAT Overall quality factor Verify3d Averaged 3D-1D score>0.2 Prove Z-score average Z-score RMS
蛋白质三级结构预测(swiss-model同源建模)
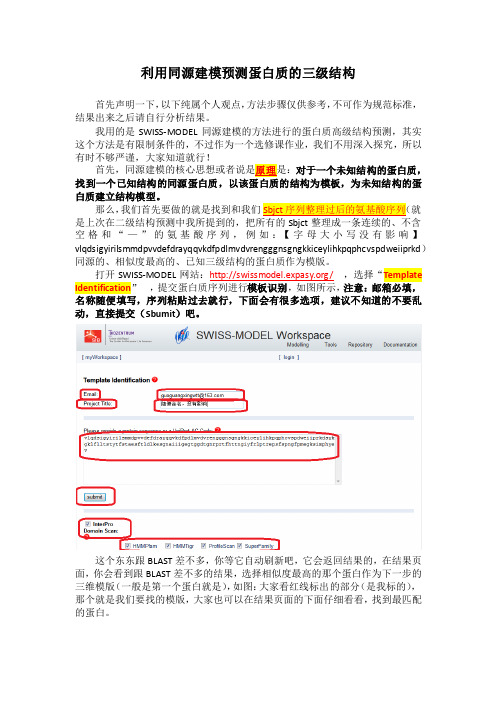
利用同源建模预测蛋白质的三级结构首先声明一下,以下纯属个人观点,方法步骤仅供参考,不可作为规范标准,结果出来之后请自行分析结果。
我用的是SWISS-MODEL同源建模的方法进行的蛋白质高级结构预测,其实这个方法是有限制条件的,不过作为一个选修课作业,我们不用深入探究,所以有时不够严谨,大家知道就行!对于一个未知结构的蛋白质,白质建立结构模型。
那么,我们首先要做的就是找到和我们空格和“—”的氨基酸序列,例如:【字母大小写没有影响】vlqdsigyirilsmmdpvvdefdrayqqvkdfpdlmvdvrengggnsgngkkiceylihkpqphcvspdweiiprkd)同源的、相似度最高的、已知三级结构的蛋白质作为模版。
打开SWISS-MODEL网站:/,选择“Template Identification,提交蛋白质序列进行模板识别,如图所示,注意:邮箱必填,名称随便填写,序列粘贴过去就行,下面会有很多选项,建议不知道的不要乱动,直接提交(Sbumit)吧。
这个东东跟BLAST差不多,你等它自动刷新吧,它会返回结果的,在结果页面,你会看到跟BLAST差不多的结果,选择相似度最高的那个蛋白作为下一步的三维模版(一般是第一个蛋白就是),如图:大家看红线标出的部分(是我标的),那个就是我们要找的模版,大家也可以在结果页面的下面仔细看看,找到最匹配的蛋白。
这里还有一点要作说明,就是上图标出的代码是PDB编号,前四个表示PDB- Code,最后一位表示Chain-ID,具体什么意思,大家有兴趣就去了解一些吧。
接下来,去NCBI串串门吧,在NCBI中搜索上面查到的蛋白的PDB号,一般输入前四位就行啦,注意:搜索蛋白库(Protein)。
找到以后,以FASTA格式显示。
接下来,我们再回到SWISS-MODEL,接下来就是重点和难点啦,在线提交序列进行同源建模分析,这个在线提交不是大家想象的那么容易,这个耗费了我大部分的时间,说到这里我就想画个圈圈诅咒它,大家注意啦~~~~~~~~~~~SWISS-MODEL 是一个自动化的蛋白质比较建模服务器,该服务器提供用户三种模式可选择:Automatic mode(简捷模式): 用于建模的氨基酸序列或是Swiss-Prot/TrEMBL (/sprot )编目号(accession)可以直接通过web界面提交。
蛋白质结构预测方法评价及分析准确性对比

蛋白质结构预测方法评价及分析准确性对比蛋白质是生物体中重要的生物分子,它们在细胞机器中扮演着关键角色。
了解蛋白质的结构对于理解其功能和相互作用至关重要。
然而,实验方法并不总能够完全揭示蛋白质的三维结构,这就需要借助计算方法进行预测。
本文将评价和分析不同蛋白质结构预测方法的准确性。
蛋白质结构预测方法主要分为两类:实验方法和计算方法。
实验方法包括X射线晶体学、NMR等,它们可以直接测定蛋白质的三维结构。
然而,实验方法存在一些限制,如费时、昂贵和技术限制等。
相比之下,计算方法更具实用性和效率。
计算方法中最主要的两种蛋白质结构预测方法是同源建模(homology modeling)和脱新GDT-TS(template-free GDT-TS)。
同源建模基于已知的同源蛋白质的结构,通过比对序列相似性和结构保守性来预测目标蛋白质的结构。
脱新GDT-TS是一种无模板方法,它通过分析蛋白质的序列和物理化学性质来推测其结构。
在评价这些结构预测方法的准确性时,常用的衡量指标是GDT-TS(Global Distance Test-Total Score)和TM分数(Template Modeling Score)。
GDT-TS是一种评估预测结构与真实结构之间相似性的分数,取值范围为0到100。
TM分数则是一种计算目标蛋白质与模板蛋白质之间结构相似性的分数。
同源建模方法在模板蛋白质与目标蛋白质之间具有较高的序列相似性时,其预测准确性通常较高。
然而,当序列相似性较低时,预测准确性会显著下降。
对于那些缺乏亲源模板的目标蛋白质,脱新GDT-TS方法可能会更有优势。
此外,脱新GDT-TS方法还可以帮助研究人员预测目标蛋白质的结构空间分布以及功能域等信息。
除了同源建模和脱新GDT-TS方法之外,还有其他一些蛋白质结构预测方法,如串联建模(ab initio modeling)、分子动力学模拟(molecular dynamics simulation)和遗传算法等。
同源模建的方法与结果分析

同源模建的方法与结果分析Version 1.0.2-------------------------------------------------------序言:作为一个以实验为主的生化工作者来说,很多时候可以通过分子生物学手段获取自己需要的目的基因,并在各种表达载体和宿主中进行对应蛋白的表达,随后对于这些蛋白的特性进行研究,这也是一般酶学研究的特定套路。
而近十几年来,人们开始思考是否能够将特性与蛋白质的三级结构进行关联,从分子水平理解蛋白质与底物之间的相互作用呢?于是类似于蛋白结构模建、分子对接、分子动力学模拟、量化计算等多种手段相继被创造以及应用。
在这些方法中,同源模建无疑是最基础也是最重要的一个步骤,因为其质量的好坏直接决定了后续工作是否可信。
因此,本文打算就同源模建的基本原理、常用软件及服务器以及结果分析与改进提供一些个人的经验,并希望各位朋友能够给予批评指正。
1.同源模建的原理及应用限制两点基本原理:1.一个蛋白质的结构由其氨基酸序列唯一的决定。
知道其一级序列,至少在理论上足以获取其结构2. 结构在进化中更稳定,变化比序列层面的变化要缓慢许多。
应用限制:模板蛋白和目标蛋白的序列一致性需要大于30%,且越大建模准确性越有保障。
了解了基本的原理,我们需要知道在实际操作中,同源模建都需要怎么样进行。
同源模建的过程从实践中可分为以下7个步骤:1.模板识别和初始比对在序列一致性比较高的时候,可以通过简单的序列比对程序如BLAST获取目标蛋白的结构(将比对的数据库选择为PDB数据库)。
2.比对结果的校正用以上的方法确定一个或多个建模模板后,应该采用更为精确的方法已取得更优的比对结果。
有时在序列一致性较低的区域比对两条序列可能会具有困难,这个时候,我们可以采取其他同源蛋白序列一起参与比对来找到解决的办法。
3.主链生成比对完成后,就可以开始实际的建模过程了,相对与后面几步来说,主链建模时最没有难度的一步了,因为大部分软件都是通过简单的拷贝模板蛋白的主链坐标来实现这一目的的。
蛋白质的评价方法

蛋白质的评价方法蛋白质是构成生物体重要组成部分的大分子有机化合物,它在细胞功能的调控和维持生命活动中起着关键作用。
因此,对蛋白质进行准确的评价是非常重要的。
目前,蛋白质的评价方法主要包括定量分析和功能评估两个方面。
定量分析是评价蛋白质含量和组成的基本方法。
常用的定量分析方法有生物化学方法、免疫学方法和质谱法。
生物化学方法包括测定总蛋白质含量的Lowry法、Bradford法和BCA法等,通过比色反应定量测定蛋白质含量。
免疫学方法常用的是酶联免疫吸附法(ELISA)和免疫印记法(Western blotting),通过检测蛋白质与特异性抗体的相互作用来定量分析。
质谱法包括质谱分析和质谱成像技术,可以直接测定蛋白质的分子量和序列,为蛋白质研究提供了更加准确和全面的信息。
功能评估是评价蛋白质生物活性和功能的方法。
常用的功能评估方法有结构与功能关系研究、酶活性分析、蛋白质相互作用研究和功能验证等。
结构与功能关系研究利用蛋白质的结构信息来预测其功能,包括比较模建法、同源建模法和分子动力学模拟等。
酶活性分析通过测定酶催化底物的转化速率来评估蛋白质的催化活性。
蛋白质相互作用研究利用蛋白质与其他分子之间的相互作用来推断其功能,常用的方法有蛋白质-蛋白质相互作用分析和蛋白质-小分子相互作用分析。
功能验证是通过基因敲除、基因过表达、体内和体外实验等手段验证蛋白质的功能。
除了以上方法,近年来还出现了一些新的评价蛋白质的方法,如蛋白质组学技术和单细胞蛋白质组学技术。
蛋白质组学技术通过高通量的蛋白质质谱分析,可以研究蛋白质在整个生物体内的表达水平和修饰情况,从而揭示蛋白质的功能和调控机制。
单细胞蛋白质组学技术可以在单个细胞水平上分析蛋白质的表达和变化,为研究细胞异质性和个体差异提供了新的途径。
综上所述,蛋白质的评价方法包括定量分析和功能评估两个方面,通过多种技术手段可以全面、准确地评价蛋白质的含量、结构和功能,为蛋白质研究提供了重要的工具和方法。
同源建模的实验报告(3篇)

第1篇一、实验目的1. 熟悉同源建模的基本原理和方法;2. 掌握同源建模的实验步骤和操作技巧;3. 通过同源建模预测蛋白质的三级结构,并验证预测结果的准确性。
二、实验原理同源建模是一种基于生物信息学的方法,利用已知蛋白质的三维结构作为模板,通过计算机模拟和计算,预测未知蛋白质的三维结构。
该方法基于以下两个原理:1. 蛋白质的结构由其氨基酸序列唯一决定,知道其一级序列,在理论上就可以获取其二级结构以及三级结构;2. 蛋白质的三级结构在进化中更稳定或者说更保守。
如果两个蛋白质的氨基酸序列有50%相同,那么约有90%的α-碳原子的位置偏差不超过3,这是同源模型化方法在结构预测方面成功的保证。
三、实验材料与仪器1. 实验材料:蛋白质序列、NCBI数据库、SwissModel在线平台、MOE软件;2. 实验仪器:计算机、网络连接。
四、实验步骤1. 收集蛋白质序列:在NCBI数据库中搜索目标蛋白质的序列,获取其一级结构信息。
2. 模板搜寻:使用SwissModel在线平台进行模板搜寻,寻找与目标蛋白质序列具有较高同源性的已知蛋白质结构作为模板。
3. 模型构建:根据模板蛋白质的三维结构,使用SwissModel在线平台构建目标蛋白质的三维结构模型。
4. 模型优化:使用MOE软件对模型进行优化,包括分子力学优化、能量最小化等。
5. 模型验证:通过比对实验结果和预测结构,验证同源建模的准确性。
6. 结果分析:分析预测结构的合理性,解释蛋白质的功能和作用机理。
五、实验结果与分析1. 模板搜寻:在SwissModel在线平台中,成功找到与目标蛋白质序列具有较高同源性的模板蛋白质。
2. 模型构建:根据模板蛋白质的三维结构,成功构建了目标蛋白质的三维结构模型。
3. 模型优化:使用MOE软件对模型进行优化,优化后的模型具有更低的能量。
4. 模型验证:通过比对实验结果和预测结构,验证同源建模的准确性。
预测结构在关键区域与实验结果基本一致。
蛋白同源建模模板要求

蛋白同源建模模板要求蛋白同源建模模板是一种用于预测蛋白质结构的方法。
它基于已知的蛋白质结构,通过比对目标蛋白质序列和已知蛋白质序列的相似性,预测目标蛋白质的结构。
在蛋白质结构预测领域,蛋白同源建模模板是一种常用的方法,因为它可以预测高精度的蛋白质结构。
蛋白同源建模模板的要求主要包括以下几个方面:1. 数据库选择蛋白同源建模模板需要使用已知的蛋白质结构作为模板,因此需要选择一个可靠的蛋白质结构数据库。
目前,常用的蛋白质结构数据库包括PDB、SCOP和CATH等。
在选择数据库时,需要考虑数据库的更新频率、数据质量和可靠性等因素。
2. 序列比对蛋白同源建模模板需要将目标蛋白质序列与已知蛋白质序列进行比对,以确定它们之间的相似性。
在进行序列比对时,需要选择一个可靠的序列比对工具,如BLAST、ClustalW和MUSCLE等。
同时,需要考虑序列比对的参数设置,以获得最佳的比对结果。
3. 模板选择蛋白同源建模模板需要选择一个合适的模板,以预测目标蛋白质的结构。
在选择模板时,需要考虑模板与目标蛋白质的相似性、模板的分辨率和可靠性等因素。
同时,需要使用多个模板进行比对,以获得最佳的模板选择结果。
4. 结构建模蛋白同源建模模板需要使用模板的结构信息,以预测目标蛋白质的结构。
在进行结构建模时,需要选择一个可靠的结构建模工具,如MODELLER、Rosetta和I-TASSER等。
同时,需要考虑结构建模的参数设置,以获得最佳的结构预测结果。
5. 结果评估蛋白同源建模模板需要对预测结果进行评估,以确定预测结果的可靠性。
在进行结果评估时,需要使用一些评估指标,如RMSD、GDT和QMEAN等。
同时,需要将预测结果与实验测定的结构进行比对,以确定预测结果的准确性。
总之,蛋白同源建模模板是一种可靠的蛋白质结构预测方法,但它需要满足一定的要求,才能获得高质量的预测结果。
在进行蛋白同源建模模板时,需要选择可靠的数据库、序列比对工具、模板选择工具、结构建模工具和结果评估指标,以获得最佳的预测结果。
同源建模_精品文档

开始
同源性对比
选择模板
靶序列与模板结构 进行比对 使用模板结构的信 息建立靶序列的模型
评价模型
模型 满意?
结束
同源建模流程图
同源建模的评价
RMSD
是评价最普遍的标 准, 它是目标蛋白 与模板蛋白结构之 间对应原子间的均 方差。
PROSA
是模型能量方面的 评价, 用于评价每 个残基之间的相互述及原理
使用这种方法有两个前提条件: 首先,目标序列的同源蛋白质中某一个或者几个结 构已经被解析(x射线或者NMR结构均可); 其次,目标序列与该蛋白质的同源性足够高,一般 50%以上的同源性能够得到比较合理的构象。 原理是序列相似则结构相似,即存在同源关系的两条 序列具有相似的结构。 特点同源建模是目前从序列预测结构的最切实可行、 应用最广泛的一种方法。 工具SWISS-MODEL, CPH models
随着计算机硬件和预测方法的发展, 同源建模将会在蛋白质设 计和计算机辅助药物设计等方面应用广泛。在后基因组时代, 蛋 白质结构同源建模将起着关键桥梁作用, 是基因组、蛋白质组等 信息应用于生物工程中。
Thank You!
Verify3D
比较模型和氨基酸 一级结构的关系
人感染甲型 H 7 N 9 亚型禽流感病毒血凝 素蛋白同源建模
模板选择和序列比对
在PDB数据库中搜索模板 以 12 条人感染 H 7N 9 亚型流感 病毒血凝素作全
序列, 分别 构建两组参 比序列数 据集 。第一组 参 与比序列数 据集是 用 BL A ST 程序 在G enBank 数据库中搜索到的同新发 H 7N 9 亚 型毒株血凝 素接近 的序列。第二组参比序列 是在 Gen—Bank 数据库中采用分层抽样的方法 , 在 H 1~H 16亚型 中每个亚型随机抽取 2 或 3 条序列。用 M U SC L E 软件 对上述数据集进行多重序列比对。
生物学中的蛋白质结构预测与分类

生物学中的蛋白质结构预测与分类蛋白质是细胞内最基本的分子机器,负责生命活动的各个方面。
由于蛋白质的结构和功能密切相关,因此对蛋白质结构的解析和预测具有极其重要的意义。
在过去的几十年中,人们利用各种方法进行了大量的蛋白质结构预测和分类研究,为生物学、医学和药物研发等领域奠定了坚实的基础。
一、蛋白质结构的预测方法蛋白质结构预测是指根据蛋白质的序列信息,通过一系列的计算方法,预测出其可能的三维结构。
目前,蛋白质结构预测的方法主要包括以下几种。
1.同源建模法(Homology modeling)同源建模法是目前最为成熟的蛋白质结构预测方法之一。
该方法基于已知蛋白质结构的同源性,通过比对相似蛋白质序列的氨基酸组成和位置信息,预测出目标蛋白质的结构。
该方法的准确度和成功率都很高,但需要一定的同源性条件。
2.蒙特卡罗模拟法(Monte Carlo Simulation)蒙特卡罗模拟法是一种基于统计力学的方法,通过对不同构象的能量计算和比较,预测出最为可能的蛋白质结构。
该方法的随机性较大,预测结果具有一定的不确定性。
3.碎片拼装法(Fragment Assembly)碎片拼装法是一种基于碎片库的方法,将大量已知的二面角片段拼接成一条完整的氨基酸链,进而预测出蛋白质的结构。
该方法的成功率较高,但需要大量的计算和有效的碎片库。
二、蛋白质结构的分类方法蛋白质结构的分类是指将不同的蛋白质结构按照其结构特征和功能进行归类。
目前,蛋白质结构的分类方法主要包括以下几种。
1.基于结构域的分类(Domain-based classification)基于结构域的分类方法是将蛋白质结构的不同结构域进行分类,并通过结构域之间的相似性来判断蛋白质结构之间的关系。
该方法的分类结果较为准确,但无法涵盖所有的蛋白质结构。
2.基于结构重复单元的分类(Fold-based classification)基于结构重复单元的分类方法是将蛋白质结构划分为一定数量的结构重复单元,并通过这些单元之间的差异和相似性来进行分类。
基于同源建模的蛋白质结构预测方法的研究

基于同源建模的蛋白质结构预测方法的研究
蛋白质结构预测是当前生物水平端建模(BLM)编程中重要的一部分,它可以提供关
于蛋白质结构的准确信息。
随着基因组计算机技术的发展,蛋白质结构预测已成为关键问题。
基于同源建模是现有蛋白质结构预测方法中研究最为深入的一种,也是最为成功的一种。
基于同源建模的蛋白质结构预测方法是一种可以通过对已知的同源蛋白的结构和序列
进行比较,从而预测新蛋白结构的方法。
它使用高度耦合的模型从软件层面实现了基于比
较结构信息的蛋白质相似性评估。
它将已知结构改变至和未知蛋白结构尽可能相似的水平,以实现完美的拷贝,然后对拷贝进行细节调整,以获得更好的预测结果。
研究表明,基于同源建模的蛋白质结构预测方法的准确性要好于其他预测方法,如随
机折叠和统计力学模拟,它能有效预测分子大小在50万到100万范围内的蛋白质结构。
另外,虽然在基于同源建模的蛋白质结构预测方法中,它需要大量的计算量以便找到一致
的拷贝,但是,计算量能够在可控范围内。
基于同源建模的蛋白质结构预测方法一直是当前蛋白结构预测领域的重要研究热点。
不仅可以有效地预测蛋白质结构,而且还可以分析正确模型后出现的功能性变化,在蛋白
质交互作用和蛋白质工程设计中发挥作用。
此外,这种方法还可以提供关于X射线衍射和
核磁共振结构解析中,蛋白质吸引形状的重要信息。
使用生物大数据技术进行蛋白质结构预测的方法

使用生物大数据技术进行蛋白质结构预测的方法生物大数据技术在蛋白质结构预测方面发挥着重要的作用。
蛋白质是细胞中最重要的生物分子之一,它们的结构决定了它们的功能和相互作用方式。
然而,实验确定蛋白质的结构是非常昂贵和耗时的过程。
因此,利用生物大数据技术进行蛋白质结构预测已成为一种广泛应用的技术。
生物大数据技术用于蛋白质结构预测的方法有多种,其中包括蛋白质同源建模、蛋白质折叠动力学模拟和深度学习等。
蛋白质同源建模是一种常用的蛋白质结构预测方法。
这种方法基于两个假设:1)相似的序列通常具有相似的结构;2)结构相似的蛋白质通常具有相似的功能。
当一个蛋白质的序列与已知结构的蛋白质序列相似时,可以通过将已知结构的蛋白质应用到目标蛋白质上,从而预测目标蛋白质的结构。
蛋白质同源建模方法的预测准确性随着目标蛋白质与已知结构蛋白质的相似度而增加。
蛋白质折叠动力学模拟是通过模拟蛋白质在原子水平上的动力学过程来预测蛋白质的结构。
该方法基于物理力学原理和模拟算法,通过计算蛋白质分子的势能和运动方程来模拟蛋白质的折叠过程。
蛋白质折叠动力学模拟方法的优点是可以同时考虑蛋白质的结构、动力学和能量等多种因素,并且可以提供对蛋白质折叠可能路径的研究。
深度学习是一种基于神经网络的机器学习方法,近年来在蛋白质结构预测方面取得了显著的突破。
深度学习方法通过训练大量的蛋白质序列和结构数据,学习序列和结构之间的相关性。
然后,通过将未知序列输入已训练好的模型中,推断未知序列的结构。
深度学习方法的优点是可以处理大规模的数据,并且在预测准确性方面表现出色。
尽管使用生物大数据技术进行蛋白质结构预测具有很大的潜力,但仍然存在一些挑战和限制。
首先,蛋白质结构的预测精度仍然不够高,尤其是对于与已知结构相比较低的蛋白质。
其次,生物大数据技术需要大量的计算资源和存储空间,对于一些研究实验室和个人来说可能有限。
此外,生物大数据的质量和可靠性也是一个重要的问题,需要采取有效的方法来处理和验证数据。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
其他几个工具结果:
Verify3D 图像曲线:
除以上常用方法外,Molprobity 除以上常用方法外,Molprobity 也是一 种结构评估方法: Molprobity是个在线模型分析工具,地址 Molprobity是个在线模型分析工具,地址 为: / index.php 几乎所有的蛋白结构在发表之 前都会经过这个工具检测,目前成为结构 生物学中一个比较全面的蛋白结构检测工 具。
步骤一:上传文件 步骤一:上传文件
步骤二:加氢原子 步骤二:
步骤三:开始检测模型
步骤四:选择要分析的参数,一般采用默认 步骤四:选择要分析的参数, 值
第五步:查看结果, 第五步:查看结果,这里有图形分析和具体表格分 析。 图形分析讲形象地告诉你那些部位的残基存在问 题; 表格分析则给出具体残基的编号。
在线评估网址: Errat: Errat:/ERRATv2/ WhatWhat-if: http://swift.cmbi.ru.nl/servers/html/index.html 工具比较全的一个网址: /SAVES/ Molprobity : /i PROCHECK, PROCHECK,WHT_CHECK,ERRAT,VERIF Y_3D,PROVE等几个工具; Y_3D,PROVE等几个工具;
将同源建模所得的pdb文件进行上传,并运行, 将同源建模所得的pdb文件进行上传,并运行,即可得到 所要的结果: 其中ERRAT , 得到的打分值与使用ERRAT 自己的网址的 其中 ERRAT, 得到的打分值与使用 ERRAT自己的网址的 分值是相同的。 分值是相同的。 在这里展示一下单独ERRAT得到的评估结果: 在这里展示一下单独ERRAT得到的评估结果:
Errat平均打分值为91,同源结果打分值为 Errat平均打分值为91,同源结果打分值为 89.3,略微偏低。 89.3,略微偏低。
Procheck结果: Procheck结果:
Procheck所得到的 Procheck所得到的Ramachandran图:
其中红色区域represent the residues in most favoured regions; 黄色区域represent the residues in additional allowed regions; 浅 黄 色 区 域 represent the residues in generously allowed regions;白色区域则是disall区域。
以得到的同源建模结果为例: 图形分析结果:
说明: 途中红色部分为较为严重的过近接触 atom clashes 绿色的为不合理的二面角 紫色的球为不合理的Cb 紫色的球为不合理的Cb 橘黄色的残基为不合理的rotamer 橘黄色的残基为不合理的rotamer
表格分析: 表格分析:
说明: 说明: 点击表格第一行蓝色的各个项,即可现实详细信息 可以看到不合理的几个残基
同源建模模型评估结果
翟耀绪 20102010-7-26
modeller同源建模后,使用chimera对模 modeller同源建模后,使用chimera对模 型做了1500步的最速下降法,得到最终模 型做了1500步的最速下降法,得到最终模 型。 在使用该模型前,需要对所得到的模型进 行评估;
最终的同源结构模型需要通过评估来确保其结构的特性符 合其生理生化规律。这包括检验Ф 合其生理生化规律。这包括检验Ф—ψ角度的异常性,键 长,紧密接触面等。另一种检验蛋白质结构模型质量的方 法是固定性考虑这些立体化学特性。这种方法可通过编辑 实验得到的蛋白质结构的空间特性和相互作用能的统计谱, 来发现模型的错误。通过比对统计参数与构建的模型,可 以知道哪些区域折叠正常,哪些是异常的。 常用的评估程序有:Procheck 检测常用生理化学参数; 常用的评估程序有:Procheck 检测常用生理化学参数; WHAT IF 检验化学正确性;ANOLEA 使用统计算的方法; 检验化学正确性; Verify3D Verify3D 使用统计计算的方法。 使用统计计算的方法。 注意 ANOLEA 和Verify3D 虽然都使用统计方法,但它 们的阈值不同,并且分数高低代表的含义也不同: ANOLEA 分数越低模型越正确,而Verify3D 中分数越高 分数越低模型越正确,而Verify3D 越正确。