生物信息学作业(一)
生物信息学作业A版

生物信息学作业CDK4:Reference:[01] Aggarwal P, V aites LP: Nuclear cyclin D1/CDK4 kinase regulates CUL4 expression and triggers neoplastic growth via activation of the PRMT5 methyltransferase. Oct .2010[02] Zhang X: MicroRNA-related genetic variations as predictors for risk of second primary tumor and/or recurrence in patients with early-stage head and neck cancer. Sep.2010[03] Lang JM: A Flexible Multiplex Bead-Based Assay for Detecting Germline CDKN2A and CDK4 V ariants in Melanoma-Prone Kindreds. Nov.2010查找序列:登陆NCBI数据库↓“search”框选“N ”,“for”框填“CDK4”↓点“mRNA LINKS”查看碱基序列基因序列:/nuccore/NM_000075.21 agccctccca gtttccgcgc gcctctttgg cagctggtca catggtgagg gtgggggtga61 gggggcctct ctagcttgcg gcctgtgtct atggtcgggc cctctgcgtc cagctgctcc121 ggaccgagct cgggtgtatg gggccgtagg aaccggctcc ggggccccga taacgggccg181 cccccacagc accccgggct ggcgtgaggg tctcccttga tctgagaatg gctacctctc241 gatatgagcc agtggctgaa attggtgtcg gtgcctatgg gacagtgtac aaggcccgtg301 atccccacag tggccacttt gtggccctca agagtgtgag agtccccaat ggaggaggag361 gtggaggagg ccttcccatc agcacagttc gtgaggtggc tttactgagg cgactggagg421 cttttgagca tcccaatgtt gtccggctga tggacgtctg tgccacatcc cgaactgacc481 gggagatcaa ggtaaccctg gtgtttgagc atgtagacca ggacctaagg acatatctgg541 acaaggcacc cccaccaggc ttgccagccg aaacgatcaa ggatctgatg cgccagtttc601 taagaggcct agatttcctt catgccaatt gcatcgttca ccgagatctg aagccagaga661 acattctggt gacaagtggt ggaacagtca agctggctga ctttggcctg gccagaatct721 acagctacca gatggcactt acacccgtgg ttgttacact ctggtaccga gctcccgaag781 ttcttctgca gtccacatat gcaacacctg tggacatgtg gagtgttggc tgtatctttg841 cagagatgtt tcgtcgaaag cctctcttct gtggaaactc tgaagccgac cagttgggca901 aaatctttga cctgattggg ctgcctccag aggatgactg gcctcgagat gtatccctgc961 cccgtggagc ctttcccccc agagggcccc gcccagtgca gtcggtggta cctgagatgg 1021 aggagtcggg agcacagctg ctgctggaaa tgctgacttt taacccacac aagcgaatct 1081 ctgcctttcg agctctgcag cactcttatc tacataagga tgaaggtaat ccggagtgag1141 caatggagtg gctgccatgg aaggaagaaa agctgccatt tcccttctgg acactgagag 1201 ggcaatcttt gcctttatct ctgaggctat ggagggtcct cctccatctt tctacagaga1261 ttactttgct gccttaatga cattcccctc ccacctctcc ttttgaggct tctccttctc1321 cttcccattt ctctacacta aggggtatgt tccctcttgt ccctttccct acctttatat1381 ttggggtcct tttttataca ggaaaaacaa aacaaagaaa taatggtctt tttttttttt1441 ttaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaa引物设计:登陆Primer3网页/primer593/input.htm↓粘贴源序列,选择参数↓点击pick primer获得引物/cgi-bin/primer3-web-cgi-bin-0.4.0/primer3_results.cgi得到最匹配引物序列及相应参数:OLIGO start len tm gc% any 3' seqLEFT PRIMER 394 20 59.99 55.00 3.00 1.00 gaaactctgaagccgaccag RIGHT PRIMER 606 20 60.02 50.00 5.00 1.00 aggcagagattcgcttgtgt SEQUENCE SIZE: 660INCLUDED REGION SIZE: 660BLASTN:登陆NCBI的BLAST网址/Blast.cgi点击nucleotide blast,粘贴序列及范围选择,点击进行BLAST结果输出匹配系列列表:序列排列情况:>ref|NM_000075.2| Homo sapiens cyclin-dependent kinase 4(CDK4), mRNALength=1474GENE ID: 1019 CDK4 | cyclin-dependent kinase 4 [Homo sapiens] (Over 100 PubMed links)Score = 394 bits (213), Expect = 1e-107Identities = 213/213 (100%), Gaps = 0/213 (0%)Strand=Plus/PlusQuery 394 AGGTGGCTTTACTGAGGCGACTGGAGGCTTTTGAGCATCCCAATGTTGTCCGGCTGATGG 453||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||Sbjct 394 AGGTGGCTTTACTGAGGCGACTGGAGGCTTTTGAGCATCCCAATGTTGTCCGGCTGATGG 453 Query 454 ACGTCTGTGCCACATCCCGAACTGACCGGGAGATCAAGGTAACCCTGGTGTTTGAGCATG 513||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||Sbjct 454 ACGTCTGTGCCACATCCCGAACTGACCGGGAGATCAAGGTAACCCTGGTGTTTGAGCATG 513 Query 514 TAGACCAGGACCTAAGGACATATCTGGACAAGGCACCCCCACCAGGCTTGCCAGCCGAAA 573||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||Sbjct 514 TAGACCAGGACCTAAGGACATATCTGGACAAGGCACCCCCACCAGGCTTGCCAGCCGAAA 573 Query 574 CGATCAAGGATCTGATGCGCCAGTTTCTAAGAG 606|||||||||||||||||||||||||||||||||Sbjct 574 CGATCAAGGATCTGATGCGCCAGTTTCTAAGAG 606>ref|NT_029419.12| Homo sapiens chromosome 12 genomic contig, GRCh37 reference primaryassemblyLength=71516776蛋白质分析:利用ProtParam查询蛋白质基本性质信息http://www.expasy.ch/tools/protparam.html↓↓http://www.expasy.ch/cgi-bin/protparamNumber of amino acids: 303 Molecular weight: 33729.8 Theoretical pI: 6.52Amino acid composition:Ala (A) 22 7.3%Arg (R) 23 7.6%Asn (N) 7 2.3%Asp (D) 17 5.6%Cys (C) 4 1.3%Gln (Q) 8 2.6%Glu (E) 19 6.3%Gly (G) 24 7.9%His (H) 9 3.0%Ile (I) 11 3.6%Leu (L) 33 10.9%Lys (K) 11 3.6%Met (M) 8 2.6%Phe (F) 13 4.3%Pro (P) 25 8.3%Ser (S) 15 5.0%Thr (T) 15 5.0%Trp (W) 3 1.0%Tyr (Y) 9 3.0%Val (V) 27 8.9%Pyl (O) 0 0.0%Sec (U) 0 0.0%(B) 0 0.0%(Z) 0 0.0%(X) 0 0.0%Total number of negatively charged residues (Asp + Glu): 36 Total number of positively charged residues (Arg + Lys): 34 Atomic composition:Carbon C 1515Hydrogen H 2381Nitrogen N 419Oxygen O 430Sulfur S 12Formula: C1515H2381N419O430S12Total number of atoms: 4757Extinction coefficients:Extinction coefficients are in units of M-1 cm-1, at 280 nm measured in water.Ext. coefficient 30160Abs 0.1% (=1 g/l) 0.894, assuming all pairs of Cys residues form cystinesExt. coefficient 29910Abs 0.1% (=1 g/l) 0.887, assuming all Cys residues are reduced Estimated half-life:The N-terminal of the sequence considered is M (Met).The estimated half-life is: 30 hours (mammalian reticulocytes, in vitro).>20 hours (yeast, in vivo).>10 hours (Escherichia coli, in vivo).Instability index:The instability index (II) is computed to be 39.16This classifies the protein as stable.Aliphatic index: 89.74Grand average of hydropathicity (GRAVY): -0.167结果分析:我们由以上可以获得氨基酸的数目,分子量等电点,消光系数,预计的半衰期,不稳定指数,组成元素,亲脂性指数等参数。
生物信息学-课堂练习作业生物信息学蛋白质序列分析-课堂练习
生物信息学蛋白质序列分析-课堂练习ZNF395, 全称为Zinc Finger Protein395, 又被称为PBF ,PRF1,DBP2,PRF-1,Si-1-8-14或DKFZp434K1210。
其氨基酸序列为(一)分析蛋白质的一级结构ZNF395蛋白的理论等电点为7.17,分子式C 2417H 3775N 679O 741S 23,原子总数为7635,总平均亲水性(GRA VY )为-0.451,脂肪指数64.54,不稳定指数69.57,序列N 末端是M (Met ),估计半衰期是:30小时(哺乳动物网状细胞,离体);>20小时(酵母,体内);>10小时(大肠杆菌,体内)。
在编码的513个氨基酸中,包括48个带负电的氨基酸(天冬氨酸+谷氨酸),33个带正电荷的氨基酸(精氨酸+赖氨酸)。
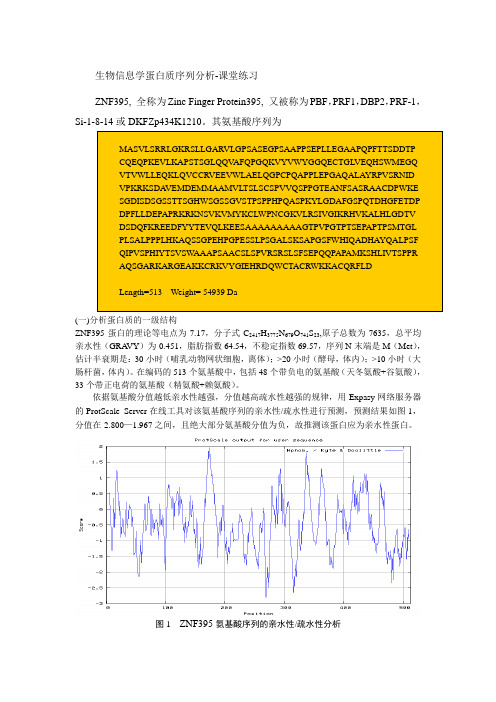
依据氨基酸分值越低亲水性越强,分值越高疏水性越强的规律,用Expasy 网络服务器的ProtScale Server 在线工具对该氨基酸序列的亲水性/疏水性进行预测,预测结果如图1,分值在-2.800—1.967之间,且绝大部分氨基酸分值为负,故推测该蛋白应为亲水性蛋白。
图1 ZNF395氨基酸序列的亲水性/疏水性分析(二)分析蛋白质的二级结构利用SOPMA在线工具对二级结构进行预测,如图2,α螺旋99个占19.30%,延伸链66个占12.87%,β-转角18个占3.51%,无规卷曲330个占64.33%,其二级结构主要由无规卷曲组成。
图2 ZNF395蛋白二级结构预测注:蓝色表示α螺旋;红色表示延伸链;紫色表示无规则卷曲(三)分析膜蛋白质利用在线分析工具TMHMM Server 2.0,对ZNF395氨基酸跨膜结构域进行在线预测和分析,结果表明,该序列编码的蛋白非跨膜蛋白(见图3)。
利用Signal P 3.0 Server在线预测工具对ZNF395蛋白质进行信号肽预测,无信号肽存在(图4)。
生物信息学作业(一)

生物信息学实验作业一1、了解NCBI、DDBJ、EMBL上网的方法自学各网站相关介绍。
答:(1)、NCBI: (National Center of Biotechnology Information,简称NCBI)美国国立生物技术信息中心。
其主页为:。
NCBI 是在NIH的国立医学图书馆(NLM)的一个分支。
NLM是因为它在创立和维护生物信息学数据库方面的经验被选择的,而且这可以建立一个内部的关于计算分子生物学的研究计划。
NCBI的任务是发展新的信息学技术来帮助对那些控制健康和疾病的基本分子和遗传过程的理解。
NCBI有一个多学科的研究小组包括计算机科学家,分子生物学家,数学家,生物化学家,实验物理学家,和结构生物学家,集中于计算分子生物学的基本的和应用的研究。
他们一起用数学和计算的方法研究在分子水平上的基本的生物医学问题。
这些问题包括基因的组织,序列的分析,和结构的预测。
在1992年10月,NCBI承担起对GenBank DNA序列数据库的责任。
NCBI 受过分子生物学高级训练的工作人员通过来自各个实验室递交的序列和同国际核酸序列数据库(EMBL和DDBJ)交换数据建立起数据库。
同美国专利和商标局的安排使得专利的序列信息也被整合。
BLAST是一个NCBI开发的序列相似搜索程序,还可作为鉴别基因和遗传特点的手段。
BLAST能够在小于15秒的时间内对整个DNA数据库执行序列搜索。
NCBI提供的附加的软件工具有:开放阅读框寻觅器(ORF Finder),电子PCR,和序列提交工具,Sequin和BankIt。
所有的NCBI数据库和软件工具可以从WWW 或FTP来获得。
NCBI还有E-mail服务器,提供用文本搜索或序列相似搜索访问数据库一种可选方法。
主要任务:(1)建立关于分子生物学,生物化学,和遗传学知识的存储和分析的自动系统(2)实行关于用于分析生物学重要分子和复合物的结构和功能的基于计算机的信息处理的,先进方法的研究(3)加速生物技术研究者和医药治疗人员对数据库和软件的使用。
生物信息学作业
乳腺癌易感基因BRCA1的研究班级:5061专业:药剂学姓名:孙建梅一、实验目的:(1)掌握中文文献全文的检索和获得方法。
(2)掌握Pubmed数据库文献的检索和交大图书馆英文数据库全文的获得方法。
(3)掌握核酸序列搜索的方法。
(4)掌握核酸序列相似性分析的方法。
(5)掌握PCR引物设计软件的原理、使用及特点。
(6)掌握蛋白质序列搜索的方法。
(7)掌握蛋白质序列分析常用软件的使用方法。
二、研究背景:乳腺癌易感基因(BRCA1)的突变率与35%~40%的家族性乳腺癌和卵巢癌有关。
该基因常以染色体显性方式遗传,并有很高的外显率。
外显率在乳腺癌为60%~80%,卵巢癌也可达15%~40%。
该基因作为一种抑癌基因, 不仅能抑制细胞生长, 还参与细胞周期调控、基因转录调节、DNA 损伤修复及其凋亡等重要细胞活动, 在维持基因稳定性中起重要作用。
BRCA1是目前所发现的最重要的乳腺癌易感基因之一,本人选择其为研究对象。
三、实验方法、步骤及结果:1.在中国知网(CNKI)中查找中文文献:2.在PubMed中查找英文文献:3 在Genbank中查找BRCA1基因及其序列:登陆NCBI主页,网址:/guide/,选择gene数据库4. 使用NCBI网站中的BLAST工具进行序列比对登陆/,选择核酸序列比对nucleotide BLAST,界面显示如下,输入登录号,NM-007294.3,点击“BLAST”。
结果如下:与其匹配的核苷酸序列和基因组序列如下:1, mRNA”,登录号:NM_007294.3。
variant 2, mRNA”,登录号:NM_007300.3。
5.蛋白质序列的比对检索页面:结果输出:6. 根据序列,设计PCR引物:(1)利用peimer3进行引物设计登陆引物设计软件primer3网址/primer3/。
输入FASTA格式的核苷酸序列,运算得到:上游引物:5’caccctctgctctgggtaaa 3’下游引物:5’aagctcattcttggggtcct 3’产物:5680bp。
生物信息学试题

生物信息学试题一、选择题1. 生物信息学主要研究的是:A. 生物实验技术B. 生物统计学C. 生物大数据分析与计算D. 生物体内生化反应2. 在生物信息学中,常用的序列比对工具是:A. BLASTB. PCRC. ELISAD. SDS-PAGE3. 下列哪个数据库主要用于存储核酸序列信息?A. PDBB. GenBankC. UniProtD. KEGG4. 以下哪种方法不是用于蛋白质结构预测的?A. 同源建模B. 折叠识别C. 从头预测D. 实验测定5. 生物信息学中的“基因家族”是指:A. 一组具有相似序列和功能的基因B. 一组来自同一物种的基因C. 一组通过基因复制产生的基因D. 一组控制同一生物过程的基因二、简答题1. 简述生物信息学在现代医学研究中的应用。
2. 描述PCR技术的原理及其在分子生物学中的重要性。
3. 解释什么是基因编辑技术,以及CRISPR-Cas9系统是如何工作的。
三、论述题1. 论述生物信息学在新药发现和开发中的作用。
2. 分析比较RNA测序技术与DNA测序技术的优势和局限性。
四、计算题1. 给定一个DNA序列:“ATGCGATACCTGAGCTG”,计算其碱基组成的比例。
2. 假设某种生物的基因组大小为200 Mb,每个碱基对的平均质量为650 Da,计算该基因组的大致质量。
五、案例分析题1. 根据给定的某种疾病的基因组数据,分析可能的致病基因,并讨论其可能的生物机制。
2. 通过分析某物种的转录组数据,探讨其在特定环境下的适应性变化。
请注意,以上试题仅供参考,具体题目应根据实际教学大纲和考试要求进行调整。
在实际考试中,题目可能会包含更多的细节和复杂性,要求考生具备扎实的生物信息学知识和分析能力。
生物信息学作业1.doc

生物信息学实验作业试验一一.找到编码拟南芥(arabidopsis)phyA(光敏色素A)基因的核酸序列编号, 并记录查找过程。
GI:224576211步骤1.进入NCBI主页2.搜索arabidopsis phyA3.Arabidopsis thaliana phytochrome A (PHYA) gene, partial cds4.VERSION:GI:224576211二.以phyA为检索词,在pubmed数据库中分别检索在题目和关键词字段中含有该检索词的文献,记录检索出的条目数目。
Results: 614三.仔细阅读所查询核酸序列在NCBI和EMBL数据库中格式的解释,理解各字段的含义,并比较NCBI 与EMBL中序列格式的异同。
实验二一.分析你感兴趣核酸序列的分子质量、碱基组成。
Composition 35 A; 25 C; 35 G; 15 T; 0 OTHERPercentage: 32% A; 23% C; 32% G; 14% T; 0%OTHERMolecular Weight (kDa): ssDNA: 34.26 dsDNA: 67.8二.列出你所分析核酸序列(或部分序列)的互补序列、反向序列、反向互补序列、DNA双链序列和RNA 序列。
R S1 ACTACTCGAG AAGCAGCGAC AGAGGCGTTA GCCCGCTCAG CAGACTGGCA GTTCTCTACC61 GACAAAAAAG AGGTAGGAGG CACAGTAATG ATACAGGCGT AGCAGGAGGGC S1 CCCTCCTGCT ACGCCTGTAT CATTACTGTG CCTCCTACCT CTTTTTTGTC GGTAGAGAAC61 TGCCAGTCTG CTGAGCGGGC TAACGCCTCT GTCGCTGCTT CTCGAGTAGTR C S1 TGATGAGCTC TTCGTCGCTG TCTCCGCAAT CGGGCGAGTC GTCTGACCGT CAAGAGATGG61 CTGTTTTTTC TCCATCCTCC GTGTCATTAC TATGTCCGCA TCGTCCTCCCD DNA S1 GGGAGGACGA TGCGGACATA GTAATGACAC GGAGGATGGA GAAAAAACAG CCATCTCTTGCCCTCCTGCT ACGCCTGTAT CATTACTGTG CCTCCTACCT CTTTTTTGTC GGTAGAGAAC61 ACGGTCAGAC GACTCGCCCG ATTGCGGAGA CAGCGACGAA GAGCTCATCATGCCAGTCTG CTGAGCGGGC TAACGCCTCT GTCGCTGCTT CTCGAGTAGTRNA S1 GGGAGGACGA UGCGGACAUA GUAAUGACAC GGAGGAUGGA GAAAAAACAG CCAUCUCUUG61 ACGGUCAGAC GACUCGCCCG AUUGCGGAGA CAGCGACGAA GAGCUCAUCA三.列出核酸序列的限制性酶切位点分析结果(酶及识别位点)。
生物信息学作业

结论一:这是什么基因1.该基因为人的CD226 抗原分子(CD226),染色体定位18号染色体67624232 -67530192基因标识符:NM_006566.22.功能:细胞粘附功能,整合素结合,蛋白结合,蛋白激酶结合;参与细胞粘合,细胞识别,细胞因子产生,正向调控Fc受体介导的刺激性信号通路,正向调控免疫球蛋白介导的免疫反应,正向调控肥大细胞的活化正向调控NK细胞介导的细胞毒性,正向调控NK细胞介导的针对肿瘤细胞靶标的细胞毒活性,调节免疫反应,信号转导等途径。
结论二:编码的蛋白质序列是怎样的蛋白标识符:"NP_006557.2" 336 aa蛋白序列为:MDYPTLLLAL LHVYRALCEE VLWHTSVPFA ENMSLECVYP SMGILTQVEWFKIGTQQDSI AIFSPTHGMV IRKPYAERVY FLNSTMASNN MTLFFRNASE DDVGYYSCSL YTYPQGTWQK VIQVVQSDSF EAAVPSNSHI VSEPGKNVTL TCQPQMTWPV QAVRWEKIQP RQIDLLTYCN LVHGRNFTSK FPRQIVSNCS HGRWSVIVIP DVTVSDSGLY RCYLQASAGE NETFVMRLTV AEGKTDNQYT LFVAGGTVLL LLFVISITTI IVIFLNRRRR RERRDLFTES WDTQKAPNNY RSPISTSQPT NQSMDDTRED IYVNYPTFSR RPKTRV结论三:有没有功能保守的结构序列?该蛋白有Ig的保守结构序列结论四;:它的功能是?功能:细胞黏附相关受体,淋巴细胞信号转导,CTL和NK介导的细胞毒性和淋巴因子分泌亚单元结构:与PVR和PVRL2相互作用亚细胞定位:细胞膜,Ⅰ类信号传播膜蛋白组织特异性:外周血T细胞表达序列:包含2个Ig-like C2型(免疫球蛋白样)结构域结论五:在真核生物中保守吗?在酵母中不存在其同源物,在一些灵长类动物存在一些同源性较高的序列,在其他的哺乳动物如:褐家鼠,野猪等中也存在一些同源性较高的序列。
生物信息学作业

生物信息学作业
一、Blast搜索
首先在NCBI的网页上打开Blast的网页,找到需要的数据库类型。
查询的序列直接粘贴到序列框中
1、可在该页面Algorithm parameters”栏目中更改相关的参数
2、点击BLAST以及Show results in a new window选择用新窗口展示分析结
果
3、点击“Formatting options”,在新网页选择变换格式
如:将其改变为Pairwise with dots for identities”格式
4、通过选择几个需要比较的序列,然后点击Distance tree of results”显示检索到的序列之间的同源关系
5、结果显示为:
6、保存:选择需要的序列,按Download保存
二、在记事本中可得到结果
比对法
一:ClustalW比对法
1、进入http://www.expasy.ch网页
2、在查找框中找到Find resources 以及ClustalW,得到页面
3、点击Clastw得
4、可在该页面上进行先关参数的设计,同时可在框中输入需要比对的序列,按下Run Clustalw可得比对结果
(由于网速问题只能进行到该阶段)
二、CLUSTAL X对比法
1、打开相应软件
将需要比对的序列从软件中导入
2、可对相关的参数进行设计:即按Alignment中Alignment Paramenter下的Multiple Alignment Paramenter即可进行
3、比对:按下Alignment中Do Complete Alignment即可得到比对结果
4、保存:。
生物信息学_复习题及答案(打印)(1)

生物信息学_复习题及答案(打印)(1)一、名词解释:1.生物信息学:研究大量生物数据复杂关系的学科,其特征是多学科交叉,以互联网为媒介,数据库为载体。
利用数学知识建立各种数学模型; 利用计算机为工具对实验所得大量生物学数据进行储存、检索、处理及分析,并以生物学知识对结果进行解释。
2.二级数据库:在一级数据库、实验数据和理论分析的基础上针对特定目标衍生而来,是对生物学知识和信息的进一步的整理。
3.FASTA序列格式:是将DNA或者蛋白质序列表示为一个带有一些标记的核苷酸或者氨基酸字符串,大于号(>)表示一个新文件的开始,其他无特殊要求。
4.genbank序列格式:是GenBank 数据库的基本信息单位,是最为广泛的生物信息学序列格式之一。
该文件格式按域划分为4个部分:第一部分包含整个记录的信息(描述符);第二部分包含注释;第三部分是引文区,提供了这个记录的科学依据;第四部分是核苷酸序列本身,以“//”结尾。
5.Entrez检索系统:是NCBI开发的核心检索系统,集成了NCBI 的各种数据库,具有链接的数据库多,使用方便,能够进行交叉索引等特点。
6.BLAST:基本局部比对搜索工具,用于相似性搜索的工具,对需要进行检索的序列与数据库中的每个序列做相似性比较。
P947.查询序列(query sequence):也称被检索序列,用来在数据库中检索并进行相似性比较的序列。
P988.打分矩阵(scoring matrix):在相似性检索中对序列两两比对的质量评估方法。
包括基于理论(如考虑核酸和氨基酸之间的类似性)和实际进化距离(如PAM)两类方法。
P299.空位(gap):在序列比对时,由于序列长度不同,需要插入一个或几个位点以取得最佳比对结果,这样在其中一序列上产生中断现象,这些中断的位点称为空位。
P2910.空位罚分:空位罚分是为了补偿插入和缺失对序列相似性的影响,序列中的空位的引入不代表真正的进化事件,所以要对其进行罚分,空位罚分的多少直接影响对比的结果。
生物信息学作业

生物信息学作业生物信息学试题1、构建分子系统树的主要方法有哪些?并简要说明构建分子进化树的一般步骤。
(20分)答:(1)构建进化树的方法包括两种:一类是序列类似性比较,主要是基于氨基酸相对突变率矩阵(常用PAM250)计算不同序列差异性积分作为它们的差异性量度(序列进化树);另一类在难以通过序列比较构建序列进化树的情况下,通过蛋白质结构比较包括刚体结构叠合和多结构特征比较等方法建立结构进化树(2)序列比对——选取所需序列——软件绘制具体如下:a测序获取序列或者在NCBI上搜索所需的目的序列b在NCBI上做blast:比对相似度较高的基因,并以fast格式下载,整合在*txt文档中。
c比对序列,比对序列转化成*meg格式d打开保存的*meg格式文件,构建系统进化树2、氨基酸序列打分矩阵PAM和BLOSUM中序号有什么意义?它们各自的规律是什么?(10分)(1)PAM矩阵:基于进化的点突变模型,如果两种氨基酸替换频繁,说明自然界接受这种替换,那么这对氨基酸替换得分就高。
一个PAM就是一个进化的变异单位, 即1%的氨基酸改变。
BLOSUM矩阵:首先寻找氨基酸模式,即有意义的一段氨基酸片断,分别比较相同的氨基酸模式之间氨基酸的保守性(某种氨基酸对另一种氨基酸的取代数据),然后,以所有60%保守性的氨基酸模式之间的比较数据为根据,产生BLOSUM60;以所有80%保守性的氨基酸模式之间的比较数据为根据,产生BLOSUM80。
(2)PAM用于家族内成员相比,然后把所有家族中对某种氨基酸的比较结果加和在一起,产生“取代”数据(PAM-1 );PAM-1自乘n次,得PAM-n。
PAM-n中,n 越小,表示氨基酸变异的可能性越小;相似的序列之间比较应该选用n值小的矩阵,不太相似的序列之间比较应该选用n 值大的矩阵。
PAM-250用于约 20%相同序列之间的比较。
BLOSUM-n中,n越小,表示氨基酸相似的可能性越小;相似的序列之间比较应该选用n 值大的矩阵,不太相似的序列之间比较应该选用n值小的矩阵。
生物信息学作业

B7家族成员生物信息学分析作业
T细胞最适活化除了需要TCR传导的第一信号外,还需共刺激分子传导的第二信号。
其中研究最多、最清楚、认为最有意义的共刺激分子是B7-1和B7-2分子,它们与受体CD28分子或CTLA-4分子相互作用,在T细胞生长、分化和死亡中起重要作用。
在寻找B7-1和B7-2的同源分子过程中,最近发现了B7家族的新成员B7H1(B7 homolog 1)、B7H2(B7 homolog 2)、B7H3(B7 homolog 3)和B7H4(B7 homolog 4)分子。
作业要求对B7家族的这几个成员进行了生物信息学分析(主要包括:人源、鼠源B7家族成员基因cDNA序列之间、氨基酸序列之间的同源性比较及发生树分析、蛋白质结构分析、三级结构预测等),并对扩增和检测B7H4基因进行了引物和探针设计。
B7家族人源、鼠源等自己从相关数据库获得的、相关生物信息学分析越多加分。
作业提供了B7家族人源、鼠源的相关序列文件。
数据获得、利用软件分析、思路等需要文字说明和截图说明,不详细的扣分。
作业在开学第一周交,即2月28日之前。
发到jiabinxj@。
《生物信息学》题集

《生物信息学》题集一、选择题(每题3分,共30分)1.生物信息学的主要研究对象是什么?A. 蛋白质结构B. 基因序列C. 生态系统D. 细胞代谢2.下列哪项技术不是生物信息学中常用的数据库技术?A. BLASTB. GenBankC. PubMedD. SWISS-PROT3.在生物信息学中,进行多序列比对时常用的软件是什么?A. MATLABB. ClustalWC. ExcelD. PowerPoint4.哪种算法常用于基因表达数据的聚类分析?A. K-meansB. DijkstraC. A*D. Floyd5.生物信息学中,下列哪项不是常用的序列分析技术?A. PCRB. 测序C. 质谱分析D. 芯片技术6.下列哪项不是生物信息学在医学领域的应用?A. 疾病诊断B. 药物设计C. 天气预报D. 个性化医疗7.下列哪项技术常用于生物大分子的结构预测?A. NMRB. X射线衍射C. 同源建模D. 质谱分析8.在生物信息学中,下列哪项不是基因注释的内容?A. 基因功能B. 基因表达水平C. 基因在染色体上的位置D. 基因的长度9.下列哪项技术不是高通量测序技术?A. Sanger测序B. Illumina测序C. 454测序D. SOLiD测序10.下列哪项不是生物信息学在农业领域的应用?A. 作物育种B. 病虫害防治C. 土壤成分分析D. 农产品品质改良二、填空题(每题2分,共20分)1.生物信息学是一门交叉学科,它主要涉及______、计算机科学和数学等领域。
2.在生物信息学中,______技术常用于基因序列的相似性搜索。
3.生物信息学在药物研发中的主要应用包括______和药物靶点的预测。
4.在基因表达数据分析中,______是一种常用的数据标准化方法。
5.生物信息学中,______技术常用于蛋白质结构的预测和分析。
6.在生物信息学数据库中,GenBank主要存储的是______数据。
生物信息学课后题及答案

生物信息学课后习题及答案(由10级生技一、二班课代表整理)一、绪论1.你认为,什么是生物信息学?采用信息科学技术,借助数学、生物学的理论、方法,对各种生物信息(包括核酸、蛋白质等)的收集、加工、储存、分析、解释的一门学科。
2.你认为生物信息学有什么用?对你的生活、研究有影响吗?(1)主要用于:在基因组分析方面:生物序列相似性比较及其数据库搜索、基因预测、基因组进化和分子进化、蛋白质结构预测等在医药方面:新药物设计、基因芯片疾病快速诊断、流行病学研究:SARS、人类基因组计划、基因组计划:基因芯片。
(2)指导研究和实验方案,减少操作性实验的量;验证实验结果;为实验结果提供更多的支持数据等材料。
3.人类基因组计划与生物信息学有什么关系?人类基因组计划的实施,促进了测序技术的迅猛发展,从而使实验数据和可利用信息急剧增加,信息的管理和分析成为基因组计划的一项重要的工作。
而这些数据信息的管理、分析、解释和使用促使了生物信息学的产生和迅速发展。
4简述人类基因组研究计划的历程。
通过国际合作,用15年时间(1990-2005)至少投入30亿美元,构建详细的人类基因组遗传图和物理图,确定人类DNA的全部核苷酸序列,定位约10万基因,并对其他生物进行类似研究。
1990,人类基因组计划正式启动。
1996,完成人类基因组计划的遗传作图,启动模式生物基因组计划。
1998完成人类基因组计划的物理作图,开始人类基因组的大规模测序。
Celera公司加入,与公共领域竞争启动水稻基因组计划。
1999,第五届国际公共领域人类基因组测序会议,加快测序速度。
2000,Celera公司宣布完成果蝇基因组测序,国际公共领域宣布完成第一个植物基因组——拟南芥全基因组的测序工作。
2001,人类基因组“中国卷”的绘制工作宣告完成。
2003,中、美、日、德、法、英等6国科学家宣布人类基因组序列图绘制成功,人类基因组计划的.目标全部实现。
2004,人类基因组完成图公布。
生物信息学作业题

生物信息学作业题生物信息学作业题绪论1.什么是生物信息学?2.生物信息学有哪些主要研究领域?第一章生物信息学的分子生物学基础1.DNA的双螺旋结构要点是什么?2.什么是基因组和蛋白质组?对它们的研究有何意义?第二章生物信息学的计算机基础1.简述网络操作系统的类型。
第三章核酸序列分析1.什么是全局比对?2.什么是局部比对?有哪些优点?第四章分子进化分析1.分子进化分析具有哪些优点?2. 简述分子进化的中性学说。
第五章基因组分析1. 什么是基因组学?其主要研究内容是什么?2.简述基因预测分析的一般步骤。
第六章蛋白质组分析1. 蛋白质组学的概念和主要研究的大致方向是什么?2. 蛋白质组功能预测的程序是怎样的?第七章生物芯片数据分析1. 什么是生物芯片?2. 生物芯片有哪些方面的应用?第八章核酸与蛋白质结构预测1. RNA二级结构典型的预测方法有哪些?2. 基于统计学的预测蛋白质二级结构的方法有哪些?第九章生物信息学平台与工具软件1. 请利用Clustal X软件对下列6条蛋白质序列进行多重比对(比对结果用BioEdit软件打开,用“截图”方式显示比对结果)。
>1mqngkvkwfn sekgfgfiev eggedvfvhf saiqgegfkt leegqevtfe veqgnrgpqatnvnkk>2mqgkvkwfnn ekgfgfieie gaddvfvhfs aiqgegykal eegqevsfdi tegnrgpqaanvvkl>3mqngkvkwfn sekgfgfiev eggedvfvhf saiqgegfkt leegqevtfe veqgnrgpqatnvnkk>4mqgkvkwfnn ekgfgfieie gaddvfvhfs aiqgegykal eegqevsfdi tegnrgpqaanvvkl>5mqngkvkwfn sekgfgfiev eggedvfvhf saiqgegfkt leegqevtfe veqgnrgpqatnvnkk>6mqgkvkwfnn ekgfgfieie gaddvfvhfs aiqgegykal eegqevsfdi tegnrgpqaanvvkl2. 现有一ZmPti1b蛋白质序列,请用DNAMAN软件分析其二级结构,给出分析结果。
《生物信息学》大作业参考模板-2016

《生物信息学》大作业 参考答案
芍药 ACS 基因的生物信息学分析
姓名: 班级: 学号: 2016 年 4 月 11 日
一、芍药 ACS 基因序列及其编码的蛋白的功能 乙烯是存在于植物体内的唯一的一种气态植物激素,调控植物花、果和叶片的衰老进程。乙烯的合成 主要在转录水平上受到 ACS(ACC synthase,ACC 合酶)和 ACC 氧化酶的调控,ACS 将 SAM(S-adenosyl
a r t i c l e i n f o
Article history: Received 17 November 2015 Accepted 23 November 2015 Available online 26 November 2015 Keywords: ACC synthase Ethylene biosynthesis Flower senescence Oncidium Gower Ramsey Gene cloning Expression analysis
Biochemical and Biophysical Research Communications 469 (2016) 20vailable at ScienceDirect
Biochemical and Biophysical Research Communications
图 1 芍药 ACS 基因的核苷酸序列及其编码的氨基酸序列 下载的论文“Molecular cloning and expression analysis of an 1-aminocyclopropane-1-carboxylate synthase gene from Oncidium Gower Ramsey” 为 2016 年发表于 Biochemical and Biophysical Research Communications 的最新英文文章(见下页) 。 (2 分)
生物信息学作业1---开放读码框与预测序列启动子(何婷,学号;1302008)

何婷学号:1302008 专业:病理学与病理生理学
1.使用Entrez信息查询系统检索与自己课题相关的基因核酸序列,预测开放读码框,并使用PromoterScan预测该序列中的启动子。
①查询HPV的核酸序列:输入网址:/,打开NCBI主页,在
检索窗口的选择数据库的下拉菜单选中Nucleotide项,在它右侧的文本输入栏输入检索词“HPV”,再点击“Search”按钮。
如下图所示:
搜索结果,如下图所示:
显示结果,如下图所示:
② 预测开放读码框:输入网址:/gorf/gorf.html ,打开NCBI
的ORF Finder 软件,输入HPV 的核酸序列的GI 号,最后点击“OrfFind ”按钮,如下图所示:
结果如下图所示:
点击“正链+1”,显示结果如下:
③使用PromoterScan预测该序列中的启动子:输入网址为
/molbio/proscan/,打开PromoterScan的在线操作页面,复制粘贴上述的HPV的核酸序列到指定的框中,点击submit按钮提交序列后,注意使用
软件时不需要设置任何参数,如下图所示:
输出结果为:
何婷学号:1302008 专业:病理学与病理生理学。
《生物信息学》上机作业

《生物信息学》上机作业题目:对人血红蛋白(HBA1)编码基因序列的生物信息分析目录引言 .............................................................................................................................................. - 1 -1 正文......................................................................................................................................... - 2 -1.1 NCBI上对相关核苷酸序列的查找............................................................................ - 2 -1.2 BLAST运行及其结果.................................................................................................. - 2 -1.3 BLASTX运行及其结果................................................................................................ - 6 -2 其他软件的运行及其结果..................................................................................................... - 8 -2.1 Clustal W运行及其结果 ............................................................................................. - 9 -2.2 MEGA4.0运行及其结果............................................................................................. - 10 -结论 ............................................................................................................................................ - 10 -引言血红蛋白又称血色素,是红细胞的主要组成部分,能与氧结合,运输氧和二氧化碳。
生物信息学试题及答案

生物信息学试题及答案一、单项选择题(每题2分,共20分)1. 生物信息学的主要研究对象是()。
A. 生物数据B. 生物实验C. 生物模型D. 生物技术答案:A2. 下列哪项不是生物信息学中的常用数据库()。
A. GenBankB. Swiss-ProtC. PubMedD. Google Scholar答案:D3. 蛋白质序列比对的主要目的是()。
A. 确定蛋白质的三维结构B. 预测蛋白质的功能C. 比较蛋白质的氨基酸序列D. 计算蛋白质的分子量答案:B4. 在生物信息学中,以下哪种算法不是用于序列比对的()。
A. BLASTB. FASTAC. Smith-WatermanD. Hidden Markov Model答案:D5. 下列哪种生物信息学工具主要用于基因表达分析()。
A. ClustalWB. Primer3C. R语言D. PDB答案:C6. 以下哪种技术不是用于蛋白质结构预测的()。
A. 同源建模B. 从头预测C. 序列比对D. 折叠识别答案:C7. 以下哪种生物信息学工具主要用于基因组注释()。
A. BLASTC. GATKD. Primer3答案:B8. 在生物信息学中,以下哪种方法不用于基因表达数据的聚类分析()。
A. K-meansB. Hierarchical clusteringC. Principal component analysisD. Multiple sequence alignment答案:D9. 下列哪种生物信息学工具主要用于蛋白质-蛋白质相互作用网络分析()。
A. STRINGB. BLASTD. Primer3答案:A10. 在生物信息学中,以下哪种数据库不包含蛋白质结构信息()。
A. PDBB. UniProtC. RCSBD. GenBank答案:D二、多项选择题(每题3分,共15分)11. 生物信息学中常用的序列比对工具包括()。
A. BLASTB. FASTAC. ClustalWD. Pfam答案:ABC12. 以下哪些是生物信息学中常用的基因表达分析软件()。
《生物信息学》练习题及答案

《生物信息学》练习题及答案1、在Genbank中查找以下6个植物蛋白序列:protein1:NP_974673.2;protein2:NP_187969.1;protein3: NP_190855.1;protein4:NP_565618.1;protein5: NP_200511.1;protein6:NP_191407.1(以FASTA格式)。
(1)用EBI上的ClustalW2工具对其进行多序列比对,分析各蛋白序列之间的同源性。
序列比对结果比对结果表明:protein1:NP_974673.2和protein4: NP_565618.1的亲缘关系最近。
(2)利用Phylip软件,选择距离法构建其进化树(要求写出具体的建树步骤)。
1.将蛋白序列保存为FASTA格式,存于txt文档;2.用Clustalx打开txt文本,保存为*.phy文件;3.用seqboot程序打开phy文件,输出结果文件*_seqboot4.用protdist程序打开*_seqboot文件,输出为*_protdist文件5.用neighbor程序打开*_protdist文件,输出为*_neighbor 文件6.用consense程序打开*_neighbor文件,输出为*_consense 文件7.用dratree程序打开*_consense文件得到进化树。
(注:由于seqboot软见无法正常运行,因此进化树无法显示)(3)任意选取其中的一个蛋白进行蛋白质一级序列分析、二级结构预测及三维结构的模拟。
选择protein3:NP_190855.1一级结构网址:/doc/479b86d06edb6f1afe001f6e.html /tools/protparam.htmlNumber of amino acids:456氨基酸数目Molecular weight:51154.5相对分子质量Theoretical pI:8.69理论pI值Amino acid composition氨基酸组成Ala(A)306.6%Arg(R)286.1%Asn(N)153.3%Asp(D)275.9%Cys(C)51.1%Gln(Q)183.9%Glu(E)286.1%Gly(G)378.1%His(H)163.5%Ile(I)163.5%Leu(L)429.2%Lys(K)327.0%Met(M)51.1%Phe(F)173.7%Pro(P)163.5%Ser(S)4610.1%Thr(T)214.6%Trp(W)81.8%Tyr(Y)194.2%Val(V)306.6%Pyl(O)00.0%Sec(U)00.0%(B)00.0%(Z)00.0%(X)00.0%正/负电荷残基数Total number of negatively charged residues(Asp+Glu): 55Total number of positively charged residues(Arg+Lys): 60Atomic composition:原子组成Carbon C2270Hydrogen H3531Nitrogen N645Oxygen O686Sulfur S10Formula:C2270H3531N645O686S10分子式Total number of atoms:7142总原子数Extinction coefficients:消光系数Extinction coefficients are in units of M-1cm-1,at280 nm measured in water.Ext.coefficient72560Abs0.1%(=1g/l)1.418,assuming all pairs of Cys residues form cystines Ext.coefficient72310Abs0.1%(=1g/l) 1.414,assuming all Cys residues are reducedEstimated half-life:半衰期The N-terminal of the sequence considered is M(Met). The estimated half-life is:30hours(mammalian reticulocytes,in vitro).>20hours(yeast,in vivo).>10hours(Escherichia coli,in vivo).Instability index:不稳定系数The instability index(II)is computed to be48.99This classifies the protein as unstable.Aliphatic index:75.26脂肪系数Grand average of hydropathicity(GRAVY):-0.554总平均亲水性蛋白质亲疏水性分析所用氨基酸标度信息Ala:1.800Arg:-4.500Asn:-3.500Asp:-3.500Cys:2.500 Gln:-3.500Glu:-3.500Gly:-0.400His:-3.200Ile:4.500 Leu:3.800Lys:-3.900Met:1.900Phe:2.800Pro:-1.600 Ser:-0.800Thr:-0.700Trp:-0.900Tyr:-1.300Val: 4.200:-3.500:-3.500:-0.490分析所用参数信息Weights for window positions1,..,9,using linear weight variation model:1234567891.001.001.001.001.001.001.001.001.00edge center edge跨膜结构预测结果(没有跨膜结构)信号肽分析:二级结构预测三级结构预测网站/doc/479b86d06edb6f1afe001f6e.html/~phyre2、在拟南芥基因组数据库中(/doc/479b86d06edb6f1afe001f6e.ht ml/)查找编号分别为At4G33050,At3G13600,At3G52870或At2G26190基因,针对所查找的基因进行初步的生物信息学分析(每人任选其中一个基因)。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
生物信息学实验作业一1、了解NCBI、DDBJ、EMBL上网的方法自学各网站相关介绍。
答:(1)、NCBI: (National Center of Biotechnology Information,简称NCBI)美国国立生物技术信息中心。
其主页为:。
NCBI 是在NIH的国立医学图书馆(NLM)的一个分支。
NLM是因为它在创立和维护生物信息学数据库方面的经验被选择的,而且这可以建立一个内部的关于计算分子生物学的研究计划。
NCBI的任务是发展新的信息学技术来帮助对那些控制健康和疾病的基本分子和遗传过程的理解。
NCBI有一个多学科的研究小组包括计算机科学家,分子生物学家,数学家,生物化学家,实验物理学家,和结构生物学家,集中于计算分子生物学的基本的和应用的研究。
他们一起用数学和计算的方法研究在分子水平上的基本的生物医学问题。
这些问题包括基因的组织,序列的分析,和结构的预测。
在1992年10月,NCBI承担起对GenBank DNA序列数据库的责任。
NCBI 受过分子生物学高级训练的工作人员通过来自各个实验室递交的序列和同国际核酸序列数据库(EMBL和DDBJ)交换数据建立起数据库。
同美国专利和商标局的安排使得专利的序列信息也被整合。
BLAST是一个NCBI开发的序列相似搜索程序,还可作为鉴别基因和遗传特点的手段。
BLAST能够在小于15秒的时间内对整个DNA数据库执行序列搜索。
NCBI提供的附加的软件工具有:开放阅读框寻觅器(ORF Finder),电子PCR,和序列提交工具,Sequin和BankIt。
所有的NCBI数据库和软件工具可以从WWW 或FTP来获得。
NCBI还有E-mail服务器,提供用文本搜索或序列相似搜索访问数据库一种可选方法。
主要任务:(1)建立关于分子生物学,生物化学,和遗传学知识的存储和分析的自动系统(2)实行关于用于分析生物学重要分子和复合物的结构和功能的基于计算机的信息处理的,先进方法的研究(3)加速生物技术研究者和医药治疗人员对数据库和软件的使用。
(4)全世界范围内的生物技术信息收集的合作努力。
教育和训练:NCBI通过赞助会议,研讨会,和系列演讲来培养在应用于分子生物学和遗传学的计算机领域的科学交流。
一个科学访问学者项目已经成立,来培养同外部科学家的合作。
作为NIH内部的部分研究项目,也提供博士后工作位置。
(2)、DDBJ: (DNA Data Bank of Japan,简称DDBJ)日本DNA数据库。
其主页www.ddbj.nig.ac.jp/。
其于1984年建立,是世界三大DNA 数据库之一。
DDBJ 开发了SQmateh工具,用来搜索基因或蛋白质中短的碱基或氨基酸序列区域,并建立了简便且易操作的SOAP(simple object aeeess protoco1)服务器。
它的数据主要通过Sakura和MST工具来完成。
与NCBI,EBM共同构成DNA三大数据库,这三大数据中心各自收集序列数据,并通过网络每天进行数据交换。
近来三大数据库合作的项目主要包括TPA(tird pannotation)、CON(struct)或CON(tig)和XML数据交换格式的建立。
TPA是一种基于已有数据库中的核酸序列产生的注释数据,它的格式与传统的GenBank一样,只是包含了“TPA”标签。
CON(struct)或CON(tig)用于存储一些片段的拼接信息,这些片段是序列长度大于350 000 bp的核酸被分割而产生的,但这种长度限制在2004年6月就被取消。
DDBJ主要向研究者收集DNA序列信息并赋予其数据存取号,信息来源主要是日本的研究机构,亦接受其他国家呈递的序列,数据库通过WWW环球网,匿名FTP,e-mail或Gopher方式为广大研究人员服务。
(3)、EMBL: (The European Molecular Biology Laboratory,简称EMBL)欧洲分子生物学实验室。
其主页为: 。
EMBL于1974年由欧洲14个国家加上亚洲的以色列共同发起建立,现在由欧洲30个成员国政府支持组成,目的在于促进欧洲国家之间的合作来发展分子生物学的基础研究和改进仪器设备、教育工作等。
分7个部分:结构、分化、物理仪器、生化仪器、生物仪器、计算机和应用数学。
包括一个位于德国Heidelberg的核心实验室,及三个位于德国Hamburg,法国Grenoble及英国Hinxton的研究分部。
EMBL的研究主要集中在以下几个方面:1. 生化实验技术质谱分析(Mass Spectrometry)等。
2.细胞生物学(Cell Biology),研究细胞膜上蛋白和脂肪的分布,包括膜运输、微管网络、细胞核及细胞周期,焦点是Rab蛋白。
3.细胞生物物理(Cell Biophysics),重点是理论创新和实际应用的研究,尤其是光学显微镜的完善使用。
4.分化(Differentiation),集中研究果蝇的早期发育。
5.基因表达(Gene Expression),研究基因到蛋白质信息传递的过程,尤其是核糖体合成在整个细胞生命过程中的重要作用。
6.结构生物学(Structure Biology),在过去9年中建立了cDNA测序技术、生物计算、蛋白工程、晶体学、电子显微镜(EM)及核磁共振(VMR),研究肌肉巨型蛋白分子Titin。
7.Grenoble研究分部,主要研究蛋白质合成过程,尤其揭示了G-蛋白-鸟苷酸交换因子偶联物的结构。
8.Hamburg研究分部,有关长期的分子生物学国际合作研究历史,着重于结构生物学研究,如光学测量系统、晶体学、X-线吸收光谱及小角散射。
9.Hinxton研究分部EBI(European Bioinformatics Institute,欧洲生物信息学研究所),重点是与世界上其他分子生物学数据库进行合作研究,最主要的有EMBL核酸序列数据库,于1980年开始建立,随后参予了与日内瓦大学共同进行的SWISS-PROT的建设。
在SWISS-PROT与EMBL核苷酸序列库之间的数据转移的基础上,产生了新的数据库TREMBL(Translation from EMBL),即使核苷酸序列库的核苷酸序列自动翻译成SWISS-PROT蛋白序列库中的蛋白序列。
10.放射性杂交数据库(Radiation Hybrid Database)。
11.Monterotondo研究中心组,EMBL和欧洲其他研究组一起,加入到哺乳类生物学和生物医学的研究行列,中心位于意大利罗马北部的Monterotondo。
EMBL 着重于鼠遗传学研究。
2、了解北大生物信息学中心等几大中文生物信息学网站。
答:北大生物信息中心,简称CBI。
CBI的主页为北大生物信息中心成立于1997年,是欧洲分子生物学网络组织EMBnet的中国国家节点。
几年来,已经与多个国家的生物信息中心建立了合作关系,其中包括:欧洲生物信息学研究所(EBI)、国际蛋白质数据库和分析中心(ExPASy)、国际遗传工程和生物技术研究所、德国生物工程研究所、英国基因组资源中心、英国基因组研究中心(Sanger Centre)、荷兰生物信息中心、澳大利亚基因组信息中心、新加坡生物信息中心等等。
目前是国内数据库种类最多,数据量最大的生物信息站点,为国内外用户提供了多项生物信息服务。
上海生物信息技术研究中心:成立于2002年8月,上海市科学技术委员会依托中国科学院上海生命科学研究院、国家人类基因组南方研究中心、复旦大学、上海交通大学、上海第二医科大学、上海医药工业研究院和中国科学院上海有机化学研究所等单位,整合上海生物信息学主要研究力量,投入1140万元资金,正式组建了上海生物信息技术研究中心。
上海生物信息技术研究中心是国内第一个以推动我国生物信息学数据共享为目的,完全从事生命科学数据库建设、生物信息学软件开发的地方政府支持的、自收自支的独立事业法人单位。
上海生物信息技术研究中心旨在开展和促进生物信息技术领域的原始性创新研究,建立具有广泛应用前景和国际先进水平的生物信息分析、数据挖掘和知识发现的技术体系,促进上海乃至全国生命科学、生物技术和生物医药产业的发展。
3、了解一些生物论坛中有关生物信息学的部分。
如:Biooo和Bioon。
答:生物谷:生物谷创建于2001年,生物谷是目前国内最大的生物医药类门户网站。
生物谷开创了生物行业新闻发布标准,开创了生物专业信息的多级分类的先河,生物医药类详细的信息分类几乎全部起源于生物谷,后被大量网站效仿,一举成为行业分类的标准。
生物谷信息内容注重丰富性、科学性、专业性和权威性,及时、全面、快速的把生物医药最新资讯与动态整理并发布于生物谷上,内容包括基础生物学、生物技术产业、生物医药产业、趋势、人物与企业等核心版块,每一版块又有针对当前热点领域进行细分栏目,这一分类模式以及生命科学领域新闻信息的发布标准,均被国内同行广泛采用和接受。
同时,通过综合服务发展模式,面向终端客户提供完善的服务体系。
中国生物技术信息网:生物通:生物信息学论坛:4、利用NCBI的查询系统检索1-3条核酸或蛋白质序列(自选),并对照所学复习各字段的含义。
答:Mouse的蛋白质序列分析:LOCUS 3UNH_B 261 aa linear ROD 28-MAR-2012DEFINITION Chain B, Mouse 20s Immunoproteasome.ACCESSION 3UNH_BVERSION 3UNH_B GI:378792444DBSOURCE pdb: molecule 3UNH, chain 66, release Mar 28, 2012;deposition: Nov 15, 2011;class: Hydrolase;source: Mol_id: 1; Organism_scientific: Mus Musculus;Organism_common: Mouse; Organism_taxid: 10090; Mol_id: 2;Organism_scientific: Mus Musculus; Organism_common: Mouse; Organism_taxid: 10090; Mol_id: 3; Organism_scientific: MusMusculus; Organism_common: Mouse; Organism_taxid: 10090; Mol_id: 4;Organism_scientific: Mus Musculus; Organism_common: Mouse; Organism_taxid: 10090; Mol_id: 5; Organism_scientific: MusMusculus; Organism_common: Mouse; Organism_taxid: 10090; Mol_id: 6;Organism_scientific: Mus Musculus; Organism_common: Mouse; Organism_taxid: 10090; Mol_id: 7; Organism_scientific: MusMusculus; Organism_common: Mouse; Organism_taxid: 10090; Mol_id: 8;Organism_scientific: Mus Musculus; Organism_common: Mouse; Organism_taxid: 10090; Mol_id: 9; Organism_scientific: MusMusculus; Organism_common: Mouse; Organism_taxid: 10090; Mol_id:10; Organism_scientific: Mus Musculus; Organism_common: Mouse;Organism_taxid: 10090; Mol_id: 11; Organism_scientific: MusMusculus; Organism_common: Mouse; Organism_taxid: 10090; Mol_id:12; Organism_scientific: Mus Musculus; Organism_common: Mouse;Organism_taxid: 10090; Mol_id: 13; Organism_scientific: MusMusculus; Organism_common: Mouse; Organism_taxid: 10090; Mol_id:14; Organism_scientific: Mus Musculus; Organism_common: Mouse;Organism_taxid: 10090;Exp. method: X-Ray Diffraction.KEYWORDS .SOURCE Mus musculus (house mouse)ORGANISM Mus musculusEukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi;Mammalia; Eutheria; Euarchontoglires; Glires; Rodentia;Sciurognathi; Muroidea; Muridae; Murinae; Mus; Mus.REFERENCE 1 (residues 1 to 261)AUTHORS Huber,E.M., Basler,M., Schwab,R., Heinemeyer,W., Kirk,C.J.,Groettrup,M. and Groll,M.TITLE Immuno- and constitutive proteasome crystal structures revealdifferences in substrate and inhibitor specificityJOURNAL Cell 148 (4), 727-738 (2012)PUBMED 22341445REFERENCE 2 (residues 1 to 261)AUTHORS Huber,E., Basler,M., Schwab,R., Heinemeyer,W., Kirk,C.,Groettrup,M. and M,M.TITLE Direct SubmissionJOURNAL Submitted (15-NOV-2011)COMMENT 2 Proteasome Subunit Alpha Type-4.FEATURES Location/Qualifierssource 1..261/organism="Mus musculus"/db_xref="taxon:10090"Region 1..237/region_name="PTZ00246"/note="proteasome subunit alpha; Provisional"/db_xref="CDD:173491"Region 3..216/region_name="proteasome_alpha_type_4"/note="proteasome_alpha_type_4. The 20S proteasome,multisubunit proteolytic complex, is the central enzyme ofnonlysosomal protein degradation in both the cytosol andnucleus. It is composed of 28 subunits arranged as fourhomoheptameric rings that stack on...; cd03752"/db_xref="CDD:48450"Siteorder(7..10,12..13,15..16,20,23,26..27,30,38,53,55..56,80..82,84..85,116,119,122..123,126..130,148,153..154,156,158..159,161)/site_type="other"/note="alpha subunit interaction site [polypeptidebinding]"/db_xref="CDD:48450"SecStr 19..30/sec_str_type="helix"/note="helix 7"Site order(32,48,50,64,166)/site_type="active"/db_xref="CDD:48450"SecStr 32..40/note="strand 11"SecStr 41..49/sec_str_type="sheet" /note="strand 12"SecStr 64..68/sec_str_type="sheet" /note="strand 13"SecStr 71..79/sec_str_type="sheet" /note="strand 14"SecStr 80..100/sec_str_type="helix" /note="helix 8"SecStr 107..123/sec_str_type="helix" /note="helix 9"SecStr 131..141/sec_str_type="sheet" /note="strand 15"SecStr 142..145/sec_str_type="sheet" /note="strand 16"SecStr 146..152/sec_str_type="sheet" /note="strand 17"SecStr 154..159/note="strand 18"SecStr 160..166/sec_str_type="sheet"/note="strand 19"SecStr 169..178/sec_str_type="helix"/note="helix 10"SecStr 186..200/sec_str_type="helix"/note="helix 11"SecStr 210..220/sec_str_type="sheet"/note="strand 20"SecStr 221..228/sec_str_type="sheet"/note="strand 21"SecStr 230..246/sec_str_type="helix"/note="helix 12"ORIGIN1 msrrydsrtt ifspegrlyq veyameaigh agtclgilan dgvllaaerr nihklldevf61 fsekiyklne dmacsvagit sdanvltnel rliaqryllq yqepipceql vtalcdikqa121 ytqfggkrpf gvsllyigwd khygfqlyqs dpsgnyggwk atcignnsaa avsmlkqdyk181 egemtlksal alavkvlnkt mdvsklsaek veiatltres gktvirvlkq keveqlikkh241 eeeeakaere kkekeqrekd kMouse的核苷酸序列分析:LOCUS JN596232 679 bp DNA linear MAM 02-MAY-2012DEFINITION Rhinopoma hardwickii taste receptor type 1 member 1 (Tas1r1)pseudogene, partial sequence.ACCESSION JN596232VERSION JN596232.1 GI:358680483KEYWORDS .SOURCE Rhinopoma hardwickii (Lesser mouse-tailed bat) ORGANISM Rhinopoma hardwickiiEukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi;Mammalia; Eutheria; Laurasiatheria; Chiroptera; Microchiroptera;Rhinopomatidae; Rhinopoma.REFERENCE 1 (bases 1 to 679)AUTHORS Zhao,H., Xu,D., Zhang,S. and Zhang,J.TITLE Genomic and genetic evidence for the loss of umami taste in batsJOURNAL Genome Biol Evol 4 (1), 73-79 (2012)PUBMED 22117084REFERENCE 2 (bases 1 to 679)AUTHORS Zhao,H.TITLE Direct SubmissionJOURNAL Submitted (18-AUG-2011) Ecology andEvolutionary Biology,University of Michigan, 830 North University, Ann Arbor, mi 48109,USAFEATURES Location/Qualifierssource 1..679/organism="Rhinopoma hardwickii"/mol_type="genomic DNA"/specimen_voucher="RH-112"/db_xref="taxon:124756"/country="United Kingdom"/collection_date="10-Apr-2007"/collected_by="Huabin Zhao"gene <1..>679/gene="Tas1r1"/note="taste receptor type 1 member 1; umami tastereceptor"/pseudoORIGIN1 acccctgtgg tgaggtcggc tgggggcagg ctttgcttcc tcatgctggt ctcccaggca61 gtgggcagct gcagcctcta tggctttttt gggaaaccca cgctgcccat gtgcttgctg121 tgccaaggcc tcttggccct cggttttgtt atcttcctgt cctacctgac aatccactcc181 tcccaactgg tcttcatctt caagttttct gccaaggtat ccaccttcta ccatgcctgg241 gtccaaaaac acggggctag cctctttgta gggatcagct cagtggccca gctatttatc301 tgtctaactt ggcttgcggt gtggacccca atgcccatta gagaatacca tccctttcct361 cagctggtgg cgcttgactg cacagaggct aactcactgg gcctcacgct gccttttgcc421 tacaaacgtc tcctctccgt cagcgcctct gcctgcaggt acgtggacaa ggacctgcca481 gagaactaca aggccttatg tgtcaccttc aacctgctcc tcaactttgt gtcctgggtc541 gccttcttca tcactgccgg ctgccaacgt gttggccatg ctgagcagcc ttagtggctg601 cttcagcggt tttttcctcc ccaagtgcta tgtgatccgg tacggctcag atctcaacag661 cacggagcac ttccgggcc。