生物信息学作业1.doc
生物信息学作业(一)

生物信息学实验作业一1、了解NCBI、DDBJ、EMBL上网的方法自学各网站相关介绍。
答:(1)、NCBI: (National Center of Biotechnology Information,简称NCBI)美国国立生物技术信息中心。
其主页为:。
NCBI 是在NIH的国立医学图书馆(NLM)的一个分支。
NLM是因为它在创立和维护生物信息学数据库方面的经验被选择的,而且这可以建立一个内部的关于计算分子生物学的研究计划。
NCBI的任务是发展新的信息学技术来帮助对那些控制健康和疾病的基本分子和遗传过程的理解。
NCBI有一个多学科的研究小组包括计算机科学家,分子生物学家,数学家,生物化学家,实验物理学家,和结构生物学家,集中于计算分子生物学的基本的和应用的研究。
他们一起用数学和计算的方法研究在分子水平上的基本的生物医学问题。
这些问题包括基因的组织,序列的分析,和结构的预测。
在1992年10月,NCBI承担起对GenBank DNA序列数据库的责任。
NCBI 受过分子生物学高级训练的工作人员通过来自各个实验室递交的序列和同国际核酸序列数据库(EMBL和DDBJ)交换数据建立起数据库。
同美国专利和商标局的安排使得专利的序列信息也被整合。
BLAST是一个NCBI开发的序列相似搜索程序,还可作为鉴别基因和遗传特点的手段。
BLAST能够在小于15秒的时间内对整个DNA数据库执行序列搜索。
NCBI提供的附加的软件工具有:开放阅读框寻觅器(ORF Finder),电子PCR,和序列提交工具,Sequin和BankIt。
所有的NCBI数据库和软件工具可以从WWW 或FTP来获得。
NCBI还有E-mail服务器,提供用文本搜索或序列相似搜索访问数据库一种可选方法。
主要任务:(1)建立关于分子生物学,生物化学,和遗传学知识的存储和分析的自动系统(2)实行关于用于分析生物学重要分子和复合物的结构和功能的基于计算机的信息处理的,先进方法的研究(3)加速生物技术研究者和医药治疗人员对数据库和软件的使用。
生物信息学作业

生物信息学试题
1、构建分子系统树的主要方法有哪些?并简要说明构建分子进化树
的一般步骤。
(20分)
答:(1)构建进化树的方法包括两种:一类是序列类似性比较,主
要是基于氨基酸相对突变率矩阵(常用PAM250)计算不同序列差异性积分作为它们的差异性量度(序列进化树);另一类在难以通过序
列比较构建序列进化树的情况下,通过蛋白质结构比较包括刚体结构
叠合和多结构特征比较等方法建立结构进化树
(2)序列比对——选取所需序列——软件绘制
具体如下:
a测序获取序列或者在NCBI上搜索所需的目的序列
b在NCBI上做blast:比对相似度较高的基因,并以fast格式下载,整合在*txt文档中。
c比对序列,比对序列转化成*meg格式
d打开保存的*meg格式文件,构建系统进化树
2、氨基酸序列打分矩阵PAM和BLOSUM中序号有什么意义?它们各自
的规律是什么?(10分)
(1)PAM矩阵:基于进化的点突变模型,如果两种氨基酸替换频繁,说明
自然界接受这种替换,那么这对氨基酸替换得分就高。
一个PAM就是一个进化的变异单位, 即1%的氨基酸改变。
BLOSUM矩阵:首先寻找氨基酸模式,即有意义的一段氨基酸片断,分别比
较相同的氨基酸模式之间氨基酸的保守性(某种氨基酸对另一种氨基酸的取代数据),然后,以所有60%保守性的氨基酸模式之间的比较数据为根据,产生BLOSUM60;以所有80%保守性的氨基酸模式之间的比较数据为根据,产生。
生物信息学习题(2010-7)

生物信息学练习题(2009-2010学年第2学期)姓名:性别:班级:学号:说明:(1)此作业主要是让大家熟悉一下生物信息学的基本知识点,并真正练习一下生物信息软件的使用。
(2)此作业将作为我们的成绩,不交者将没有成绩,请认真对待;(3)作业统一用A4纸打印,并装订;(4)在7月10日前,各班学委收起后,交到新生化大楼C615房间;(5)如有问题可与我联系,一.问答题:1. 当今世界上主要的三大生物数据库是指哪些数据库?答:当今世界上主要的三大生物数据库是美国国家生物技术信息中心NCBI(National Center for Biotechnology Information),EBI(European Bioinformatics Institute)欧洲生物信息研究所,DDBJ(DNA Data Bank of Japan)日本核酸数据库2. 人类基因组计划的完成将绘制出“四张图“,请问这四张图是指哪些图?答:人类基因组计划的完成将绘制出“四张图“是指:1遗传图谱,又称连锁图谱(linkage map),它是以具有遗传多态性(在一个遗传位点上具有一个以上的等位基因,在群体中的出现频率皆高于1%)的遗传标记为“路标”,以遗传学距离(在减数分裂事件中两个位点之间进行交换、重组的百分率,1%的重组率称为1cM)为图距的基因组。
2物理图谱,是以一段已知核酸序列的片段STS序列为路标,以碱基对数目的多少为图距来表示两个遗传标记之间的物理距离[基本单位是Mb、kb、bp]的图谱。
3序列图谱,是分别将各染色体全部碱基序列绘制的图谱。
包括转录序列和非转录序列。
4转录图谱谱也叫基因表达图谱,以表达序列标签(expressed sequence tag , EST )为位标,反映基因在不同条件下的表达情况的图谱。
3. 生物信息学的定义有狭义与广义之分,请问狭义的生物信息学定义是什么?答:目前生物信息学可以狭义地定义为:将计算机科学和数学应用于生物大分子信息的获取、加工、存储、分类、检索与分析,以达到理解这些生物大分子信息的生物学意义的交叉学科。
生物信息学作业

结论一:这是什么基因1.该基因为人的CD226 抗原分子(CD226),染色体定位18号染色体67624232 -67530192基因标识符:NM_006566.22.功能:细胞粘附功能,整合素结合,蛋白结合,蛋白激酶结合;参与细胞粘合,细胞识别,细胞因子产生,正向调控Fc受体介导的刺激性信号通路,正向调控免疫球蛋白介导的免疫反应,正向调控肥大细胞的活化正向调控NK细胞介导的细胞毒性,正向调控NK细胞介导的针对肿瘤细胞靶标的细胞毒活性,调节免疫反应,信号转导等途径。
结论二:编码的蛋白质序列是怎样的蛋白标识符:"NP_006557.2" 336 aa蛋白序列为:MDYPTLLLAL LHVYRALCEE VLWHTSVPFA ENMSLECVYP SMGILTQVEWFKIGTQQDSI AIFSPTHGMV IRKPYAERVY FLNSTMASNN MTLFFRNASE DDVGYYSCSL YTYPQGTWQK VIQVVQSDSF EAAVPSNSHI VSEPGKNVTL TCQPQMTWPV QAVRWEKIQP RQIDLLTYCN LVHGRNFTSK FPRQIVSNCS HGRWSVIVIP DVTVSDSGLY RCYLQASAGE NETFVMRLTV AEGKTDNQYT LFVAGGTVLL LLFVISITTI IVIFLNRRRR RERRDLFTES WDTQKAPNNY RSPISTSQPT NQSMDDTRED IYVNYPTFSR RPKTRV结论三:有没有功能保守的结构序列?该蛋白有Ig的保守结构序列结论四;:它的功能是?功能:细胞黏附相关受体,淋巴细胞信号转导,CTL和NK介导的细胞毒性和淋巴因子分泌亚单元结构:与PVR和PVRL2相互作用亚细胞定位:细胞膜,Ⅰ类信号传播膜蛋白组织特异性:外周血T细胞表达序列:包含2个Ig-like C2型(免疫球蛋白样)结构域结论五:在真核生物中保守吗?在酵母中不存在其同源物,在一些灵长类动物存在一些同源性较高的序列,在其他的哺乳动物如:褐家鼠,野猪等中也存在一些同源性较高的序列。
信息生物学作业

浅谈基因芯片2009221107100174 09试点基因芯片(microarray),又称DNA芯片或DNA微阵列,是指通过微加工技术和微电子技术,将成千上万与生命相关的探针分子以预先设计好的排列方式同化在固相支持物(硅片、玻片、聚丙烯酞胺凝胶、尼龙膜等载体)的表面,组成密集二维分子排列,通过检测每个探针分子的杂交信号强度进而获取样品分子的数量和序列信息,以达到对样本基因的表达水平、突变和多态性进行快速、并行、准确、高效的检测分析。
它是近几年发展起来的又一新的分子生物学研究工具,被广泛应用于基因的表达和调控、新基因的功能发现、疾病的诊断和预后、药靶的发现、毒理学、微生物检测、农林业生产、食品、环境保护和检测等领域li-3]。
它综合了分子生物学、半导体微电子技术、激光、化学、计算机科学等众多学科领域的相关技术,使其具有高通量、快速、并行化采集生物信息的特点问;同时,大规模、高通量的信息获得也对“海量”数据的分析及信息的提取提出了新的挑战,如基因芯片数据的标准化、样本(或基因)间距离的度量以及样本(或基因)的监督和非监督分类等分析方法,力图将无机的信息数据和有机的生命活动结合起来阐释生命特征及基因功能,已经成为生物信息学的研究课题嘲。
这些探索基因功能的新技术和新方法是目前研究的重点,新的分析工具和方法不断产生。
本文就基因芯片实验数据现有的基础分析方法做一个概括的总结,并介绍一些新的分析方法。
1.基因芯片数据的获取及标准化芯片上的每个点包含2个数值成分:信号值和背景值。
信号值是指芯片数据的真实强度值,这些数值能提供相关基因表达的量化信息;背景值则指那些因不真实的生化反应导致的强度值。
生物芯片通过探针与经荧光标记的目标样品进行生物反应,使用芯片专用检测系统,经荧光共聚焦扫描进行荧光信号的采集,通过图形分析软件产生每个点信号及其背景的数据,最终转化成可计算的数字信息;但标记物的差异、标记效率、空间位置的差异、荧光标记检测效能的差异以及样品RNA的原始浓度的差异等,都可能对基因表达结果产生影响。
生物信息学作业

B7家族成员生物信息学分析作业
T细胞最适活化除了需要TCR传导的第一信号外,还需共刺激分子传导的第二信号。
其中研究最多、最清楚、认为最有意义的共刺激分子是B7-1和B7-2分子,它们与受体CD28分子或CTLA-4分子相互作用,在T细胞生长、分化和死亡中起重要作用。
在寻找B7-1和B7-2的同源分子过程中,最近发现了B7家族的新成员B7H1(B7 homolog 1)、B7H2(B7 homolog 2)、B7H3(B7 homolog 3)和B7H4(B7 homolog 4)分子。
作业要求对B7家族的这几个成员进行了生物信息学分析(主要包括:人源、鼠源B7家族成员基因cDNA序列之间、氨基酸序列之间的同源性比较及发生树分析、蛋白质结构分析、三级结构预测等),并对扩增和检测B7H4基因进行了引物和探针设计。
B7家族人源、鼠源等自己从相关数据库获得的、相关生物信息学分析越多加分。
作业提供了B7家族人源、鼠源的相关序列文件。
数据获得、利用软件分析、思路等需要文字说明和截图说明,不详细的扣分。
作业在开学第一周交,即2月28日之前。
发到jiabinxj@。
生物信息学作业1.doc

生物信息学实验作业试验一一.找到编码拟南芥(arabidopsis)phyA(光敏色素A)基因的核酸序列编号, 并记录查找过程。
GI:224576211步骤1.进入NCBI主页2.搜索arabidopsis phyA3.Arabidopsis thaliana phytochrome A (PHYA) gene, partial cds4.VERSION:GI:224576211二.以phyA为检索词,在pubmed数据库中分别检索在题目和关键词字段中含有该检索词的文献,记录检索出的条目数目。
Results: 614三.仔细阅读所查询核酸序列在NCBI和EMBL数据库中格式的解释,理解各字段的含义,并比较NCBI 与EMBL中序列格式的异同。
实验二一.分析你感兴趣核酸序列的分子质量、碱基组成。
Composition 35 A; 25 C; 35 G; 15 T; 0 OTHERPercentage: 32% A; 23% C; 32% G; 14% T; 0%OTHERMolecular Weight (kDa): ssDNA: 34.26 dsDNA: 67.8二.列出你所分析核酸序列(或部分序列)的互补序列、反向序列、反向互补序列、DNA双链序列和RNA 序列。
R S1 ACTACTCGAG AAGCAGCGAC AGAGGCGTTA GCCCGCTCAG CAGACTGGCA GTTCTCTACC61 GACAAAAAAG AGGTAGGAGG CACAGTAATG ATACAGGCGT AGCAGGAGGGC S1 CCCTCCTGCT ACGCCTGTAT CATTACTGTG CCTCCTACCT CTTTTTTGTC GGTAGAGAAC61 TGCCAGTCTG CTGAGCGGGC TAACGCCTCT GTCGCTGCTT CTCGAGTAGTR C S1 TGATGAGCTC TTCGTCGCTG TCTCCGCAAT CGGGCGAGTC GTCTGACCGT CAAGAGATGG61 CTGTTTTTTC TCCATCCTCC GTGTCATTAC TATGTCCGCA TCGTCCTCCCD DNA S1 GGGAGGACGA TGCGGACATA GTAATGACAC GGAGGATGGA GAAAAAACAG CCATCTCTTGCCCTCCTGCT ACGCCTGTAT CATTACTGTG CCTCCTACCT CTTTTTTGTC GGTAGAGAAC61 ACGGTCAGAC GACTCGCCCG ATTGCGGAGA CAGCGACGAA GAGCTCATCATGCCAGTCTG CTGAGCGGGC TAACGCCTCT GTCGCTGCTT CTCGAGTAGTRNA S1 GGGAGGACGA UGCGGACAUA GUAAUGACAC GGAGGAUGGA GAAAAAACAG CCAUCUCUUG61 ACGGUCAGAC GACUCGCCCG AUUGCGGAGA CAGCGACGAA GAGCUCAUCA三.列出核酸序列的限制性酶切位点分析结果(酶及识别位点)。
生物信息学习题

GTATCACACG ACTCAGCGCA GCATTTGCCC
GTATCACATA GCTCAGCGCA GCATTTGCCC
6、对于下列距离矩阵,用 UPGMA 构建系统发生树。
ABCDE
A0
B3 0
C6 5 0
D 9 9 10 0
E 12 11 13 9 0 7、对下面距离矩阵,用 UPGMA 法构建系统发生树
1、蛋白质得分矩阵类型有 、
、、
和
等。
2、对位排列主要有局部比对和 三、运算题 1、画出下面两条序列的简单点阵图。将第一条序列放在 x 坐标轴上,将第二条序列放在 y
坐标轴上。 TGAACTCCCTCAGATATTA CGAACCCTCACATATTAGCG
2、对两个核酸序列 ACACACTA 和 AGCACACA 进行全局比对
第八章 后基因组时代的生物信息学(问题与练习)
1、 比较生物还原论与生物综合论的异同 2、 简述“后基因组生物信息学”的基本研究思路 3、 后基因组生物信息学的主要挑战是什么? 4、 功能基因组系统学的基本特征是什么? 5、 说明后基因组生物信息学对信息流动的最新理解 6、 列举几种预测蛋白质-蛋白质相互作用的理论方法 7、 解释从基因表达水平关联预测蛋白质-蛋白质相互作用的理论方法 8、 解释基因保守近邻法预测蛋白质-蛋白质相互作用的理论方法 9、 解释基因融合法预测蛋白质-蛋白质相互作用的理论方法 10、解释种系轮廓发生法预测蛋白质-蛋白质相互作用的理论方法
正确的树的可能性比前一种情况大还是小?
5、对于下列 5 条序列的比对构造一个距离矩阵,其中序列之间的距离值为比对中失配的碱
基数目,但是颠换的权值为转换的两倍。
GTGCTGCACG GCTCAGTATA GCATTTACCC
生物信息学题库.doc

生物信息学题库一、名词解释1、生物信息学:生物分子信息的获取、存贮、分析和利用;以数学为基础,应用计算机技术,研究生物学数据的科学。
2、相似性(similarity):相似性是指序列比对过程中用来描述检测序列和目标序列之间相同DNA碱基或氨基酸残基顺序所占比例的高低。
3、同源性(homology):生物进化过程中源于同一祖先的分支之间的关系。
4、BLAST(Basic Local Alignment Search Tool):基本局部比对搜索工具,用于相似性搜索的工具,对需要进行检索的序列与数据库中的每个序列做相似性比较。
5、HMM隐马尔可夫模型:是蛋白质结构域家族序列的一种严格的统计模型,包括序列的匹配,插入和缺失状态,并根据每种状态的概率分布和状态间的相互转换来生成蛋白质序列。
6、一级数据库:一级数据库中的数据直接来源于实验获得的原始数据,只经过简单的归类整理和注释(投稿文章首先要将核苷酸序列或蛋白质序列提交到相应的数据库中)7、二级数据库:对原始生物分子数据进行整理、分类的结果,是在一级数据库、实验数据和理论分析的基础上针对特定的应用目标而建立的。
8、GenBank: 是具有目录和生物学注释的核酸序列综合公共数据库,由NCBI构建和维护。
9、EMBL:EMBL 实验室——欧洲分子生物学实验室,EMBL 数据库——是非盈利性学术组织 EMBL 建立的综合性数据库,EMBL 核酸数据库是欧洲最重要的核酸序列数据库,它定期地与美国的 GenBank、日本的 DDBJ 数据库中的数据进行交换,并同步更新。
10、DDBJ: 日本核酸序列数据库,是亚洲唯一的核酸序列数据库。
11、Entrez:是由 NCBI 主持的一个数据库检索系统,它包括核酸,蛋白以及Medline 文摘数据库,在这三个数据库中建立了非常完善的联系。
12、SRS(sequence retrieval system):序列查询系统,是 EBI 提供的多数据库查询工具之一。
生物信息学作业题

生物信息学作业题生物信息学作业题绪论1.什么是生物信息学?2.生物信息学有哪些主要研究领域?第一章生物信息学的分子生物学基础1.DNA的双螺旋结构要点是什么?2.什么是基因组和蛋白质组?对它们的研究有何意义?第二章生物信息学的计算机基础1.简述网络操作系统的类型。
第三章核酸序列分析1.什么是全局比对?2.什么是局部比对?有哪些优点?第四章分子进化分析1.分子进化分析具有哪些优点?2. 简述分子进化的中性学说。
第五章基因组分析1. 什么是基因组学?其主要研究内容是什么?2.简述基因预测分析的一般步骤。
第六章蛋白质组分析1. 蛋白质组学的概念和主要研究的大致方向是什么?2. 蛋白质组功能预测的程序是怎样的?第七章生物芯片数据分析1. 什么是生物芯片?2. 生物芯片有哪些方面的应用?第八章核酸与蛋白质结构预测1. RNA二级结构典型的预测方法有哪些?2. 基于统计学的预测蛋白质二级结构的方法有哪些?第九章生物信息学平台与工具软件1. 请利用Clustal X软件对下列6条蛋白质序列进行多重比对(比对结果用BioEdit软件打开,用“截图”方式显示比对结果)。
>1mqngkvkwfn sekgfgfiev eggedvfvhf saiqgegfkt leegqevtfe veqgnrgpqatnvnkk>2mqgkvkwfnn ekgfgfieie gaddvfvhfs aiqgegykal eegqevsfdi tegnrgpqaanvvkl>3mqngkvkwfn sekgfgfiev eggedvfvhf saiqgegfkt leegqevtfe veqgnrgpqatnvnkk>4mqgkvkwfnn ekgfgfieie gaddvfvhfs aiqgegykal eegqevsfdi tegnrgpqaanvvkl>5mqngkvkwfn sekgfgfiev eggedvfvhf saiqgegfkt leegqevtfe veqgnrgpqatnvnkk>6mqgkvkwfnn ekgfgfieie gaddvfvhfs aiqgegykal eegqevsfdi tegnrgpqaanvvkl2. 现有一ZmPti1b蛋白质序列,请用DNAMAN软件分析其二级结构,给出分析结果。
《生物信息学》大作业参考模板-2016

《生物信息学》大作业 参考答案
芍药 ACS 基因的生物信息学分析
姓名: 班级: 学号: 2016 年 4 月 11 日
一、芍药 ACS 基因序列及其编码的蛋白的功能 乙烯是存在于植物体内的唯一的一种气态植物激素,调控植物花、果和叶片的衰老进程。乙烯的合成 主要在转录水平上受到 ACS(ACC synthase,ACC 合酶)和 ACC 氧化酶的调控,ACS 将 SAM(S-adenosyl
a r t i c l e i n f o
Article history: Received 17 November 2015 Accepted 23 November 2015 Available online 26 November 2015 Keywords: ACC synthase Ethylene biosynthesis Flower senescence Oncidium Gower Ramsey Gene cloning Expression analysis
Biochemical and Biophysical Research Communications 469 (2016) 20vailable at ScienceDirect
Biochemical and Biophysical Research Communications
图 1 芍药 ACS 基因的核苷酸序列及其编码的氨基酸序列 下载的论文“Molecular cloning and expression analysis of an 1-aminocyclopropane-1-carboxylate synthase gene from Oncidium Gower Ramsey” 为 2016 年发表于 Biochemical and Biophysical Research Communications 的最新英文文章(见下页) 。 (2 分)
实用生物信息技术第一次作业

实用生物信息技术课程第1 次作业网络书籍、文档和文献资源检索和应用一、分子月报(Molecular of the Month):1) 按功能分类,网站的生物大分子分为哪几大类?1、DNA, RNA, and Protein Synthesis - building the molecules of life2、Enzymes - the cell's chemists3、Molecular Infrastructure - supporting cells, tissues and organisms4、Transport - delivering the cell's resources5、Biological Energy - capturing and converting sources of power6、Molecules and the Environment - proteins with global impact7、Photosynthesis - capturing energy from the sun8、Molecular Motors - directed motion at the molecular level9、Cellular Signaling - sending and receiving molecular message二、2000 年12 篇月报中描述的蛋白质分子,你熟悉的有哪些?Pepsin胃蛋白酶)Nucleosome 核小体DNA PolymeraseMyoglobin 肌红蛋白三、有关DNA、rRNA、tRNA 的月报有哪几篇?1、DNA2、Transfer Ribonucleic Acid (tRNA)3、Transfer-Messenger RNA四、该网站中和DNA 复制相关的蛋白质分子有哪些?1、DNA Helicase 解旋酶2、DNA Polymerase 聚合酶3、Sliding Clamps4、DNA ligase 连接酶五、该网站中介绍的病毒有哪些?HIVAdenovirus腺病毒Ebola Virus Proteins 埃博拉病毒Bacteriophage phiX174 噬菌体病毒Dengue Virus 登革病毒HIV艾滋病毒Poliovirus and Rhinovirus脊髓灰质炎病毒Simian Virus 40Tobacco Mosaic Virus烟草花叶病毒六、该网站中你熟悉的酶分子有哪些?DNA PolymeraseRNA PolymerasecAMP-dependent Protein Kinase (PKA)Acetylcholinesterase乙酰胆碱酯酶Pepsin胃蛋白酶Topoisomerases拓扑异构酶Aminoacyl-tRNA Synthetases氨酰tRNA合成酶DNA Ligase连接酶七、阅读该网站关于血红蛋白的短文,说明其结合和释放氧气时不同亚基之间如何协同作用。
研究生《生物信息学》作业模板

研究生《医学生物信息学》作业班级:专业:姓名:一、实验目的:(1)掌握中文文献全文的检索和获得方法。
(2)掌握Pubmed数据库文献的检索和交大图书馆英文数据库全文的获得方法。
(3)掌握核酸序列搜索的方法。
(4)掌握核酸序列相似性分析的方法。
(5)掌握PCR引物设计软件的原理、使用及特点。
(6)掌握蛋白质序列搜索的方法。
(7)掌握蛋白质序列分析常用软件的使用方法。
二、研究背景:AIB1基因为近年来发现的p160类固醇受体转录共激活因子SRC-1家族成员,是新定义的一个原癌基因[1]。
该基因表达的蛋白在许多生物学过程中发挥重要作用,如细胞生长,增殖,分化,性成熟,女性生殖功能等[2]。
近年发现,该基因的表达异常与多种肿瘤的发生发展有关,以在乳腺癌中研究最多。
AIB1基因的高表达与乳腺癌的发生和发展有关[3]。
AIB1蛋白通过与雌激素受体相互作用,能强烈地增强雌激素受体的促进靶基因转录的效应,进而引起细胞增殖和肿瘤形成,此外,AIB1蛋白还在多条信号传导通路中发挥作用[4]。
AIB1基因(amplified in breast cancer1)又称为ACTR,TRAM1,RAC3,SRC3,NCoA3,P/CIP等。
本人选择其为研究对象。
三、实验方法、步骤及结果:1.在中国知网(CNKI)中查找中文文献:2.在PubMed中查找英文文献:登陆NCBI主页,网址:/guide/,选择gene数据库4. 使用NCBI网站中的BLAST工具进行序列比对登陆/,选择核酸序列比对nucleotide BLAST,界面显示如下,输入登录号,AF012108,点击“BLAST”。
结果如下:共有2条核苷酸序列和2条基因组序列和其匹配:第一条核苷酸序列为“Homo sapiens nuclear receptor coactivator 3 (NCOA3), transcript variant 2, mRNA”,登录号:NM_006534。
生物信息学作业1---开放读码框与预测序列启动子(何婷,学号;1302008)

何婷学号:1302008 专业:病理学与病理生理学
1.使用Entrez信息查询系统检索与自己课题相关的基因核酸序列,预测开放读码框,并使用PromoterScan预测该序列中的启动子。
①查询HPV的核酸序列:输入网址:/,打开NCBI主页,在
检索窗口的选择数据库的下拉菜单选中Nucleotide项,在它右侧的文本输入栏输入检索词“HPV”,再点击“Search”按钮。
如下图所示:
搜索结果,如下图所示:
显示结果,如下图所示:
② 预测开放读码框:输入网址:/gorf/gorf.html ,打开NCBI
的ORF Finder 软件,输入HPV 的核酸序列的GI 号,最后点击“OrfFind ”按钮,如下图所示:
结果如下图所示:
点击“正链+1”,显示结果如下:
③使用PromoterScan预测该序列中的启动子:输入网址为
/molbio/proscan/,打开PromoterScan的在线操作页面,复制粘贴上述的HPV的核酸序列到指定的框中,点击submit按钮提交序列后,注意使用
软件时不需要设置任何参数,如下图所示:
输出结果为:
何婷学号:1302008 专业:病理学与病理生理学。
生物信息学作业
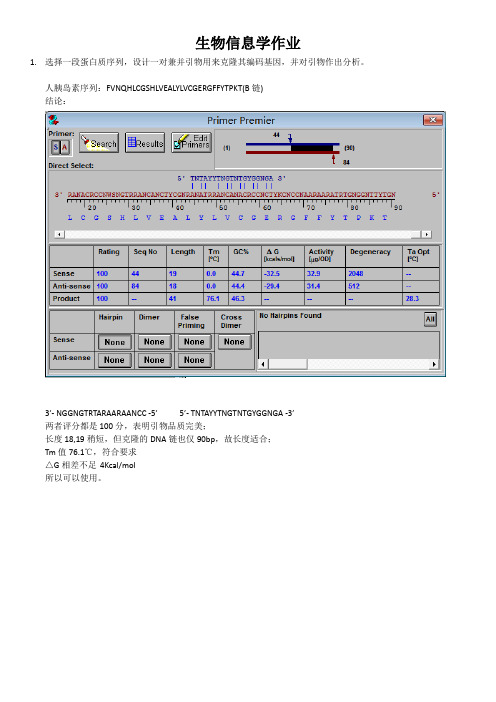
生物信息学作业1.选择一段蛋白质序列,设计一对兼并引物用来克隆其编码基因,并对引物作出分析。
人胰岛素序列:FVNQHLCGSHLVEALYLVCGERGFFYTPKT(B链)结论:3’- NGGNGTRTARAARAANCC -5’ 5’- TNTAYYTNGTNTGYGGNGA -3’两者评分都是100分,表明引物品质完美;长度18,19稍短,但克隆的DNA链也仅90bp,故长度适合;Tm值76.1℃,符合要求△G相差不足4Kcal/mol所以可以使用。
Step1:打开primer premier 5.0 输入蛋白质链,转化为DNA链。
获得DNA链。
2.选择一段基因,预测期编码RNA的二级结构,并分析功能。
取一段基因:ACGCG GGCGG GCATG TGGGC AGCTT TACCC AGTGC TACTG TGCTG GCCAGCACTG AAACA GGGGC ACTGG TTTGG GGTGG ATGAA GGGTA GAAGT GCAAGTTCCA TTGCC TGTGC AATCC CTGCC TTGCT CAGAC CCTGC TCACT CCTCAGGCCC CATCA GCCCC TCAAC TCTGC TAACC ATGGT GGTAG AAATC AGCTACAATA AACCC TGGAG CCAGT AAAAA AAAAA AAAAA AAAAA AAAAA AAAGT点击Fold as RNA点击START点击Draw Stuclture得到RNA二级结构RNA功能预测打开网址http://sidirect2.rnai.jp/输入DNA序列得出结论:。
《生物信息学》课程期末作业

山东大学生命科学学院2012~2013学年第一学期期末考试试卷(研究实践型)考试科目: 《生物信息学》适用类别: 本科院系:生命科学学院专业:年级:2010级姓名:kengnidiancom学号:第1页,共22页考试说明和要求1.试卷内容布局包括七部分:目录、引言、实践资源(使用的软件和数据库)、实践方法、实践结果和讨论、参考文献、心得与致谢。
(要求使用此论文模板创建规范统一答卷,详见模板使用说明,请从此模板第3页开始答卷;参考文献要求借助专业软件按《微生物学报》样式统一进行参考文献格式化;请于12月23日前提交电子版(发至邮箱:lzf-204@)和请于12月25-26日提交纸质版(微生物楼北楼玻璃房377室)答卷;团队讨论或受他人帮助请在致谢中说明体现)2.实践素材:完整目的基因groel被克隆入表达载体PET-32a的Nde I与Hind III酶切位点切点中,得到重组克隆PET-32a-groel,利用载体通用引物“T7 promoter”和“T7 terminator primer #69337-3”对重组克隆进行测序,得到序列采集结果“PET-32a-groel__T7.ab1”和“PET-32a-groel__T7ter.scf”。
3.实践要求:(实践方法和结果部分请提供说明问题的关键截图)●对序列采集结果“PET-32a-groel__T7.ab1”和“PET-32a-groel__T7ter.scf”进行基本处理,得到无污染的完整目的基因groel,结果中展示最终的contig装配截图;●设计合适的引物,实现将完整目的基因groel克隆插入表达载体PET-32a的Nde I与HindIII酶切位点的切点中,结果中展示引物参数信息及待送公司合成的引物序列订单;●参考“pET-32a”图谱及其序列文件,绘制(用作测序模板的)重组克隆PET-32a-groel的载体图谱,生成重组载体图谱PET-32a-groel.vec并在结果中展示导出的PET-32a-groel 图谱;●参考“groel information.txt”信息,将得到的完整目的基因groel模拟提交数据库,生成groel.sqn文件并在结果中展示完整的内容信息。
生物信息学试题及答案

生物信息学试题及答案一、单项选择题(每题2分,共20分)1. 生物信息学的主要研究对象是()。
A. 生物数据B. 生物实验C. 生物模型D. 生物技术答案:A2. 下列哪项不是生物信息学中的常用数据库()。
A. GenBankB. Swiss-ProtC. PubMedD. Google Scholar答案:D3. 蛋白质序列比对的主要目的是()。
A. 确定蛋白质的三维结构B. 预测蛋白质的功能C. 比较蛋白质的氨基酸序列D. 计算蛋白质的分子量答案:B4. 在生物信息学中,以下哪种算法不是用于序列比对的()。
A. BLASTB. FASTAC. Smith-WatermanD. Hidden Markov Model答案:D5. 下列哪种生物信息学工具主要用于基因表达分析()。
A. ClustalWB. Primer3C. R语言D. PDB答案:C6. 以下哪种技术不是用于蛋白质结构预测的()。
A. 同源建模B. 从头预测C. 序列比对D. 折叠识别答案:C7. 以下哪种生物信息学工具主要用于基因组注释()。
A. BLASTC. GATKD. Primer3答案:B8. 在生物信息学中,以下哪种方法不用于基因表达数据的聚类分析()。
A. K-meansB. Hierarchical clusteringC. Principal component analysisD. Multiple sequence alignment答案:D9. 下列哪种生物信息学工具主要用于蛋白质-蛋白质相互作用网络分析()。
A. STRINGB. BLASTD. Primer3答案:A10. 在生物信息学中,以下哪种数据库不包含蛋白质结构信息()。
A. PDBB. UniProtC. RCSBD. GenBank答案:D二、多项选择题(每题3分,共15分)11. 生物信息学中常用的序列比对工具包括()。
A. BLASTB. FASTAC. ClustalWD. Pfam答案:ABC12. 以下哪些是生物信息学中常用的基因表达分析软件()。
《生物信息学》练习题及答案

《生物信息学》练习题及答案1、在Genbank中查找以下6个植物蛋白序列:protein1:NP_974673.2;protein2:NP_187969.1;protein3: NP_190855.1;protein4:NP_565618.1;protein5: NP_200511.1;protein6:NP_191407.1(以FASTA格式)。
(1)用EBI上的ClustalW2工具对其进行多序列比对,分析各蛋白序列之间的同源性。
序列比对结果比对结果表明:protein1:NP_974673.2和protein4: NP_565618.1的亲缘关系最近。
(2)利用Phylip软件,选择距离法构建其进化树(要求写出具体的建树步骤)。
1.将蛋白序列保存为FASTA格式,存于txt文档;2.用Clustalx打开txt文本,保存为*.phy文件;3.用seqboot程序打开phy文件,输出结果文件*_seqboot4.用protdist程序打开*_seqboot文件,输出为*_protdist文件5.用neighbor程序打开*_protdist文件,输出为*_neighbor 文件6.用consense程序打开*_neighbor文件,输出为*_consense 文件7.用dratree程序打开*_consense文件得到进化树。
(注:由于seqboot软见无法正常运行,因此进化树无法显示)(3)任意选取其中的一个蛋白进行蛋白质一级序列分析、二级结构预测及三维结构的模拟。
选择protein3:NP_190855.1一级结构网址:/doc/479b86d06edb6f1afe001f6e.html /tools/protparam.htmlNumber of amino acids:456氨基酸数目Molecular weight:51154.5相对分子质量Theoretical pI:8.69理论pI值Amino acid composition氨基酸组成Ala(A)306.6%Arg(R)286.1%Asn(N)153.3%Asp(D)275.9%Cys(C)51.1%Gln(Q)183.9%Glu(E)286.1%Gly(G)378.1%His(H)163.5%Ile(I)163.5%Leu(L)429.2%Lys(K)327.0%Met(M)51.1%Phe(F)173.7%Pro(P)163.5%Ser(S)4610.1%Thr(T)214.6%Trp(W)81.8%Tyr(Y)194.2%Val(V)306.6%Pyl(O)00.0%Sec(U)00.0%(B)00.0%(Z)00.0%(X)00.0%正/负电荷残基数Total number of negatively charged residues(Asp+Glu): 55Total number of positively charged residues(Arg+Lys): 60Atomic composition:原子组成Carbon C2270Hydrogen H3531Nitrogen N645Oxygen O686Sulfur S10Formula:C2270H3531N645O686S10分子式Total number of atoms:7142总原子数Extinction coefficients:消光系数Extinction coefficients are in units of M-1cm-1,at280 nm measured in water.Ext.coefficient72560Abs0.1%(=1g/l)1.418,assuming all pairs of Cys residues form cystines Ext.coefficient72310Abs0.1%(=1g/l) 1.414,assuming all Cys residues are reducedEstimated half-life:半衰期The N-terminal of the sequence considered is M(Met). The estimated half-life is:30hours(mammalian reticulocytes,in vitro).>20hours(yeast,in vivo).>10hours(Escherichia coli,in vivo).Instability index:不稳定系数The instability index(II)is computed to be48.99This classifies the protein as unstable.Aliphatic index:75.26脂肪系数Grand average of hydropathicity(GRAVY):-0.554总平均亲水性蛋白质亲疏水性分析所用氨基酸标度信息Ala:1.800Arg:-4.500Asn:-3.500Asp:-3.500Cys:2.500 Gln:-3.500Glu:-3.500Gly:-0.400His:-3.200Ile:4.500 Leu:3.800Lys:-3.900Met:1.900Phe:2.800Pro:-1.600 Ser:-0.800Thr:-0.700Trp:-0.900Tyr:-1.300Val: 4.200:-3.500:-3.500:-0.490分析所用参数信息Weights for window positions1,..,9,using linear weight variation model:1234567891.001.001.001.001.001.001.001.001.00edge center edge跨膜结构预测结果(没有跨膜结构)信号肽分析:二级结构预测三级结构预测网站/doc/479b86d06edb6f1afe001f6e.html/~phyre2、在拟南芥基因组数据库中(/doc/479b86d06edb6f1afe001f6e.ht ml/)查找编号分别为At4G33050,At3G13600,At3G52870或At2G26190基因,针对所查找的基因进行初步的生物信息学分析(每人任选其中一个基因)。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
生物信息学实验作业试验一一.找到编码拟南芥(arabidopsis)phyA(光敏色素A)基因的核酸序列编号, 并记录查找过程。
GI:224576211步骤1.进入NCBI主页2.搜索arabidopsis phyA3.Arabidopsis thaliana phytochrome A (PHYA) gene, partial cds4.VERSION:GI:224576211二.以phyA为检索词,在pubmed数据库中分别检索在题目和关键词字段中含有该检索词的文献,记录检索出的条目数目。
Results: 614三.仔细阅读所查询核酸序列在NCBI和EMBL数据库中格式的解释,理解各字段的含义,并比较NCBI 与EMBL中序列格式的异同。
实验二一.分析你感兴趣核酸序列的分子质量、碱基组成。
Composition 35 A; 25 C; 35 G; 15 T; 0 OTHERPercentage: 32% A; 23% C; 32% G; 14% T; 0%OTHERMolecular Weight (kDa): ssDNA: 34.26 dsDNA: 67.8二.列出你所分析核酸序列(或部分序列)的互补序列、反向序列、反向互补序列、DNA双链序列和RNA 序列。
R S1 ACTACTCGAG AAGCAGCGAC AGAGGCGTTA GCCCGCTCAG CAGACTGGCA GTTCTCTACC61 GACAAAAAAG AGGTAGGAGG CACAGTAATG ATACAGGCGT AGCAGGAGGGC S1 CCCTCCTGCT ACGCCTGTAT CATTACTGTG CCTCCTACCT CTTTTTTGTC GGTAGAGAAC61 TGCCAGTCTG CTGAGCGGGC TAACGCCTCT GTCGCTGCTT CTCGAGTAGTR C S1 TGATGAGCTC TTCGTCGCTG TCTCCGCAAT CGGGCGAGTC GTCTGACCGT CAAGAGATGG61 CTGTTTTTTC TCCATCCTCC GTGTCATTAC TATGTCCGCA TCGTCCTCCCD DNA S1 GGGAGGACGA TGCGGACATA GTAATGACAC GGAGGATGGA GAAAAAACAG CCATCTCTTGCCCTCCTGCT ACGCCTGTAT CATTACTGTG CCTCCTACCT CTTTTTTGTC GGTAGAGAAC61 ACGGTCAGAC GACTCGCCCG ATTGCGGAGA CAGCGACGAA GAGCTCATCATGCCAGTCTG CTGAGCGGGC TAACGCCTCT GTCGCTGCTT CTCGAGTAGTRNA S1 GGGAGGACGA UGCGGACAUA GUAAUGACAC GGAGGAUGGA GAAAAAACAG CCAUCUCUUG61 ACGGUCAGAC GACUCGCCCG AUUGCGGAGA CAGCGACGAA GAGCUCAUCA三.列出核酸序列的限制性酶切位点分析结果(酶及识别位点)。
Restriction analysis on USMethylation: dam-No dcm-NoScreened with 117 enzymes, 5 sites foundEcl136II 1 GAG/CTC103EcoICRI 1 GAG/CTC103SacI 1 GAGCT/C105SapI 1 GCTCTTCN/93SstI 1 GAGCT/C105List by Site Order93 SapI 103 Ecl136II 105 SstI 105 SacI103 EcoICRINon Cut EnzymesAatII Acc65I AccIII AclI AflII AgeIAhaIII Alw44I AlwNI ApaBI ApaI ApaLIAscI Asp718I AsuII AvrII BalI BamHIBbeI BbvII BclI BglI BglII Bpu1102IBsc91I BsiI BsmI Bsp1407I BspHI BspMIBspMII BssHII BstD102I BstEII BstXI Bsu36IClaI Csp45I CspI CvnI DraI DraIIIDrdI EagI Eam1105I Eco31I Eco47III Eco52IEco56I Eco57I Eco72I EcoNI EcoRI EcoRVEheI EspI FseI HindIII HpaI I-PpoIKpnI MfeI Mlu113I MluI MscI MstIMstII NaeI NarI NcoI NdeI NheINotI NruI NsiI PacI PflMI PinAIPmaCI PmeI PstI PvuI PvuII RleAISacII SalI SauI ScaI SciI SfiISgrAI SmaI SnaBI SpeI SphI SplISpoI SrfI SspI SstII StuI SunISwaI Tth111I VspI XbaI XcmI XhoIXmaI XmaIII XmnI XorIIRestriction sites on US1 GGGAGGACGATGCGGACATAGTAATGACACGGAGGATGGAGAAAAAACAGCCATCTCTTGSacISstIEcl136IISapI EcoICRI61 ACGGTCAGACGACTCGCCCGATTGCGGAGACAGCGACGAAGAGCTCATCA四.分析一对你所设计的引物,并对其进行综合评判。
2 GGAGGACGATGCGGACATAOligo: 5'-GGAGGACGATGCGGACATA-3'Primer1: 19 basesComposition 6 A; 3 C; 8 G; 2 T; 0 OTHERPercentage: 31% A; 15% C; 42% G; 10% T; 0%OTHERMW=5.99 kDaHybridization: D:DSalt: 50 mMFormamide: 0%Mismatch: 0 bpThermo Tm = 62.0 Hybridization Tm = 52.1 GC+AT Tm = 60.0 Primer-US(1-110) complementarity.First complementarity in continuous: 19 bp5'-GGAGGACGATGCGGACATA-3' Primer|||||||||||||||||||3'-CCTCCTGCTACGCCTGTAT-5' (20) Strand -No second possible complementarityMax complementarity in discontinuous: 19 bp5'-GGAGGACGATGCGGACATA-3' Primer|||||||||||||||||||3'-CCTCCTGCTACGCCTGTAT-5' (20) Strand -105 AGCTCTTCGTCGCTGTCTCCOligo: 5'-AGCTCTTCGTCGCTGTCTCC-3'Primer1: 20 basesComposition 1 A; 8 C; 4 G; 7 T; 0 OTHER Percentage: 5% A; 40% C; 20% G; 35% T; 0%OTHER MW=6.07 kDaHybridization: D:DSalt: 50 mMFormamide: 0%Mismatch: 0 bpThermo Tm = 62.2 Hybridization Tm = 54.5 GC+AT Tm = 64.0 Primer-US(1-110) complementarity.First complementarity in continuous: 20 bp5'-AGCTCTTCGTCGCTGTCTCC-3' Primer||||||||||||||||||||3'-TCGAGAAGCAGCGACAGAGG-5' (86) Strand +No second possible complementarityMax complementarity in discontinuous: 20 bp5'-AGCTCTTCGTCGCTGTCTCC-3' Primer||||||||||||||||||||3'-TCGAGAAGCAGCGACAGAGG-5' (86) Strand +五.运用Sequin软件进行序列提交,并打印你完成的序列提交文件(后缀为.sqn)。
LOCUS GY482612 110 bp mRNA linear UNA 17-FEB-2002 DEFINITION Sequence 33 from patent US 8030290.ACCESSION GY482612VERSION GY482612.1 GI:353292184KEYWORDS .SOURCE unidentifiedORGANISM unidentifiedunclassified sequences.REFERENCE 1 (bases 1 to 110)AUTHORS chen,h.TITLE Sequence 33 from patent US 8030290JOURNAL UnpublishedREFERENCE 2 (bases 1 to 110)AUTHORS chen,h.TITLE Direct SubmissionJOURNAL Submitted (17-FEB-2002) SCAU, Bio, yucheng, yanan, sichuan, Chinai FEATURES Location/Qualifierssource 1..110/organism="unidentified"/mol_type="mRNA"/db_xref="taxon:32644"BASE COUNT 35 a 25 c 35 g 15 tORIGIN1 gggaggacga tgcggacata gtaatgacac ggaggatgga gaaaaaacag ccatctcttg61 acggtcagac gactcgcccg attgcggaga cagcgacgaa gagctcatca//。